

Abstract
A series of transition metal chloro complexes with the tetradentate tripodal tris(2-amino-oxazoline) ligand TAO have been synthesized and characterized. X-ray structural analyses of these compounds demonstrate the formation of the mononuclear complexes [MII(TAO)(Cl)]+, where MII = Cr, Mn, Fe, Co, Ni, Cu and Zn. These complexes exhibit distorted trigonal-bipyramidal geometry, coordinating the metal through an apical tertiary amine, three equatorial imino nitrogen atoms, and an axial chloride anion. All the complexes possess an intramolecular hydrogen-bonding (H-bonding) network within the cavity occupied by the metal-bound chloride ion. The metal-chloride bond distances are atypically long, which is attributed to the effects of the H-bonding network. Nuclear magnetic resonance (NMR) spectroscopy of the Zn complex suggests that the solid-state structures are representative of that observed in solution, and that the H-bonding interactions persist as well. Additionally, density functional theory (DFT) calculations were carried out to probe the electronic structures of the complexes.
Introduction
The function of metal complexes is governed by the combined effects of the primary and secondary coordination spheres. The primary sphere almost exclusively utilizes covalent bonds between metal ions and ligands, whereas the secondary sphere is often controlled with non-covalent interactions. Coordinating these effects within synthetic metal complexes is challenging, in part, because of the difficulties associated with regulating the secondary coordination sphere.1 Information emerging from structural biology shows that active sites within metalloproteins utilize a number of non-covalent interactions, particularly hydrogen bonds (H-bonds) that often form extensive networks around the coordinated metal ion(s).2 Their effects serve a variety of purposes, including tuning redox potentials, orienting external substrates, and providing a means for proton transport.2,3
In order to understand how H-bonds can manipulate the properties of metal complexes, our group1,4 and others5 have designed multidentate ligands containing rigid organic frameworks that are capable of forming intramolecular H-bonds. These ligands produce complexes having open primary coordination site(s) that can bind external species via covalent bonds to the metal center and H-bonds with groups positioned within the secondary coordination sphere.
We recently introduced the tetradentate ligand tris(2-amino-oxazoline)amine ligand (TAO, Figure 1).6 We found that TAO exists in solution in tautomeric equilibrium between the 2-amino-oxazoline and 2-imino-oxazolidine tautomers, yet preferentially binds metal ions as the imino form. This tautomerization establishes a dynamic cavity capable of accommodating an external ligand that binds to the metal ion and is involved in an intramolecular H-bonding network with the three oxazolidine groups. A relatively rigid complex is thus formed by binding an H-bond acceptor within the cavity. In this report we describe the synthesis and structure of a series of trigonal bipyramidal transition metal complexes of TAO featuring a chloride anion as the fifth ligand. These findings emphasize the versatility of the ligand TAO to accommodate a variety of metal ions and the ability of those complexes to stabilize exogenous ligands through non-covalent interactions. Quantum chemical calculations on the [MIITAO(Cl)]+ series also provide some insight into the electronic structures of these coordination complexes.
Fig. 1.
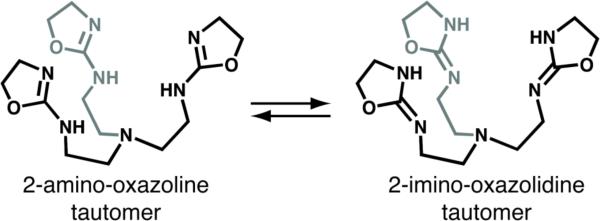
Possible tautomerization of tris(2-amino-oxazoline) ligand, TAO
General Methods
All reagents were purchased from commercial sources and used as received, unless otherwise noted. Solvents were sparged with argon and dried over columns containing Q-5 and molecular sieves. The synthesis of TAO was carried out according to literature procedures.6 The syntheses of metal complexes were conducted in a Vacuum Atmospheres, Co. drybox under an argon atmosphere.
Synthesis of the Complexes
[CrIITAO(Cl)]BPh4
To a solution of TAO (130 mg, 0.366 mmol) dissolved in 4 mL MeOH was added anhydrous CrCl2 (44.3 mg, 0.360 mmol). The deep blue solution stirred for 1 h, after which NaBPh4 (127 mg, 0.370 mmol) in 2 mL MeOH was added, forming a pale blue precipitate. After stirring for 1 h, the mixture was filtered and the solid washed with MeOH and Et2O, then dried in vacuo to yield 221 mg (81%) of a light blue powder. The product was dissolved in THF and filtered into a vial. The volume of the filtrate was reduced in vacuo to 3 mL and placed in an Et2O diffusion chamber, affording X-ray quality violet-blue crystals. Anal. calcd. for C39H47BClCrN7O3: C 61.63, H 6.23, N 12.90. Found: C 61.19, H 6.24, N 12.59%. FTIR (KBr, cm-1): ν(NH) 3372, 3353, 3280, ν(CN) 1687 and 1656 (imino, s). UV/Vis (THF): λmax/nm (ε, M-1 cm-1): 295 (1300), 415 (25), 675 (sh), 935 (42). μeff = 4.72 μB.
[MnIITAO(Cl)]BPh4
A synthetic procedure similar to that for [Cr(TAO)Cl]BPh4 was followed, using TAO (250 mg, 0.708 mmol), anhydrous MnCl2 (94.7 mg, 0.753 mmol), and NaBPh4 (250 mg, 0.731 mmol). The reaction yielded 435 mg (81 %) of a white powder. X-ray quality crystals were obtained from a DCM/pentane diffusion mixture. Anal. calcd. for C39H47BClMnN7O3: C 61.82, H 6.41, N 12.91. Found: C 61.39, H 6.21, N 12.85%. FTIR (KBr, cm-1): ν(NH) 3364, 3339, 3304, 3263, ν(CN) 1684 and 1653 (imino, s). μeff = 5.80 μB.
[FeIITAO(Cl)]BPh4
A synthetic procedure similar to that for [Cr(TAO)Cl]BPh4 was followed, using TAO (50 mg, 0.140 mmol), anhydrous FeCl2 (17.9 mg, 0.141 mmol), and NaBPh4 (48.4 mg, 0.141 mmol). The reaction yielded 75 mg (69%) of a fine white powder. X-ray quality crystals were obtained from a DCM/pentane diffusion mixture. Anal. calcd. for C39H47BClFeN7O3·0.25C5H12: C 61.82, H 6.44, N 12.54. Found: C 62.00, H 6.23, N 12.63%. FTIR (KBr, cm-1): ν(NH) 3383, 3359, ν(CN) 1685 (imino, s). μeff = 5.08 μB.
[CoIITAO(Cl)]BPh4
A synthetic procedure similar to that for [Cr(TAO)Cl]BPh4 was followed, using TAO (50 mg, 0.140 mmol), anhydrous CoCl2 (18.3 mg, 0.141 mmol), and NaBPh4 (48.4 mg, 0.141 mmol). The reaction yielded 72 mg (66%) of a fine violet powder. X-ray quality crystals were obtained from a DCM/pentane diffusion mixture. Anal. calcd. for C39H47BClCoN7O3·0.25CH2Cl2: C 59.80, H 6.07, N 12.44. Found: C 59.62, H 6.03, N 12.35%. FTIR (KBr, cm-1): ν(NH) 3325 and 3223, ν(CN) 1672 and 1643 (imino, s). UV/Vis: 489(sh), 534(76), 750(16). μeff = 3.97 μB.
[NiIITAO(Cl)]BPh4
A synthetic procedure similar to that for [Cr(TAO)Cl]BPh4 was followed, using TAO (120 mg, 0.340 mmol), NiCl2·6H2O (72.5 mg, 0.358 mmol), and NaBPh4 (122 mg, 0.357 mmol). The reaction yielded 195 mg (75 %) of a green powder. X-ray quality crystals were obtained from a DCM/pentane diffusion mixture. Anal. Calcd. for C39H47BClN7NiO3: C 61.09, H 6.18, N 12.79. Found: C 61.17, H 6.26, N 12.69. FTIR (KBr, cm-1): ν(NH) 3361 and 3291, ν(CN) 1688 and 1653 (imino, s). UV/Vis (DCM, 298K): λmax/nm (ε, M-1cm-1): 423 (75), 485 (40), 685 (45), 982 (20). μeff = 2.68 μB.
[CuIITAO(Cl)]BPh4
A synthetic procedure similar to that for [Cr(TAO)Cl]BPh4 was followed, using TAO (143 mg, 0.405 mmol), anhydrous CuCl2 (53.5 mg, 0.398 mmol), and NaBPh4 (140 mg, 0.409 mmol). The reaction yielded 264 mg (86%) of a yellow-green powder. X-ray quality crystals were obtained from a DCM/pentane diffusion mixture. Anal. calcd. for C39H47BClCuN7O3: C 60.70, H 6.14, N 12.71. Found: C 60.54, H 6.26, N 12.76%. FTIR (KBr, cm-1): ν(NH) 3357, 3347, 3272, ν(CN) 1689 and 1656 (imino, s). EPR (X-band, DCM, 4K): g = 2.09, g = 2.24. UV/Vis (DCM): λmax/nm(ε, M-1cm-1): 275 (5000), 286 (4200), 349 (sh), 784 (60), 1000 (120). μeff = 1.75 μB.
[ZnIITAO(Cl)]BPh4
A synthetic procedure similar to that for [Cr(TAO)Cl]BPh4 was followed, using TAO (139 mg, 0.393 mmol), anhydrous ZnCl2 (50.2 mg, 0.368 mmol), and NaBPh4 (140 mg, 0.410 mmol). The reaction yielded 250 mg (88%) of a white powder. X-ray quality crystals were obtained from a DCM/pentane diffusion mixture. Anal. calcd. for C39H47BClN7O3Zn: C 60.56, H 6.12, N 12.68. Found: C 60.85, H 6.19, N 12.76%. FTIR (KBr, cm-1): ν(NH) 3338, 3323, 3240, ν(CN) 1686 and 1655 (imino, s). 1H NMR (500 MHz, DMSO-d6) δH (ppm) [Zn(TAO)Cl]+ 7.94 (0.5H, s, NH), 7.60 (2.5H, s, NH), 4.49 (6H, t, J = 7.9 Hz, OCH2CH2N), 3.62 (6H, t, J = 7.9 Hz, OCH2CH2N), 3.26 (6H, t, J = 5.6 Hz, NapCH2CH2NCox) 2.57 (6H, t, J = 5.6 Hz, NapCH2CH2NCox); BPh4- 7.17 (8H, br t, o-Ar), 6.92 (8H, t, J = 7.4 Hz, m-Ar), and 6.78 (4H, t, J = 7.2 Hz, p-Ar); 13C NMR (500 MHz, DMSO-d6) δC (ppm) [Zn(TAO)Cl]+ 162.12 (N=Cox), 67.89 (OCH2CH2N), 50.89 (NCH2CH2NCox), 42.08 (NCH2CH2NCox), 41.59 (OCH2CH2N); BPh4- 163.35 (B-C1, JB-C = 195 Hz), 135.53 (C2), 125.28 (C3), and 121.51 (C4).
Physical Methods
Elemental analyses were performed on a Perkin-Elmer 2400 CHNS analyzer. Electronic absorbance spectra were recorded with a Cary 50 spectrophotometer using a 1.00 mm or 1.00 cm quartz cuvette. Fourier transform infrared (FTIR) spectra were collected on a Varian 800 Scimitar Series FTIR spectrometer with values reported in wavenumbers. 1H NMR and 13C NMR spectra were recorded on a Bruker DRX500 spectrometer. Magnetic moments of the metal salts were measured by Evan's method7 in DMSO-d6 at 298K using a DRX500 spectrometer.
DFT Methods
The hybrid density functionals PBE0,8 B3LYP,9 and TPSSh10 were used to assess the performance of DFT for this series of compounds. These functionals were chosen for their utility in a wide variety of transition metal complex calculations, and their minimal amount of parametrization. All calculations were performed with TURBOMOLE v6.3.11 The crystal structure geometries of each compound (excluding BPh4- and solvent molecules) were optimized first using double-zeta quality split-valence basis sets with polarization functions (def2-SVP)12 in C1 symmetry. The geometries were further relaxed with larger triple-zeta basis sets (def2-TZVP)13 for all atoms, and only the geometric data from the final optimizations are reported. The bond length differences between basis sets were generally less than 0.02 Å. Ideal C3 symmetric structures for each metal were constructed using Ecce Builder14 and optimized following the same procedure. Complications arose for the FeII and NiII compounds since the highest beta (“down”) spin electron can occupy two degenerate e-type orbitals. These Jahn-Teller systems were therefore optimized using fractional occupation. Structures optimized from the experimental data were confirmed to be minima through the absence of imaginary frequencies in the calculated vibrational spectra, computed using second analytical derivatives15 employing SVP basis sets. Fine quadrature grids (size m4)16 were used throughout. Validation of the semi-local DFT results for ZnII were obtained via additional geometry optimizations with RIMP217 using TZVP basis sets and single-point energy calculations with the recently developed RI-RPA method. 18,19
Crystallographic Data Collection
Crystals suitable for X-ray diffraction were obtained for all salts in the series. For all salts except [CrIITAO(Cl)]BPh4, crystals were grown by slow diffusion of pentane into concentrated dichloromethane solutions. Crystals for [CrIITAO(Cl)]BPh4 were obtained from vapor diffusion of ether into a concentrated THF solution. Crystallographic data for the [MIITAO(Cl)]BPh4 complexes are provided in Table S1, and selected bond lengths and angles are contained in Table 1. For the salts, a single crystal was mounted on a glass fiber and transferred to a Bruker SMART APEX II diffractometer. The APEX220 program package was used to determine the unit-cell parameters and for data collection (25-30 sec/frame scan time for a sphere of diffraction data). The raw frame data was processed using SAINT21 and SADABS22 to yield the reflection data file. Subsequent calculations were carried out using the SHELXTL23 program. Full crystallographic details can be found in the supporting information.
Table 1.
Selected Bond Distances (Å), Angles (deg), and Trigonality Parametersa for [MIITAO(Cl)]+
| Cr | Mn | Fe | Co | Ni | Cu | Zn | |
|---|---|---|---|---|---|---|---|
| M(1)-Cl(1) | 2.3668(6) | 2.4556(5) | 2.4375(4) | 2.3895(5) | 2.3866(4) | 2.2957(4) | 2.3875(5) |
| M(1)-N(1) | 2.1140(18) | 2.3040(14) | 2.2119(13) | 2.2089(15) | 2.0790(13) | 2.0614(12) | 2.2470(17) |
| M(1)-N(2) | 2.1767(18) | 2.1593(14) | 2.0828(13) | 2.0829(15) | 2.0245(13) | 2.0909(12) | 2.0649(16) |
| M(1)-N(3) | 2.1731(18) | 2.1630(15) | 2.1208(13) | 2.0610(15) | 2.0812(13) | 2.1133(13) | 2.0938(5) |
| M(1)-N(4) | 2.1604(19) | 2.1700(14) | 2.1163(13) | 2.0584(15) | 2.0786(13) | 2.0947(12) | 2.0696(17) |
| N(5)···Cl(1) | 3.272 | 3.238 | 3.169 | 3.066 | 3.129 | 3.171 | 3.205 |
| N(6)···Cl(1) | 3.142 | 3.326 | 3.231 | 3.125 | 3.176 | 3.075 | 3.075 |
| N(7)···Cl(1) | 3.195 | 3.233 | 3.215 | 3.182 | 3.167 | 3.132 | 3.131 |
| N(1)-M(1)-Cl(1) | 179.33(5) | 177.23(4) | 175.91(4) | 178.62(4) | 174.54(4) | 178.85(4) | 178.22(5) |
| N(2)-M(1)-N(3) | 117.68(7) | 108.88(6) | 107.75(5) | 118.62(6) | 106.15(5) | 119.66(5) | 118.58(7) |
| N(2)-M(1)-N(4) | 114.75(7) | 122.35(5) | 112.92(5) | 118.88(6) | 111.16(5) | 116.31(5) | 114.77(7) |
| N(3)-M(1)-N(4) | 120.75(7) | 115.78(5) | 129.02(5) | 114.41(6) | 136.15(5) | 119.50(5) | 118.40(7) |
| N(1)-M(1)-N(2) | 81.58(7) | 77.45(5) | 80.24(5) | 80.05(6) | 83.76(5) | 83.34(5) | 81.07(6) |
| N(1)-M(1)-N(3) | 80.92(7) | 78.12(5) | 78.44(5) | 80.36(6) | 79.86(5) | 82.39(5) | 79.75(6) |
| N(1)-M(1)-N(4) | 81.29(7) | 78.07(5) | 79.48(5) | 80.97(6) | 82.09(5) | 82.95(5) | 80.28(6) |
| d(M···Neq)b | 0.330 | 0.455 | 0.390 | 0.343 | 0.298 | 0.260 | 0.348 |
| τ a | 0.976 | 0.915 | 0.782 | 0.996 | 0.640 | 0.987 | 0.994 |
τ = (β - α)/60, where α and β are the two largest angles in the primary coord. sphere , and β > α.
The displacement of the metal ion from the plane formed by atoms N(2), N(3), and N(4).
Results and Discussion
Synthesis
The metal chloride complexes were synthesized by combining the ligand TAO in dry methanol with the appropriate MIICl2 precursor (Scheme 1). The complexes were readily isolated by metathesis with NaBPh4, resulting in the immediate precipitation of the tetraphenylborate salts from solution. The solids were filtered, washed, and collected in yields that ranged from 66-88%. The complexes of CrII, MnII, FeII, CoII, and NiII ions were all determined to be high-spin at room temperature in DMSO-d6 based on magnetic susceptibility measurements obtained using Evan's method.7
Scheme 1.
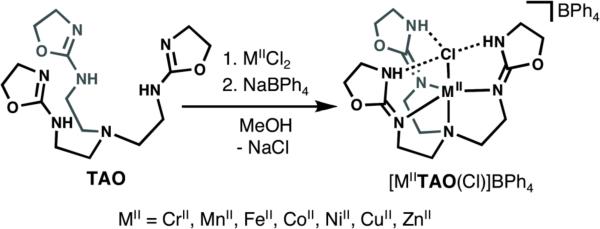
General route to [MIITAO(Cl)]BPh4 complexes
Molecular Structures
The crystal structures of the salts were determined by X-ray diffraction methods, revealing complexes that are five-coordinate with distorted trigonal bipyramidal geometries. Selected thermal ellipsoid diagrams of the structures are shown in Figure 2. The trigonal plane is defined by the imino atoms N2, N3 and N4 of the TAO ligand with the metal ions displaced toward the chloride ion with distances that range from 0.260 to 0.455 Å (Table 1). The tertiary amino nitrogen atom (N1) of TAO and an exogenous chloride ion occupy the remaining two axial coordination sites.
Fig. 2.

Thermal ellipsoid diagrams of [FeIITAO(Cl)]+ (A), [CoIITAO(Cl)]+, (B) [NiIITAO(Cl)]+ (C), and [ZnIITAO(Cl)]+ (D). The ellipsoids are drawn at 50% probability level, and only the oxazolidine hydrogen atoms are shown for clarity.
The degree of trigonality of the complexes was evaluated using the structural parameter τ (Table 1).24 Most of the structures possess τ values above 0.90, demonstrating a trigonal environment; however, [FeIITAO(Cl)]+ and [NiIITAO(Cl)]+ exhibit larger deviations from trigonal symmetry, with τ values of 0.78 and 0.64, respectively. The FeII and NiII complexes are high spin, which in trigonal symmetry would lead to partial occupation of degenerate d orbitals. Therefore, the distortion toward square pyramidal coordination geometry observed in [FeIITAO(Cl)]+ and [NiIITAO(Cl)]+ would be expected because of Jahn-Teller effects. Accordingly, large variations in the equatorial N-M-N angles are found for FeII and NiII complexes (Table 1), differences which are not observed in the other complexes. In addition, the Cl-M-N1 angles are approximately 175° for the FeII and NiII complexes, in contrast to the greater than 178° bond angles found for this angle in the other complexes. A detailed theoretical analysis of the geometries and Jahn-Teller effects can be found in the DFT discussion section. Distortion of the complex geometries also influenced packing of the molecules in the solid state: The TAO complexes with τ > 0.97 crystallized in the P21/c space group, whereas the MnII, FeII, and NiII complexes packed in the P-1 space group.
Intramolecular H-Bonding Networks
The molecular structures of [MIITAO(Cl)]+ complexes also show that the TAO ligand has adopted the 2-imino-oxazolidine tautomer. Support for this claim comes from comparing C–N bond lengths. In each complex, the bonds between the non-ring equatorial N and ring C (Neq-C) are significantly shorter than the appropriate N–C bond distance within the ring (Nox-C). For example, in [CrIITAO(Cl)]+ the average Nox-C bond length is 1.336(9) Å, whereas the Neq-C bonds possess more double-bond character with average lengths of 1.294(9) Å. These data are consistent with the formation of the 2-imino-oxazolidine tautomer for TAO in all the metal complexes. This tautomer of TAO results in the establishment of intramolecular H-bonding networks surrounding the MII–Cl units. The average distance between the oxazolidine amino nitrogen atoms and the bound chloride in each complex is less than 3.2 Å, suggesting the formation of intramolecular H-bonds.25 The lone exception was found in [MnIITAO(Cl)]+, in which the N···Cl distances were slightly longer than 3.2 Å.
A survey of structures in the Cambridge Crystallographic Database indicates that the MII–Cl bond distances in [MIITAO(Cl)]+ are unusually long in comparison to other metal-chloro complexes with trigonal bipyramidal coordination geometry.26 For example, the NiII–Cl bond distance in [NiII(Me6tren)(Cl)]+ is 2.295(2) Å, 27 in contrast to a bond length of 2.3866(4) Å in [NiIITAO(Cl)]+. Moreover, a trigonal bipyramidal complex with a ligand bearing equatorial imino N-donors, [CoII(ntb)(Cl)]+, possesses a CoII–Cl bond length of 2.296(2) Å, a value that is significantly shorter than that observed in [CoIITAO(Cl)]+ (Co1–Cl1, 2.3895(5) Å).28 The lengthening of the metal-chloride bond distances is ascribed to effects of the H-bonding network, which removes electron density from the chloride ligand and causes a weakening of the metal-chloride bond.
This premise is further supported by comparing the bond lengths in [ZnIITAO(Cl)]+ to a previously reported tren complex, [ZnII(tren)Cl]+ (Figure 3A/B).29 In the tren complex, the Zn-Cl length is 2.308(5) Å (Figure 3A), whereas the length in the TAO system is 2.3875(5) Å (Figure 3B). The Zn-apical nitrogen (Nap) distances for [ZnII(tren)Cl]+ and [ZnIITAO(Cl)]+ follow the opposite trend, with Zn-Nap lengths of 2.325(7) Å and 2.2470(17)Å, respectively. The Zn-Nap shortening is caused by the trans-influence, strengthening the Zn-Nap interaction as the Zn-Cl bond length increases. Mareque-Rivas observed similar trends in ZnII–Cl complexes containing pyridyl tripodal ligands (Figure 3 C/D).5j,30
Fig. 3.

Molecular diagrams of tren-based (A/B)29 and pyridine-based5j,30 (C/D) tripodal zinc-chloro complexes with and without intramolecular hydrogen bonds. The Zn-Cl and Zn-apical N bond lengths are displayed under each respective molecule.
H-bond networks around Fe–Cl units have also been observed within the active site of the metalloproteins. In particular, the Fe–Cl bond length of 2.4375(4) Å found in [FeIITAO(Cl)]+ is strikingly similar to the distance observed in the active site of the α-ketoglutarate-dependent non-heme halogenase SyrB2.2d,31 The active site of this enzyme features a six-coordinate iron center with an Fe–Cl bond distance of 2.44 Å. The typical FeII–Cl bond length for six-coordinate complexes is 2.30 Å;32 the elongation observed in the SyrB2 crystal structure is attributed to an H-bond network established between two water molecules in the secondary coordination sphere of the active site and the bound chloride ion. Although the iron center in SyrB2 is six-coordinate, preventing a direct comparison to [FeIITAO(Cl)]+, an octahedral iron-chloride complex from the Stack group ([FeIIPY5(Cl)]+), which lacks intramolecular H-bonds, has an Fe–Cl distance of 2.318(2) Å, suggesting that H-bonding interactions can significantly elongate M–Cl bonds.33
Solution Structure: [ZnIITAO(Cl)]+
1H NMR studies of [ZnIITAO(Cl)]BPh4 in DMSO-d6 demonstrate the presence of a mononuclear complex in solution. Four triplet resonances in the NMR spectrum correspond to methylene protons in TAO (a-d in Figure 4); comparison of the integrated ligand peaks to those of BPh4- in the spectrum indicates a 1:1 ligand to counterion ratio. Chemical shifts and integration of the methylene proton peaks in the spectrum also indicate that the ligand arms are in similar chemical environments, suggesting that the complex exhibits local three-fold symmetry in solution on the NMR timescale.
Fig. 4.

1H NMR spectrum of [ZnIITAO(Cl)]BPh4 in DMSO-d6 at 298K. All unlabeled hydrogen atoms in the molecular diagram follow the same label convention as the rightmost ligand arm. Asterisks denote solvent peaks.
The H-bonding network observed in the solid-state molecular structure also exists in solution. A single sharp resonance at 7.6 ppm integrates to three protons and is attributed to the three oxazolidine NH groups (e in Figure 4). The 1.3 ppm downfield shift of this NH resonance relative to that of free TAO suggests significant deshielding of the proton environment, consistent with a H-bonding interaction with the bound chloride ion. The assignment of the NH peak was also supported by variable temperature analyses of the solution, which showed broadening at higher temperatures.
The NMR data are similar to those of another TAO complex previously reported by our group, ZnIITAO(SO4).6 This complex was also shown to exhibit local three-fold symmetry in solution, with a single NH resonance at 9.25 ppm which integrated to 3 protons. Accordingly, the molecular structure of ZnIITAO(SO4) corroborated the existence of an intramolecular H-bonding network between the ligand NH groups and the oxygen atoms of the bound SO42- anion. Taken together, the NMR results and molecular structures of ZnIITAO(SO4) and [ZnIITAO(Cl)]+ indicate that the H-bonding networks formed in these mononuclear complexes exist in both solution and the crystalline state.
Density Functional Theory Analysis of [MIITAO(Cl)]+
To confirm the large distortion away from trigonal bipyramidal coordination is a result of electronic effects, the entire series of complexes was assessed using Kohn-Sham Density Functional Theory. The TPSSh functional was found to yield the closest overall agreement with the X-ray data, in line with the findings of previous studies on first row transition metal complexes.34 Tables of computed structural data and the coordinates of the optimized structures, in .xyz format, are provided in the supporting information.
Average structural data is well-reproduced using TZVP basis sets (see Table S2). The optimized bond lengths of the ligand support the formation of the 2–imino tautomer given the optimized non-ring N-C bond lengths are roughly 1.29 Å on average. The predicted metal-chlorine bond lengths are uniformly too long by ~2 pm, except for Mn which has an optimized bond length ~1 pm too short compared to experiment. The MII-Nap distance is generally overestimated by ~5 pm for the entire series, which is counterintuitive given the long MII-Cl bond distance. The computed average MII-Neq and N···Cl distances are also overestimated compared to the average experimental values, with errors on the order of 1 to 2 pm. RI-MP2 optimizations were performed as a consistency check for the Zn complexes, and the results were largely the same: the MII-Cl, MII-Nap, and N···Cl distances are all overestimated by 1, 8, and 2 pm, respectively, while the average MII-Neq distance is underestimated by 3 pm compared to experiment. The good agreement of the RI-MP2 structures with the semi-local DFT results suggests that the latter are accurate and that dispersion interactions do not play a major role for the structure of the present compounds. To further validate the DFT results, calculations were performed with RIRPA. These subsequent calculations, outlined in the supporting information, confirmed the semi-local and RI-MP2 results for the MII-Nap bond lengths in the ZnII compound. Natural population analysis yields large partial charges on the metal and atoms in the primary coordination sphere. These partial charges indicate a predominantly ionic MII-Cl bond for all the complexes.
The predicted SVP N-H stretching frequencies (~3400 cm-1, before scaling) are uniformly too large; scaling the frequencies according to the optimal factors reported by Merrick reduces the magnitude of the error.35 In many cases this subsequently leads to an underestimation of the computed vibrational frequency compared to experiment (Table S3). The N-H stretch of the free ligand was computed to be approximately 3600 cm-1 (before scaling); the redshift supports the presence of a H-bond network with the Cl- ligand.36 Thus, the H-bonding interactions in the binding pocket stabilize the complexes via charge transfer from the Cl- to anti-bonding N-H orbitals, which subsequently leads to an elongated MII-Cl bond and a redshift in the N-H vibrational frequency.
The CrII, MnII, CoII, CuII, and ZnII complexes exhibit a C3-symmetric minimum. However, for the FeII and NiII complexes, the C3 structure is a saddle point, and relaxing the symmetry to C1 leads to a stabilization of 12.5 and 18.4 kcal/mol, respectively, at the TPSSh/TZVP level. This symmetry breaking for the FeII and NiII compounds is a classic example of a Jahn-Teller effect caused by fractional occupation of a doubly degenerate e orbital in C3 symmetry, which has mostly metal d character. The breaking of the orbital degeneracy is accompanied by a distortion toward square pyramidal geometry, consistent with the solid state structure observations.
Plots of the pertinent frontier orbitals for [NiIITAO(Cl)]+ are included as supporting information. The distortion towards square pyramidal coordination is predicted to be larger for NiII than FeII according to the computed τ parameter, which is in line with the experimental findings. It is clear from this set of calculations that the FeII and NiII compounds exhibit strong geometric distortion due to their electronic structure, while the other metals in the series prefer a threefold-symmetric primary coordination environment.
Conclusions
In summary, our group has synthesized a series of trigonal bipyramidal first-row transition metal chloride complexes with the tetradentate ligand TAO. For each complex, the tris(2-iminooxazolidine) tautomer of TAO bound the metal ion and established intramolecular H-bonding interactions among the ring amino groups and the bound chloro ligand. The molecular structures verify the existence of the H-bonding network in the solid state, and 1H NMR studies with [ZnIITAO(Cl)]+ support the presence of these interactions in solution. Quantum chemical computations on the series indicate predominantly ionic character for the MII-Cl interaction. The MII-Cl bond lengths were found to be longer than those found in other trigonal bipyramidal complexes because of the electron-withdrawing effects of the H-bonding network. These findings further illustrate the ability of H-bonding networks to effect changes in the primary coordination spheres of metal complexes.
Supplementary Material
Acknowledgements
Acknowledgment is made to Dr. Michael Takase for help in X-Ray diffraction experiments, and the National Institutes of Health USA (GM050781, ASB), the National Science Foundation USA (0738252, FF), and EXAX Inc. (YJP) for financial support. We also thank Dr. Nathan Crawford for useful discussions and NSF grants CHE-0911266 and CHE-0840513 for financial support of the computations.
Footnotes
† Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/b000000x/
Notes and references
- 1.a Borovik AS. Acc. Chem. Res. 2005;38:54–61. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]; b Shook RL, Borovik AS. Chem. Commun. 2008:6095–6107. doi: 10.1039/b810957e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shook RL, Borovik AS. Inorg. Chem. 2010;49:3646–3660. doi: 10.1021/ic901550k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a Yang J, Kloek AP, Goldberg DE, Mathews FS. Proc. Natl. Acad. Sci. U. S. A. 1995;92:4224–4228. doi: 10.1073/pnas.92.10.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Holm RH, Kennepohl P, Solomon EI. Chem. Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]; c Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]; d Blasiak LC, Vaillancourt FH, Walsh CT, Drennan CL. Nature. 2006;440:368–371. doi: 10.1038/nature04544. [DOI] [PubMed] [Google Scholar]; e You Z, Omura S, Ikeda H, Cane DE, Jogl G. J. Biol. Chem. 2007;282:36552–36560. doi: 10.1074/jbc.M706358200. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Dumitru R, Jiang WZ, Weeks DP, Wilson MA. J. Mol. Biol. 2009;392:498–510. doi: 10.1016/j.jmb.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Umena Y, Kawakami K, Shen J-R, Kamiya N. Nature. 2011;473:55–60. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 3.a Leveque VJP, Vance CK, Nick HS, Silverman DN. Biochemistry. 2001;40:10586–10591. doi: 10.1021/bi010792p. [DOI] [PubMed] [Google Scholar]; b Thoms S. J. Theor. Biol. 2002;215:399–404. doi: 10.1006/jtbi.2002.2528. [DOI] [PubMed] [Google Scholar]; c Marshall NM, Garner DK, Wilson TD, Gao Y-G, Robinson H, Nilges MJ, Lu Y. Nature. 2009;462:113–116. doi: 10.1038/nature08551. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Dang M, Pochapsky SS, Pochapsky TC. Metallomics. 2011;3:339–343. doi: 10.1039/c0mt00065e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a MacBeth CE, Golombek AP, Young VG, Jr., Yang C, Kuczera K, Hendrich MP, Borovik AS. Science. 2000;289:938–941. doi: 10.1126/science.289.5481.938. [DOI] [PubMed] [Google Scholar]; b Lucas RL, Zart MK, Murkerjee J, Sorrell TN, Powell DR, Borovik AS. J. Am. Chem. Soc. 2006;128:15476–15489. doi: 10.1021/ja063935+. [DOI] [PubMed] [Google Scholar]; c Lacy DC, Gupta R, Stone KL, Greaves J, Ziller JW, Hendrich MP, Borovik AS. J. Am. Chem. Soc. 2010;132:12188–12190. doi: 10.1021/ja1047818. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ng GKY, Ziller JW, Borovik AS. Inorg. Chem. 2011;50:7922–7924. doi: 10.1021/ic200881t. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Park YJ, Ziller JW, Borovik AS. J. Am. Chem. Soc. 2011;133:9258–9261. doi: 10.1021/ja203458d. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Shook RL, Peterson SM, Greaves J, Moore C, Rheingold AL, Borovik AS. J. Am. Chem. Soc. 2011;133:5810–5817. doi: 10.1021/ja106564a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a Wuenschell GE, Tetreau C, Lavalette D, Reed CA. J. Am. Chem. Soc. 1992;114:3346–3355. [Google Scholar]; b Kitajima N, Tamura N, Amagai H, Fukui H, Moro-oka Y, Mizutani Y, Kitagawa T, Mathur R, Heerwegh K, et al. J. Am. Chem. Soc. 1994;116:9071–9085. [Google Scholar]; c Berreau LM, Allred RA, Makowska-Grzyska MM, Arif AM. Chem. Commun. 2000:1423–1424. [Google Scholar]; d Berreau LM, Makowska-Grzyska MM, Arif AM. Inorg. Chem. 2001;40:2212–2213. doi: 10.1021/ic001190h. [DOI] [PubMed] [Google Scholar]; e Yeh C-Y, Chang CJ, Nocera DG. J. Am. Chem. Soc. 2001;123:1513–1514. doi: 10.1021/ja003245k. [DOI] [PubMed] [Google Scholar]; f Kimblin C, Bu X, Butler A. Inorg. Chem. 2002;41:161–163. doi: 10.1021/ic010892j. [DOI] [PubMed] [Google Scholar]; g Wada A, Ogo S, Nagatomo S, Kitagawa T, Watanabe Y, Jitsukawa K, Masuda H. Inorg. Chem. 2002;41:616–618. doi: 10.1021/ic001058h. [DOI] [PubMed] [Google Scholar]; h Yamaguchi S, Wada A, Funahashi Y, Nagatomo S, Kitagawa T, Jitsukawa K, Masuda H. Eur. J. Inorg. Chem. 2003:4378–4386. doi: 10.1021/ic035080x. [DOI] [PubMed] [Google Scholar]; i Wada A, Honda Y, Yamaguchi S, Nagatomo S, Kitagawa T, Jitsukawa K, Masuda H. Inorg. Chem. 2004;43:5725–5735. doi: 10.1021/ic0496572. [DOI] [PubMed] [Google Scholar]; j Rivas JCM, Prabaharan R, Torres Martin de Rosales R, Metteau L, Parsons S. Dalton Trans. 2004:2800–2807. doi: 10.1039/B407790C. [DOI] [PubMed] [Google Scholar]; k Feng G, Mareque-Rivas JC, Williams NH. Chem. Commun. 2006:1845–1847. doi: 10.1039/b514328d. [DOI] [PubMed] [Google Scholar]; l Feng G, Natale D, Prabaharan R, Mareque-Rivas JC, Williams NH. Angew. Chem., Int. Ed. 2006;45:7056–7059. doi: 10.1002/anie.200602532. [DOI] [PubMed] [Google Scholar]; m Mareque Rivas Juan C, Hinchley Sarah L, Metteau L, Parsons S. Dalton Trans. 2006:2316–2322. doi: 10.1039/b516234c. [DOI] [PubMed] [Google Scholar]; n Metteau L, Parsons S, Mareque-Rivas JC. Inorg. Chem. 2006;45:6601–6603. doi: 10.1021/ic0612193. [DOI] [PubMed] [Google Scholar]; o Reid SD, Wilson C, Blake AJ, Love JB. Dalton Trans. 2010;39:418–425. doi: 10.1039/b909842a. [DOI] [PubMed] [Google Scholar]; p Searls CE, Kleespies ST, Eppright ML, Schwartz SC, Yap GPA, Scarrow RC. Inorg. Chem. 2010;49:11261–11263. doi: 10.1021/ic101612f. [DOI] [PubMed] [Google Scholar]
- 6.Park YJ, Sickerman NS, Ziller JW, Borovik AS. Chem. Commun. 2010;46:2584–2586. doi: 10.1039/c000160k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans DF. J. Chem. Soc. 1959:2003–2005. [Google Scholar]
- 8.Perdew JP, Ernzerhof M, Burke K. J. Chem. Phys. 1996;105:9982–9985. [Google Scholar]
- 9.Becke AD. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- 10.Perdew JP, Tao J, Staroverov VN, Scuseria GE. J. Chem. Phys. 2004;120:6898–6911. doi: 10.1063/1.1665298. [DOI] [PubMed] [Google Scholar]
- 11.Turbomole, V6.3 TURBOMOLE GmbH, Karlsruhe, 2011; available from http://www.turbomole.com.
- 12.Schaefer A, Horn H, Ahlrichs R. J. Chem. Phys. 1992;97:2571–2577. [Google Scholar]
- 13.Weigend F, Ahlrichs R. Phys. Chem. Chem. Phys. 2005;7:3297–3305. doi: 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- 14.Black G, Chase J, Chatterton J, Daily J, Elsethagen T, Feller D, Gracio D, Jones D, Keller T, Lansing C, Matsumoto S, Palmer B, Peterson M, Schuchardt K, Stephan E, Sun L, Swanson K, Taylor H, Thomas G, Vorpagel E, Windus T, Winters C. Pacific Northwest National Laboratory; Richland, Washington 99352-0999, USA: 2009. [Google Scholar]
- 15.Deglmann P, Furche F, Ahlrichs R. Chem. Phys. Lett. 2002;362:511–518. [Google Scholar]
- 16.Treutler O, Ahlrichs R. J. Chem. Phys. 1995;102:346–354. [Google Scholar]
- 17.a Haase F, Ahlrichs R. J. Comput. Chem. 1993;14:907–912. [Google Scholar]; b Weigend F, Haser M. Theor. Chem. Acc. 1997;97:331–340. [Google Scholar]
- 18.Eshuis H, Yarkony J, Furche F. J. Chem. Phys. 2010;132:234114/234111–234114/234119. doi: 10.1063/1.3442749. [DOI] [PubMed] [Google Scholar]
- 19.Self-consistent TPSS/def2-TZVP input orbitals were used for the RI-RPA calculation.
- 20.APEX2 Version 2008.3-0. Bruker AXS, Inc.; Madison, WI: 2008. [Google Scholar]
- 21.SAINT Version 7.53a. Bruker AXS, Inc.; Madison, WI: 2008. [Google Scholar]
- 22.Sheldrick GM. SADABS, Version 2008/1. Bruker AXS, Inc.; Madison, WI: 2008. [Google Scholar]
- 23.Sheldrick GM. SHELXTL, Version 2008/3. Bruker AXS, Inc.; Madison, WI: 2008. [Google Scholar]
- 24.Addison AW, Rao TN, Reedijk J, Van Rijn J, Verschoor GC. J. Chem. Soc., Dalton Trans. 1984:1349–1356. [Google Scholar]
- 25.Where a N···Cl distance of approximately 3.2 Å is generally accepted to represent an H-bond: Emsley J. Chem. Soc. Rev. 1980;9:91–124.Aullon G, Bellamy D, Orpen AG, Brammer L, Bruton EA. Chem. Commun. 1998:653–654.
- 26.a Allen F. Acta Crystallogr., Sect. B: Struct. Sci. 2002;58:380–388. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]; b Bruno IJ, Cole JC, Edgington PR, Kessler M, Macrae CF, McCabe P, Pearson J, Taylor R. Acta Crystallogr., Sect. B: Struct. Sci. 2002;58:389–397. doi: 10.1107/s0108768102003324. [DOI] [PubMed] [Google Scholar]
- 27.Colpas GJ, Kumar M, Day RO, Maroney MJ. Inorg. Chem. 1990;29:4779–4788. [Google Scholar]
- 28.Lah MS, Moon M. Bull. Korean Chem. Soc. 1997;18:406–409. [Google Scholar]
- 29.Zalkin A, Sime RJ, Dodge RP, Templeton DH. Inorg. Chem. 1971;10:537–541. [Google Scholar]
- 30.Adams H, Bailey NA, Fenton DE, He Q-Y. J. Chem. Soc., Dalton Trans. 1997:1533–1540. [Google Scholar]
- 31.Vaillancourt FH, Yin J, Walsh CT. Proc. Natl. Acad. Sci. U. S. A. 2005;102:10111–10116. doi: 10.1073/pnas.0504412102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hawker PN, Twigg MV. In: Wilkinson G, Gillard RD, McCleverty JA, editors. Vol. 4. Pergamon; Oxford: 1987. p. 1179. [Google Scholar]
- 33.Klein Gebbink RJM, Jonas RT, Goldsmith CR, Stack TDP. Inorg. Chem. 2002;41:4633–4641. doi: 10.1021/ic025617r. [DOI] [PubMed] [Google Scholar]
- 34.a Buehl M, Kabrede H. J. Chem. Theory Comput. 2006;2:1282–1290. doi: 10.1021/ct6001187. [DOI] [PubMed] [Google Scholar]; b Furche F, Perdew JP. J. Chem. Phys. 2006;124:044103/044101-044103/044127. doi: 10.1063/1.2162161. [DOI] [PubMed] [Google Scholar]; c Sousa SF, Carvalho ES, Ferreira DM, Tavares IS, Fernandes PA, Ramos MJ, Gomes JANF. J. Comput. Chem. 2009;30:2752–2763. doi: 10.1002/jcc.21304. [DOI] [PubMed] [Google Scholar]
- 35.Merrick JP, Moran D, Radom L. J. Phys. Chem. A. 2007;111:11683–11700. doi: 10.1021/jp073974n. [DOI] [PubMed] [Google Scholar]
- 36.Li X, Liu L, Schlegel HB. J. Am. Chem. Soc. 2002;124:9639–9647. doi: 10.1021/ja020213j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.