

Abstract
A tetradentate tripodal ligand containing 2-amino-oxazoline moeities has been developed. This system tautomerizes upon chelation of a metal ion, forming a flexible cavity capable of accomodating ligands via an intramolecular hydrogen bonding network.
Oxazoline rings are a desirable building block in the design of ligands because of their proven utility in developing catalysts having nitrogen donors.1 Key attributes of oxazolines are the relative synthetic ease at which they can be incorporated into multidentate ligands and excellent coordinating ability to metal ions, affording a diversity of compounds that can be used in a variety of applications. We have been investigating tripodal ligands and their ability to promote intramolecular hydrogen bonding (H-bonding) networks within the secondary coordination sphere of metal ions.2 Reported herein is the design and preparation of a new neutral tripodal ligand containing 2-amino-oxazoline moieties. We demonstrate that intramolecular H-bonding networks can be formed through locking one tautomer from the 2-amino-oxazoline/2-imino-oxazolidine equilibrium by metal ion coordination.
Most of the tripodal ligands developed that produce intramolecular H-bonds utilize pyridyl units to bind metal ions.3,4,5 Appended groups from the pyridyl rings serve as H-bond donors/acceptors that interact with additional ligands bound to the metal ion. We have developed anionic tripodal ligands containing urea groups, in which one of the urea NH groups on each arm is deprotonated to bind a metal ion, while the other NH moieties function as H-bond donors.2 During our ongoing studies on urea chemistry we serendipitously found a convenient synthetic route to 2-amino-oxazoline tripodal ligands (Scheme 1).
Scheme I.
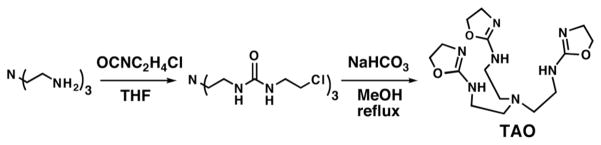
Preparative route to the tris(2-amino-oxazoline) ligand TAO.
Treating tren with 3 equiv of 1-chloro-2-isocyanatoethane afforded the tris-urea species in approximately 90% yield. Refluxing the tris-urea species in methanol in the presence of excess NaHCO3 produced the tris(2-amino-oxazoline) ligand (TAO) in 82% yield. TAO was isolated as highly viscous oil after drying but it became insoluble over time. However, we have found that solutions of TAO are stable for weeks and are the preferable method of storage.6
Because 2-amino-oxazolines can tautomerize, we anticipated that TAO could have two possible limiting structures (Fig. 1). Theoretical studies on other compounds containing 2-amino-oxazoline units showed that the amino tautomer is more stable,7 which is supported by experimental investigations. Consistent with these findings are structural studies showing that monodentate 2-amino-oxazoline ligands bind metal ions through the ring nitrogen atom in neutral conditions.8 We have further probed metal ion binding in a simpler ligand, 2-(t-butylamino)-oxazoline (BAO) that contains a single 2-amino-oxazoline unit.
Fig. 1.
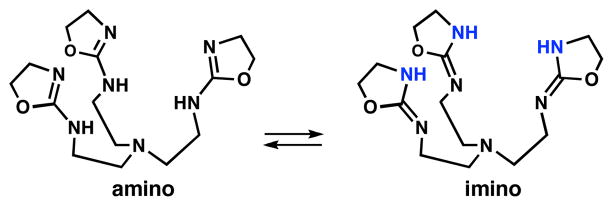
Possible tautomerization for tris(2-amino-oxazoline) ligand, TAO.
Treating BAO with Zn(OAc)2 gave single crystals of the complex [Zn(BAO)2(κ1-OAc)2], whose structure is shown in Fig. 2. The BAO ligands bind in a monodentate manner through the ring nitrogen atoms N1 and N3, supporting the presence of the amino tautomer. Additional evidence for this tautomer comes from comparing the N–C bond length within coordinated the BAO ligands: the fact that the N1–C3 and N2–C10 bond distances of ~1.307(2) Å are significantly shorter than those found for the N3–C3 and N4–C11 bonds (~1.330(2) Å) is clearly indicating the position of the double bond. Moreover, the amino nitrogen atoms N3 and N4 have protons that were located directly from the Fourier difference map and form intramolecular H-bonds to the monodentate acetate ligands, as indicated by N3···O4 and N4···O6 distances of less than 2.85 Å. NMR studies on [Zn(BAO)2(κ1-OAc)2] also suggest that the complex adopts a similar structure in solution, in which a single set of resonances are observed (Fig. S1).
Fig. 2.
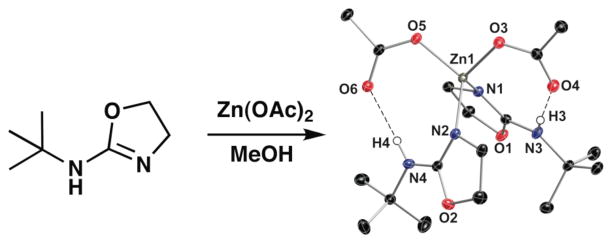
Synthesis and thermal ellipsoid diagram of [Zn(BAO)2(κ1-OAc)2]. The ellipsoids are drawn at the 50% probability level, and only the amino hydrogen atoms are depicted. Selected bond lengths (Å): Zn1–N1, 2.196(2); Zn1–N2, 2.096(2); Zn1–O3, 2.047(1); Zn1–O4, 2.134(2); Zn1–O5, 2.054(1); N3···O4, 2.821; N4···O6, 2.805.
We have investigated the solution properties of TAO using NMR spectroscopy but were unable to find which tautomer was prevalent at room temperature. The 1H NMR spectrum showed a single set of CH peaks with a broad NH signal that was often unobservable. Furthermore, we could not observe a correlation peak between the CH and NH protons through COSY experiment. Taken together, our spectral results suggested that tautomerization in TAO is fast compared to the NMR timescale and that both imino and amino forms should be present in solution.
To evaluate the ability of TAO to bind metal ions we prepared [ZnTAO(κ1-OAc)]+ following the synthetic route shown in Scheme 2. The complex was isolated as the tetraphenylborate salt and purified via recrystallization from a methylene chloride/pentane mixture.
Scheme II.

Synthetic routes to [ZnTAO(κ1-OAc)]+.
Analytical and spectroscopic measurements of [ZnTAO(κ1-OAc)]+ supported the premise that one acetate ion is coordinated to the Zn(II) center. This was further corroborated by the solid-state structure determined using X-ray diffraction methods. The molecular structure of [ZnTAO(κ1-OAc)]+ unambiguously revealed a five-coordinate Zn(II) center with a trigonal bipyramidal geometry (Fig. 3A). Unlike other metal complexes with monodentate 2-amino-oxazoline ligands, TAO tautomerized to its imino form upon binding to the metal ion, in which oxazolidine rings are formed in [ZnTAO(κ1-OAc)]+. Support for oxazolidine rings is again provided by comparing N–C bonds lengths. For instance, the non-ring N2–C3 bond length of 1.286(2) Å is significantly shorter than the N5–C3 bond distance of 1.333(2) Å. A similar trend is observed for the other rings in TAO—notice these differences in N–C bond distances are opposite the trend observed for the BAO ligand, in which the amino tautomer was present.
Fig. 3.
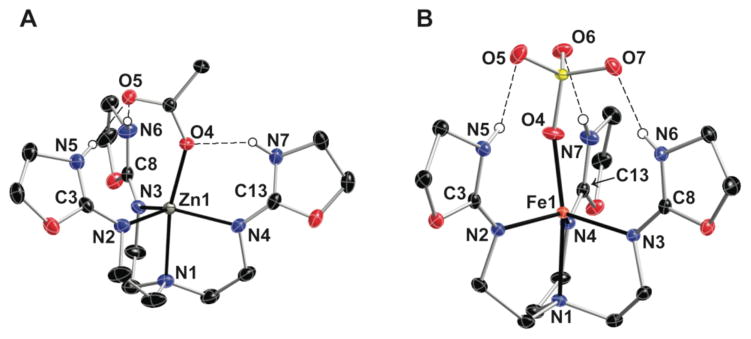
Thermal ellipsoid diagrams of [ZnTAO(κ1-OAc)]+ (A) and [FeIITAO(κ1-SO4)] (B). The ellipsoids are drawn at the 50% probability level and only the oxazolidine hydrogen atoms are shown. Selected bond lengths (Å) and angles (°): Zn1–N1, 2.196(2); Zn1–N2, 2.096(2); Zn1–N3, 2.047(1); Zn1–N3, 2.134(2); Zn1–O4, 2.054(1); N5···O5, 2.771; N6···O5, 2.871; N7···O4, 2.746; N2-Zn1-N3, 108.64(6); N3-Zn1-N4, 117.78(6); N2-Zn1-N4, 124.91(6); N1-Zn1-O4, 166.91(6); Fe1-N1, 2.2132(14); Fe1-N2, 2.1334(14); Fe1-N3, 2.0998(13); Fe1-N4, 2.1400(14); Fe1-O4, 1.9998(13); N5···O5, 2.965; N6···O7, 2.748; N7···O6, 2.847; N2-Fe1-N3, 107.72(6); N3-Fe1-N4, 112.25(6); N2-Fe1-N4, 128.90(6); N1-Fe1-O4, 168.99(6).
In [ZnTAO(κ1-OAc)]+ the N2, N4, and N6 atoms coordinate to the zinc center, having an average Zn1–N bond distance of 2.093(2) Å. These imino atoms define a trigonal plane with the Zn atom positioned 0.357 Å from the plane. The Zn1–N1 bond length is 2.196(2) Å and is nearly perpendicular to the trigonal plane. The O4 atom of the acetate completes the primary coordination sphere with a Zn1–O4 bond distance of 2.053(1) Å. In addition, the acetate ion resides within a cavity created by the oxazolidine rings of TAO and is part of an extensive H-bonding network. The acetate is positioned in such a way as to produce three intramolecular H-bonds with the NH groups of the oxazolidine rings. Besides being coordinated to the zinc center, O4 forms a proximal H-bond with one of the rings (O4···N7, 2.746(2) Å). Note however that the H-bonding network is not just centered about O4, but rather appears to adjust to accommodate the entire acetate ligand. The distal O5 atom of the acetate is involved in two H-bonds to the remaining oxazolidine NH groups with O5···N5 and O5···N6 distances of 2.771(2) and 2.871(2) Å, respectively. The H-bonding network also results in the Zn1–O4 bond being tilted toward ring containing N7 (the N1–Zn1–O4 angle is 166.91(6)°), and a constriction of the N2–Zn1–N3 angle to 108.64(6)°.
The intramolecular H-bonding network observed in the solid state appears persists in solution. NMR studies in DMSO-d6 show that in [ZnTAO(κ1-OAc)]+, the tripodal arms of the TAO ligand are not all in the same chemical environment (Fig. S2), which is expected if a similar H-bonding network observed in the solid state was still present in the solution. Two of the arms are equivalent and are assigned to the ones involved in H-bonding to the distal oxygen atom of the acetate. The protons on the third arm have different chemical shifts, consistent with a single arm interacting with the proximal oxygen atom that is bonded to the zinc center. This difference is clearly observed in the chemical shifts of the oxazolidine NH protons: the proximal H-bonded proton is at 9.5 ppm whereas those involved in the distal H-bonds are at 7.92 ppm.9 Preliminary variable-temperature NMR measurements showed that as the temperature was increased the initial two sets of CH peaks converted to a single set of resonances with a gradual disappearance of NH peaks (Fig. S3). The coalescence temperature was observed at approximately 388 K and the process was reversible upon cooling to room temperature. The symmetric spectra at higher temperatures are consistent with fast rotation of the acetate ion within the H-bonding cavity on the NMR timescale, producing indistinguishable chemical environments for each arm of the TAO ligand.10
The results on [ZnTAO(κ1-OAc)]+ suggest that the cavity formed by TAO is flexible to adopt a H-bonding network that matches the characteristics of the external ligand. To further investigate this premise we have prepared [MIITAO(κ1-SO4)] (MII = Zn and Fe) by treating TAO with MIISO4. The solid-state molecular structure of [FeIITAO(κ1-SO4)] (Fig. 3B) reveals a five-coordinate Fe(II) center with a similar trigonal bipyramidal coordination geometry as in [ZnTAO(κ1-OAc)]+. The fifth ligand in [FeIITAO(κ1-SO4)] is provided by the O4 atom of the sulfate ion with a Fe1–O4 bond length of 2.000(2) Å and an N1–Fe1–O4 angle of 168.99°. [FeIITAO(κ1-SO4)] also has three H-bonds but the network is substantially different than that found in [ZnIITAO(κ1-OAc)]+. Each distal oxygen atom (i.e., O5, O6, and O7) on the sulfate ligand forms an intramolecular H-bond with one NH group of an oxazolidine ring, producing a nearly C3 symmetric network. Unlike [ZnIITAO(κ1-OAc)]+, there is no evidence that the proximal oxygen atom O4 forms H-bonds with the TAO ligand, as indicated by O4···N distances that are greater than 3.0 Å. NMR studies on the Zn(II) analog ([ZnIITAO(κ1-SO4)]) suggested that the symmetric H-bonding network is maintained in solution. The 1H NMR spectrum of [ZnIITAO(κ1-SO4)] in DMSO-d6 at room temperature contains only one set of signals assignable to the TAO ligand, including a single NH resonance at 9.25 ppm (Fig. S4).
The spectroscopic and structural findings demonstrate that the new tripodal ligand TAO can bind metal ions and form flexible H-bonding cavities. We suggest that the chelate effect involving the apical and imino nitrogen atoms provided a suitable driving force to favor the imino tautomer and thus produced the observed structures in [ZnIITAO(κ1-OAc)]+ and [FeIITAO(κ1-SO4)]. A related type of tautomerization was recently described by Love for a bidentate pyrrole ligand,11 and together with our work, illustrates the potential of this type of process in designing new ligands that can control both the primary and secondary coordination spheres. Coupling H-bonding networks with oxazoline-based ligands could lead to metal complexes with new or enhanced function. Efforts to assess this premise are ongoing.
Supplementary Material
Acknowledgments
Acknowledgements is made to Dr. Michael Takase for help in X-ray diffraction experiments and the National Institutes of Health USA (GM050781 to ASB) and EXAX Inc. (YJP) for financial support.
Footnotes
Electronic Supplementary Information (ESI) available: [Detailed experimental procedures, crystallographic data (including cif files) and NMR spectra (1H and VT)]. See DOI: 10.1039/b000000x/
Notes and references
- 1.Reviews: Hargaden GC, Guiry PJ. Chem Rev. 2009;109:2505–2550. doi: 10.1021/cr800400z.Desimoni G, Faita G, Jørgensen KA. Chem Rev. 2006;106:3561–3651. doi: 10.1021/cr0505324.McManus HA, Guiry PJ. Chem Rev. 2004;104:4151–4202. doi: 10.1021/cr040642v.
- 2.(a) Gupta R, Borovik AS. J Am Chem Soc. 2003;125:13234–13242. doi: 10.1021/ja030149l. [DOI] [PubMed] [Google Scholar]; (b) MacBeth CE, Hammes BS, Young VG, Jr, Borovik AS. Inorg Chem. 2001;40:4733–4741. [PubMed] [Google Scholar]; (c) MacBeth CE, Gupta R, Mitchell-Koch KR, Young VG, Jr, Lushington GH, Ward WH, Hendrich MP, Borovik AS. J Am Chem Soc. 2004;126:2556–2567. doi: 10.1021/ja0305151. [DOI] [PubMed] [Google Scholar]; (d) Gupta R, MacBeth CE, Young VG, Jr, Borovik AS. J Am Chem Soc. 2002;124:1136–1137. doi: 10.1021/ja016741x. [DOI] [PubMed] [Google Scholar]; (e) Shirin Z, Young VG, Jr, Borovik AS. J Am Chem Soc. 2000;122:1836–1837. [Google Scholar]; (f) Shirin Z, Young VG, Jr, Borovik AS. Chem Commun. 1997:1967–1968. [Google Scholar]; (g) Mukherjee J, Lucas RL, Zart MK, Powell DR, Day VW, Borovik AS. Inorg Chem. 2008;47:5780–5786. doi: 10.1021/ic800048e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Wada A, Honda Y, Yamaguchi S, Nagatomo S, Kitagawa T, Jitsukawa K, Masuda H. Inorg Chem. 2004;43:5725–5735. doi: 10.1021/ic0496572. [DOI] [PubMed] [Google Scholar]; (b) Jitsukawa K, Oka Y, Yamaguchi S, Masuda H. Inorg Chem. 2004;43:8119–8129. doi: 10.1021/ic0494399. [DOI] [PubMed] [Google Scholar]; (c) Yamaguchi S, Kumagai A, Funahashi Y, Jitsukawa K, Masuda H. Inorg Chem. 2003;42:7698–7700. doi: 10.1021/ic034962t. [DOI] [PubMed] [Google Scholar]; (d) Ogo S, Yamahara R, Roach M, Suenobu T, Aki M, Ogura T, Kitagawa T, Masuda H, Fukuzumi S, Watanabe Y. Inorg Chem. 2003;41:5513–5520. doi: 10.1021/ic0200040. [DOI] [PubMed] [Google Scholar]; (e) Wada A, Ogo S, Nagamoto S, Kitagawa T, Watanabe Y, Jitsukawa K, Masuda H. Inorg Chem. 2002;41:616–618. doi: 10.1021/ic001058h. [DOI] [PubMed] [Google Scholar]; (f) Harata M, Jitsukawa K, Masuda H, Einaga H. J Am Chem Soc. 1994;116:10817–10818. [Google Scholar]; (g) Wada A, Harata M, Hasegawa K, Jitsukawa K, Masuda H, Mukai M, Kitagawa T, Einaga H. Angew Chem, Int Ed. 1998;37:798–799. doi: 10.1002/(SICI)1521-3773(19980403)37:6<798::AID-ANIE798>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]; (h) Ogo S, Wada S, Watanabe Y, Iwase M, Wada A, Harata M, Jitsukawa K, Masuda H, Einaga H. Angew Chem, Int Ed. 1998;37:2102–2104. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2102::AID-ANIE2102>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 4.Selected examples: Makowska-Grzyska MM, Jeppson PC, Allred RA, Arif AM, Berreau LM. Inorg Chem. 2002;41:4872–4887. doi: 10.1021/ic0255609.Rudzka K, Arif AM, Berreau LM. J Am Chem Soc. 2007;128:17018–17023. doi: 10.1021/ja0601336.Natale D, Mareque-Rivas JC. Chem Commun. 2008:425–437. doi: 10.1039/b709650j.Feng G, Mareque-Rivas JC, Williams NH. Chem Commun. 2006:1845–1847. doi: 10.1039/b514328d.Mareque-Rivas JC, Hinchley SL, Metteau L, Parsons S. Dalton Trans. 2006:2316–2322. doi: 10.1039/b516234c.Mareque-Rivas JC, Prabaharan R, Torres Martín de Rosales R, Metteau L, Parsons S. Dalton Trans. 2004:2800–2807. doi: 10.1039/B407790C.Mareque-Rivas JC, Torres Martín de Rosales R, Parsons S. Chem Commun. 2004:610–611. doi: 10.1039/b314616b.Mareque-Rivas JC, Prabaharan R, Parsons S. Dalton Trans. 2004:1648–1655. doi: 10.1039/b402084g.Mareque-Rivas JC, Salvagni E, Prabaharan R, Torres Martín de Rosales R, Parsons S. Dalton Trans. 2004:172–177. doi: 10.1039/b312221b.Mareque-Rivas JC, Salvagni E, Torres Martín de Rosales R, Parsons S. Dalton Trans. 2003:3339–3349.
- 5.Shook RL, Gunderson WA, Greaves J, Ziller JW, Hendrich MP, Borovik AS. J Am Chem Soc. 2008;130:8888–8889. doi: 10.1021/ja802775e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.TAO should be dissolved in an appropriate solvent (e.g. MeOH, DCM or chloroform) and stored as a solution for later use. Soild TAO eventually becomes an insoluble transparent gel after ~5 days. The time periods for gelling can vary and appears to depend on the degree of drying. Ring opening polymerization of oxazoline group might be one of the possible explanations that leads to gel formation. Lobert M, Thijs HML, Erdmenger T, Eckardt R, Ulbricht C, Hoogenboom R, Schubert US. Chem Eur J. 2008;14:10396–10407. doi: 10.1002/chem.200800671. Detailed investigations of this process are on the way.
- 7.(a) Remko M, Walsh OA, Richards WG. Phys Chem Chem Phys. 2001;3:901–907. [Google Scholar]; (b) Remko M, Walsh OA, Richards WG. Chem Phys Lett. 2001;336:156–162. [Google Scholar]
- 8.(a) Ferreira CMP, Guedes da Silva MFC, Michelin RA, Kukushkin VY, Fraústo da Silva JJR, Pombeiro AJL. Dalton Trans. 2003:3751–3756. [Google Scholar]; (b) Decken A, Gossage RA. J Inorg Biochem. 2005;99:664–667. doi: 10.1016/j.jinorgbio.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 9.An additional small peak was observed at 8.06 ppm., whose identity has yet to be determined. We suggest that this other peak does not come from impurities because it always appears at the same chemical shift and its integration ratio is constant upon repeated purifications by recrystallization or separate syntheses. Furthermore, it exhibited the same temperature dependency as the two major NH peaks in VT-NMR experiments.
- 10.A possible dissociation of the bound acetate ligand at elevated temperature could also account for these observations.
- 11.Reid SD, Wilson C, Blake AJ, Love JB. Dalton Trans. 2010 doi: 10.1039/b909842a. Advance Article. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.