

Abstract
The effects of redox-inactive metal ions on dioxygen activation were explored using a new FeII complex containing a tripodal ligand with 3 sulfonamido groups. This iron complex exhibited a faster initial rate for the reduction of O2 than its MnII analog. Increases in initial rates were also observed in the presence of group 2 metal ions for both the FeII and MnII complexes, which followed the trend NMe4+ < BaII < CaII = SrII. These studies led to the isolation of heterobimetallic complexes containing FeIII-(μ-OH)-MII cores (MII = Ca, Sr, and Ba) and one with a [SrII(OH)MnIII]+ motif. The analogous [CaII(OH)GaIII]+ complex was also prepared and its solid state molecular structure is nearly identical to that of the [CaII(OH)FeIII]+ system. Nuclear magnetic resonance studies indicated that the diamagnetic [CaII(OH)GaIII]+ complex retained its structure in solution. Electrochemical measurements on the heterobimetallic systems revealed similar one-electron reduction potentials for the [CaII(OH)FeIII]+ and [SrII(OH)FeIII]+ complexes, which were more positive than the potential observed for [BaII(OH)FeIII]+. Similar results were obtained for the heterobimetallic MnII complexes. These findings suggest that Lewis acidity is not the only factor to consider when evaluating the effects of group 2 ions on redox processes, including those within the oxygen-evolving complex of Photosystem II.
Introduction
Group 2 metal ions are known to influence a variety of chemical transformations.1 Compounds containing these metal ions make attractive components in designing reactions because they are earth-abundant Lewis acids. Furthermore, group 2 ions have been shown to affect transition metal-mediated redox processes, with a growing number of examples spanning both natural and synthetic systems.2,3 For instance, the oxygen-evolving complex (OEC) within Photosystem II represents a rare example of a CaII ion contained within an Mn4O5 cluster.4 It is widely accepted that the CaII ion is an essential cofactor for oxidation of two water molecules to dioxygen, yet the exact function(s) of the CaII ion is still debated.5,6,7 Among the various Lewis acidic cations that have a similar size as a CaII ion (e.g., SrII and CdII ions) or that are stronger Lewis acids (MgII and LaIII ions), only the SrII ion exhibits comparable function to CaII, albeit at a slower turnover rate.5 These observations have led to the suggestion that there might be a unique combination of size and Lewis acidity that is needed to promote function.8
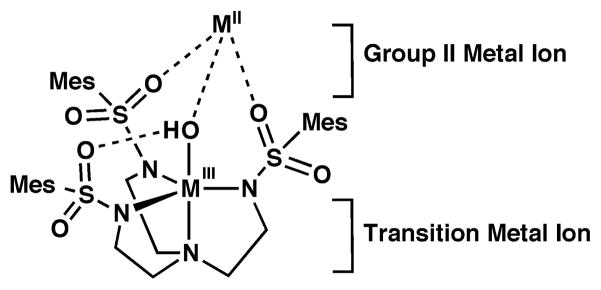
Efforts to examine these effects have been hindered because heterometallic complexes containing group 2 ions are often difficult to prepare. While notable advances have been made,2a,9 there is still a need to develop molecular systems that are capable of binding a transition metal ion(s) and one group 2 ion. We recently reported the development of the tripodal ligand, N,N′,N″-[2,2′,2″-nitrilotris(ethane-2,1-diyl)]tris-(2,4,6-trimethylbenzene-sulfonamido, [MST]3−), that allowed for the isolation of heterobimetallic complexes with MII-(μ-OH)-MnIII cores (MII = Ca and Ba, denoted: [MII(OH)MnIII]+).10,11 The [MST]3− ligand (Chart 1) displayed two metal ion binding sites: the manganese ion coordinated to the nitrogen atom donors while the group 2 metal ion bound to the two oxygen atoms of the sulfonamido groups. These heterobimetallic complexes were synthesized by treating [MnIIMST]− with dioxygen in the presence of either CaII or BaII ions, which caused an enhancement in the rate of O2 activation. We have extended this study to now include the effect of SrII ions on this reduction process, leading to the structural characterization of the [SrII(OH)MnIII]+ complex. Moreover, we have explored dioxygen reduction with the [FeIIMST]− analog and various group 2 metal ions, and found increased rates compared to those found for the manganese series. The solid-state molecular structures of the three heterobimetallic complexes [MII(OH)FeIII]+ (MII = Ca, Sr, Ba) are described, as well as their physical properties. To further probe the structural properties for this class of heterobimetallic complexes, we prepared the [CaII(OH)GaIII]+ species, a diamagnetic analog of [CaII(OH)FeIII]+. Nuclear magnetic resonance (NMR) studies of [CaII(OH)GaIII]+ indicated that it retains its heterobimetallic structure in solution in which the [CaII(15-crown-5)]2+ unit is coordinated to the [GaIIIMST(OH)]− complex. An important outcome of this work is that CaII and SrII ions have similar effects on the reactivity of the FeII and MnII complexes of [MST]3− and the electrochemical properties of the corresponding heterobimetallic complexes; these findings are similar to what is observed in the OEC and do not correlate with Lewis acidity of the group 2 metal.
Chart 1.
Experimental Section
General Methods
All reagents were purchased from commercial sources and used as received unless otherwise noted. Solvents were sparged with argon and dried over columns containing Q-5 and molecular sieves. Sodium hydride (NaH) as a 30% dispersion in mineral oil was filtered with a medium porosity glass frit and washed 5 times each with pentane and Et2O. Solid NaH was dried under a vacuum and stored under an inert atmosphere. The synthesis of the ligand was carried out in the air and the preparations of the metal complexes were conducted in a Vacuum Atmospheres, Co. drybox under an argon atmosphere. Dioxygen was dried on a Drierite gas purifier purchased from Fischer Scientific. 18O2 was obtained from Cambridge Isotopes Laboratory (CIL) (Andover, MA). H3MST, Ca(OTf)2/15-crown-5 and Ba(OTf)2/18-crown-6 were synthesized according to the previous report.10 Sr(OTf)2 was prepared by the literature procedure.12
Complex Synthesis
Preparation of [NMe4][FeIIMST]
A solution of H3MST (400 mg, 0.60 mmol) dissolved in 20 mL of anhydrous dimethylacetamide (DMA) was treated with solid NaH (42 mg, 1.8 mmol). The mixture was stirred until gas evolution ceased. Fe(OAc)2 (100 mg, 0.60 mmol) and NMe4OAc (77 mg, 0.60 mmol) were added to the pale yellow solution, after which the mixture was stirred for 3 h. After the reaction, the pale yellow turbid mixture was filtered to remove insoluble species. The filtrate was concentrated under vacuum to ca. 5 mL and treated with diethyl ether (Et2O, 30 mL) to precipitate a white solid. The white solid was collected on a medium glass-fritted funnel, washed with diethyl ether (10 mL) and dried under a vacuum to give 445 mg of product (94%). Elemental analysis calcd for [NMe4][FeIIMST], C37H57FeN5O6S3: C, 54.20; H, 7.01; N, 8.54%, found: C, 54.15; H, 6.95; N, 8.71%. FTIR (KBr disc, cm−1, selected bands): 3040, 2975, 2935, 2852, 1603, 1487, 1467, 1405, 1277, 1138, 1072, 1054, 1039, 975, 927, 847, 824, 739, 661, 604, 579, 542.
Preparation of Sr(OTf)2/15-Crown-5-Ether Complex
This complex was synthesized following the procedure previously reported10 with the following modfication: strontium trifluoromethanesulfonate (0.38 g, 1.0 mmol) and 15-crown-5 (0.22 g, 1.0 mmol) were suspended in MeCN (10 mL) and heated at 50°C (heating mantle) for 2 h. The initial suspension became clear during the reaction. The solution was cooled and filtered to remove any insoluble materials. The filtrate was concentrated under vacuum to ca. 4 mL and Et2O (10 mL) was added to precipitate the product. The white solid was filtered and washed twice with Et2O (10 mL). Drying in vacuo gave 0.56 g of Sr(OTf)2/15-crown-5 ether adduct as a white solid in 93% yield; this complex was used without further purification. Elemental analysis confirmed the 1:1 adduct. Elemental analysis calcd for Sr(OTf)2/15-crown-5, C12H20SrF6O11S2: C, 23.78; H, 3.33%, found: C, 23.31; H, 3.08%.
Preparation of [15-crown-5⊃CaII-(μ-OH)-FeIIIMST]OTf
A Schlenk flask was charged with a solution of [NMe4][FeIIMST] (50 mg, 0.060 mmol) and Ca(OTf)2/15-crown-5 adduct (34 mg, 0.060 mmol) in 10 mL of anhydrous CH2Cl2. The flask was sealed with a rubber septum and brought out from the dry box, after which excess O2 (10 mL, T = 298 K, P = 1 atm, 0.40 mmol) was injected into the headspace via syringe and the mixture was stirred overnight at room temperature. During the reaction, the initial pale yellow solution immediately became reddish-yellow in color. After the reaction was stopped, all volatiles were removed under vacuum and the reaction flask was brought into the box. The reddish-orange residue was redissolved in CH2Cl2 (5 mL) and filtered to remove any insoluble materials. The filtrate was layered with pentane. The resulting orange needle-type crystals were collected on a glass frit and dried under vacuum, affording the product in yields that ranged from 60–70%. Elemental analysis calcd for [15-crown-5⊃CaII-(μ-OH)-FeIIIMST]OTf·0.5CH2Cl2, C44.5H67CaClF3Fe-N4O15S4: C, 44.00; H, 5.56; N, 4.61%, found: C, 44.14; H, 5.31; N, 4.99%. FTIR (KBr disc, cm−1, selected bands): 3375 (OH), 3023, 2968, 2936, 2868, 2745, 1603, 1565, 1468, 1457, 1405, 1382, 1355, 1274, 1223, 1143, 1223, 1143, 1089, 1052, 1030, 954, 872, 852, 812, 741, 659, 637, 611, 573, 540, 517. (Nujol, cm−1): 3369 (OH; FWMH 104).
Preparation of [15-crown-5⊃SrII-(μ-OH)-FeIIIMST]OTf
This salt was prepared using the method described above for the Fe/Ca system with [NMe4][FeIIMST] (50 mg, 0.060 mmol) and Sr(OTf)2/15-crown-5 adduct (37 mg, 0.060 mmol) to produce the desired salt with a yield of 71%. Elemental analysis calcd for [15-crown-5⊃SrII-(μ-OH)-FeIIIMST]OTf·CH2Cl2, C45H68Cl2SrF3-FeN4O15S4: C, 41.43; H, 5.25; N, 4.29%, found: C, 41.20; H, 5.26; N, 4.62%. FTIR (KBr disc, cm−1, selected bands): 3391 (OH), 3022, 2939, 2920, 2863, 1604, 1565, 1467, 1405, 1387, 1353, 1284, 1223, 1144, 1085, 1030, 955, 865, 849, 822, 809, 739, 658, 637, 606, 576, 541, 517. (Nujol, cm−1): 3389 (OH; FWMH 70).
Preparation of [18-crown-6⊃BaII-(μ-OH)-FeIIIMST]OTf
This salt was prepared using the method described above for the Fe/Ca system with [NMe4][FeIIMST] (50 mg, 0.060 mmol) and Ba(OTf)2/18-crown-6 adduct (44 mg, 0.060 mmol) to produce the desired salt in a yield of 85%. X-ray quality crystals were obtained from THF/pentane. Elemental analysis calcd for [18-crown-6⊃BaII-(μ-OH)-FeIIIMST]OTf·THF, C50H78BaF3Fe-N4O17S4: C, 43.34; H, 5.67; N, 4.04%, found: C, 43.16; H, 5.50; N, 4.02%. FTIR (KBr disc, cm−1, selected bands): 3374 (OH), 3022, 2912, 2864, 2751, 1603, 1564, 1468, 1403, 1384, 1352, 1282, 1261, 1225, 1143, 1097, 1034, 965, 818, 742, 658, 637, 612, 576, 542, 516. (Nujol, cm−1): 3371 (OH; FWMH 59).
Preparation of [15-Crown-5⊃CaII-(μ-OH)-GaIIIMST]OTf
A suspension of H3MST (223 mg, 0.321 mmol) and NaH (23.5 mg, 0.979 mmol) in 8 mL of THF was allowed to stir for 4 h, after which the solution became clear. The reaction mixture was transferred to a vial containing 0.5 M GaCl3 in pentane (462 mg, 0.336 mmol), causing fuming, bubbling, and precipitation of a white solid. The reaction was stirred for 2 h and then filtered through a fine-porosity glass-fritted funnel to afford 2–3 equiv of NaCl. The filtrate was dried under reduced pressure, then pentane was added and the residue was scraped loose to yield a white solid. The product was collected on a medium-porosity glass-fritted funnel, washed with 10 mL each of Et2O and pentane, and dried to yield 217 mg (88 %) of a white powder. This solid, formulated as [GaIIIMST], was used without further purification. 1H NMR (500MHz, DMSO-d6, ppm): 2.20 (s, 9H), 2.55 (br t, 6H), 2.68 (s, 18H), 3.09 (br t, 6H), 6.82 (s, 6H). FTIR (KBr, selected bands, cm−1) 2977, 2940, 2875, 1604, 1565, 1460, 1405, 1382, 1290, 1143, 1092, 1061, 977, 815, 665.
A suspension of [GaIIIMST] (50.1 mg, 0.0660 mmol), Me4NOH·5H2O (12.0 mg, 0.0662 mmol), and Ca(OTf)2/15-crown-5 adduct (38.1 mg, 0.0682 mmol) in 4 mL of THF was stirred for 2 h. The reaction mixture was filtered through a fine-porosity glass-fritted funnel and volatiles were removed under reduced pressure. The residue was dissolved in 3 mL of CH2Cl2 and layered under pentane, yielding colorless crystals that formed over the next 5 d (11.5 mg, 10%). 1H NMR (500MHz, CDCl3, ppm): 2.30 (s, 9H), 2.61 (br t, 6H), 2.75 (s, 18H), 2.95 (br t, 6H), 3.80 (m, 20H), 4.73 (s, OH), 6.95 (s, 6H). 13C{1H} NMR (125 MHz, CDCl3, ppm): 21.0, 23.5, 40.7, 51.8, 68.3, 131.9, 134.7, 139.7, 141.6. FTIR (KBr, selected bands, cm−1): 3456 (OH), 2972, 2936, 2877, 1604, 1565, 1469, 1457, 1355, 1274, 1147, 1090, 1031, 977, 832, 662, 638. HRMS (ES+): Exact mass calcd for C43H66CaGaN4O12S3+ [M+], 1035.2721. Found 1035.2698.
Preparation of [15-Crown-5⊃SrII-(μ-OH)-GaIIIMST]OTf
This salt was prepared using the method described above for the [CaII(OH)GaIII]+ system with [GaIIIMST] (49.1 mg, 0.0646 mmol), Me4NOH·5H2O (12.1 mg, 0.0668 mmol), and Sr(OTf)2/15-crown-5 adduct (49 mg, 0.0809 mmol) to produce the desired salt with a crystalline yield of 30%. 1H NMR (500MHz, CDCl3, ppm): 2.31 (s, 9H), 2.61 (br t, 6H), 2.75 (s, 18H), 2.94 (br t, 6H), 3.82 (m, 20H), 4.49 (s, OH), 6.95 (s, 6H). 13C{1H} NMR (125 MHz, CDCl3, ppm): 21.0, 23.4, 40.9, 52.6, 68.6, 131.8, 135.0, 139.6, 141.5. FTIR (KBr, selected bands, cm−1) ν(OH): 3504; 2933, 2882, 2866, 1604, 1472, 1353, 1285, 1148, 1090, 1030, 978, 952, 832, 662, 638. HRMS (ES+): Exact mass calcd for C43H66GaN4O12S3Sr+ [M+], 1083.2157. Found 1083.2145.
Preparation of [15-crown-5⊃SrII-(μ-OH)-MnIIIMST]OTf
This salt was prepared using the method described for the previously reported [CaII(OH)MnIII]+ complex with [NMe4][MnIIMST] (100 mg, 0.12 mmol) and Sr(OTf)2/15-crown-5 (74 mg, 0.12 mmol) to produce the desired salt in yields ranging from 30–40%.10 FTIR (KBr disc, cm−1, selected bands): 3305 (OH), 3023, 2934, 2882, 2746, 1604, 1562, 1469, 1404, 1356, 1278, 1140, 1088, 1054, 1031, 970, 956, 867, 830, 743, 723, 662, 638, 575, 542, 517. (Nujol, cm−1): 3290 (OH; FWMH 104). UV-vis: λmax (DCM, ε M−1 cm−1) = 455(339), 638(572), 757(sh, 494). HRMS (ES+): Exact mass calcd for C43H66FeN4O12S3Sr+ [M+], 1069.2280. Found 1069.2292.
Physical Methods
Electronic absorbance spectra were recorded with a Cary 50 or an Agilent 8453 spectrophotometer using a 1.00 cm quartz cuvette. Fourier transform infrared (FTIR) spectra were collected on a Varian 800 Scimitar Series FTIR spectrometer. 1H NMR and 13C NMR spectra were recorded on a Bruker DRX500 spectrometer. Cyclic voltammetric experiments were conducted using a CHI600C electrochemical analyzer. A 2.0 mm glassy carbon electrode was used as the working electrode at scan velocities 0.1 Vs−1. A ferrocenium/ferrocene couple ([FeCp2]+/0) was used as an internal reference to monitor the reference electrode (Ag+/Ag). Perpendicular-mode X-band electron paramagnetic resonance (EPR) spectra were collected using a Bruker EMX spectrometer at 4 K using liquid helium. Elemental analyses were performed on a Perkin-Elmer 2400 CHNS analyzer. Crystals for X-ray diffraction were mounted on a Bruker SMART APEX II diffractometer and the APEX2 program package was used to determine the unit-cell parameters and for data collection.‡
Kinetic Studies
Kinetic measurements for the activation of dioxygen with [FeIIMST]− and [MnIIMST]− complexes were conducted on an Agilent 8453 UV-vis spectrometer equipped with a Unisoku Unispeks cryostat. The studies of [FeIIMST]− were performed at 20.0°C on a 0.30 mM CH2Cl2 solution that was prepared in an Ar-filled drybox. Three separate 12 mL aliquots (3.6 μmol of [FeIIMST]−) of the solution were premixed with 3 equiv of MII(OTf)2/crown (MII = Ca, Sr, Ba). A 3 mL portion was transferred to a 1.0 cm quartz cuvette, which was sealed with a rubber septum and removed from the box. Excess O2 (5 mL, T = 298 K, P = 1 atm, 0.20 mmol) was added to the headspace of the cuvette via a gas-tight syringe and the absorbance change was monitored every 3 s at 383 nm. For studies with [MnIIMST]−, a 3.0 mL portion (11 μmol of [MnIIMST]−) of a 3.5 mM solution of complex was transferred to a cuvette and sealed with a rubber septum. Immediately before addition of 5 mL of O2 into the headspace, 0.50 mL of a 21 mM MII/15-crown-5(OTf)2 solution (MII = Ca, Sr, 11 μmol, 1 equiv) were injected directly into the solution, giving a final [MnIIMST]− concentration of 3.0 mM. The absorbance change was monitored at 640 nm every 10 seconds at 25.0°C. All experiments were repeated 3 times for [MnIIMST]− and 6 times for [FeIIMST]−. Note that the kinetic experiments with [FeIIMST]− and [MnIIMST]− were conducted at different temperatures (20.0°C and 25.0°C, respectively); these temperatures were utilized because they gave the best reproducibility.
Results and Discussion
Preparative Routes
The preparation of [FeIIMST]− was achieved via the synthetic route outlined in Scheme 1. Treating [FeIIMST]− with excess O2 in the presence of 1 equiv Ca(OTf)2/15-crown-5 produced [15-crown-5⊃CaII-(μ-OH)-FeIIIMST]+ (denoted [CaII(OH)FeIII]+, Scheme 1), a complex with similar composition to its [MnIII-(μ-OH)-CaII]+ analog. Unlike the [MnIIMST]− case, which took over 10 h to reach completion, the reaction with [FeIIMST]− was finished within 5 min as monitored by UV/vis spectroscopy (Fig. 1). The major product from the reaction, [CaII(OH)FeIII]+, was obtained in yields ranging from 60 to 70% as a crystalline orange solid after recrystallization from CH2Cl2/pentane mixtures. A solution of the isolated crystals has a visible absorbance spectrum with features at λmax (εM) = 383 nm (4800) (Fig. 1, inset) that matches the final spectrum obtained from the reaction.
Scheme 1.

Preparative routes using [FeIIMST]−.
Fig. 1.

Electronic absorbance spectra for the reduction of dioxygen by [FeIIMST]− in the presence of Ca(OTf)2/15-crown-5 in CH2Cl2 at 20°C. Spectra were recorded every 30 sec. Inset is the spectrum for the isolated product, [CaII(OH)FeIII]+.
We have also prepared the analogous [SrII(OH)FeIII]+ complex via dioxygen activation using the same synthetic procedure outlined in Scheme 1. In addition, the [BaII(OH)FeIII]+ complex was isolated using 18-crown-6 as the ancillary ligand to the BaII center. The optical spectra measured on CH2Cl2 solutions of these two isolated salts had nearly identical features as their [CaII(OH)FeIII]+ counterpart. For example, the electronic absorbance spectrum for [SrII(OH)FeIII]+ contained a broad absorbance band at λmax = 385 nm (Fig. S1). EPR spectra measured at 4 K for the [MII(OH)FeIII]+ complexes are consistent with the FeIII centers having S = 5/2 spin ground states, with g-values at 9.3, 4.6, and 4.0 (Figs. S2 and S3).
The synthesis of the [CaII(OH)GaIII]+ complex required a different procedure in which the hydroxo ligand was installed using Me4NOH (Scheme 2). [GaIIIMST], prepared from GaCl3 and [MST]3− in 80% yield, was treated with Me4NOH·5H2O in the presence of one equiv of Ca(OTf)2/15-crown-5. The complex was isolated as colorless crystals from CH2Cl2/pentane.
Scheme 2.

Preparative routes using [GaIIIMST].
Vibrational Properties of the [MII(OH)FeIII]+ Complexes
The solid-state vibrational properties of the [MII(OH)FeIII]+ were assessed using FTIR spectroscopy. In particular, the ν(OH) bands were comparable, with [SrII(OH)FeIII]+ having the highest energy peak at 3389 cm−1 (Fig. 2). However, the band for [CaII(OH)FeIII]+ at ν(OH) = 3369 cm−1 was considerably broader than those in the other complexes; its full-width at half-maximum value of 104 cm−1 is larger than the 70 and 59 cm−1 found for [SrII(OH)FeIII]+ and [(OTf)BaII(OH)FeIII], respectively.13 This difference in peak widths suggests that the intramolecular H-bond in [CaII(OH)FeIII]+ should be the strongest within the series.
Fig. 2.

FTIR spectra in Nujol of [CaII(OH)FeIII]+ (grey), [SrII(OH)FeIII]+ (black) and [BaII(OH)FeIII]+ (dashed black).
We have used the [SrII(OH)FeIII]+ complex to verify that the oxygen atom of the hydroxo ligand was derived from dioxygen.14 Preparing the complexes under an 18O2 atmosphere produced [SrII(18OH)FeIII]+, which had a band for the ν(OH) at 3379 cm−1. The observed shift of 10 cm−1 agrees with the value expected from the 18OH isotopomer based on a harmonic O–H oscillator (FTIR: ν(16OH)/ν(18OH) = 1.003; calcd 1.003, Fig. S4).
Solid-State Molecular Structures
[CaII(OH)FeIII]+ Complex
The molecular structure of [15-crown-5⊃CaII-(μ-OH)-FeIIIMST]OTf was determined by X-ray diffraction methods to reveal the heterobimetallic complex with an FeIII-(μ-OH)-CaII core (Fig. 3, Table 1), which is isomorphous to that of the MnIII-(μ-OH)-CaII complex that was reported previously.10 The FeIII center has an N4O primary coordination sphere that adopts a distorted trigonal-bipyramidal geometry (τ = 0.798). The nitrogen atoms of the [MST]3− ligand coordinate to the FeIII ion with an Fe1–N1 bond length of 2.230(2) Å and an average Fe1–Neq bond distance of 2.029(2) Å. The primary coordination sphere of the iron center is completed by a hydroxo ligand having an Fe1–O7 bond distance of 1.865(2) Å and an N1-Fe1-O7 bond angle of 173.57(7)°. The configuration of the SO2Ar groups produced a cavity that formed an intramolecular H-bond between the FeIII–OH unit and O5 of the [MST]3− ligand with an O7···O5 distance of 2.700(6) Å.15 The CaII ion is coordinated to O1 and O3 of [MST]3− with bond distances of 2.344(2) and 2.370(2) Å, and to the hydroxo oxygen atom with a Ca1–O7 bond length of 2.316(2) Å. The CaII center was 6-coordinate with the oxygen atoms of the 15-crown-5 occupying the remaining binding sites on the CaII ion.
Fig. 3.

Thermal ellipsoid diagram depicting the molecular structure of [CaII(OH)FeIII]+. Ellipsoids are drawn at the 50% probability level, and only the hydroxo hydrogen atom is shown for clarity.
Table 1.
Selected metrical parameters for the MII-(μ-OH)-FeIII complexes.
| [MII(OH)FeIII]+ | MII | ||
|---|---|---|---|
| Ca | Sr | Ba | |
| Bond Distances (Å) | |||
| Fe1—N1 | 2.230(2) | 2.280(3) | 2.284(2) |
| Fe1—N2 | 2.024(2) | 2.009(3) | 2.016(2) |
| Fe1—N3 | 2.016(2) | 2.010(3) | 2.021(2) |
| Fe1—N4 | 2.046(2) | 2.024(3) | 2.048(2) |
| Fe1—O7 | 1.865(2) | 1.872(2) | 1.859(2) |
| O7···O5 | 2.700(6) | 2.685(6) | 2.692(6) |
| M1—O7 | 2.316(2) | 2.464(2) | 2.695(2) |
| M1···O1 | 2.344(2) | 2.538(3) | 2.773(2) |
| M1···O3 | 2.370(2) | 2.547(2) | 2.756(2) |
| Fe1···M1 | 3.719(2) | 3.839(2) | 4.174(2) |
| M1—Otriflate | – | – | 2.729(2) |
| Avg M1—Ocrown | 2.508(2) | 2.619(5) | 2.866(3) |
| d[M––Neq] | 0.392 | 0.399 | 0.431 |
| d[M1—Ocrown] | 1.156 | 1.334 | 0.776 |
|
| |||
| Bond Angles (°) | |||
| O7-Fe1-N1 | 173.57(7) | 175.98(9) | 172.91(9) |
| O7-Fe1-N2 | 100.87(8) | 100.45(10) | 107.41(9) |
| O7-Fe1-N3 | 106.18(8) | 110.40(11) | 103.49(8) |
| O7-Fe1-N4 | 97.14(7) | 99.67(10) | 96.40(8) |
| N1-Fe1-N2 | 78.73(8) | 79.27(10) | 78.42(9) |
| N1-Fe1-N3 | 79.88 (8) | 79.45(10) | 77.95(8) |
| N1-Fe1-N4 | 78.16(8) | 77.10(10) | 76.95(9) |
| N2-Fe1-N3 | 109.27(8) | 110.40(11) | 108.37(9) |
| N3-Fe1-N4 | 113.94(8) | 119.41(11) | 118.34(9) |
| N2-Fe1-N4 | 125.71(9) | 118.64(11) | 120.05(9) |
| Fe1-O7-M1 | 125.27(7) | 124.01(10) | 132.02(8) |
| τ value | 0.798 | 0.943 | 0.880 |
[SrII(OH)FeIII]+ and [SrII(OH)MnIII]+ Complexes
We have also structurally characterized heterobimetallic complexes that contain SrII ions in the place of CaII ions, i.e. [15-crown-5⊃SrII-(μ-OH)-FeIIIMST]OTf (Fig. 4, Table 1) and [15-crown-5⊃SrII-(μ-OH)-MnIIIMST]OTf (Fig. S5, Table S3). The molecular structures of these [SrII(OH)MIII]+ complexes are nearly identical to those with CaII ions, as all used 15-crown-5 as the ancillary ligand to the group 2 ion and contain MII-(μ-OH)-MIII cores. The transition metal center in [SrII(OH)MIII]+ was five-coordinate with a trigonal-bipyramidal geometry and the bridging hydroxo ligand also formed an intramolecular H-bond with a sulfonamido oxygen atom of the [MST]3− ligand (for instance, an O7···O5 distance of 2.685(6) Å was found in [SrII(OH)FeIII]+).
Fig. 4.

Thermal ellipsoid diagram depicting the molecular structure of [SrII(OH)FeIII]+. Ellipsoids are drawn at the 50% probability level, and only the hydroxo hydrogen atom is shown for clarity.
The bond distances from the transition metal center to the equatorial nitrogen atoms within the two [SrII(OH)MIII]+ complexes were comparable, with an average M1–Neq distance of 2.063(3) Å for the [SrII(OH)MnIII]+ complex and a slightly shorter distance of 2.014(3) Å for the [SrII(OH)FeIII]+ complex. In contrast, the axial bond distances to the apical nitrogen atom and to the oxygen atom of the hydroxo unit were longer in the [SrII(OH)FeIII]+ complex than in the [SrII(OH)MnIII]+ complex (M1–N1 = 2.049(3) Å for [SrII(OH)MnIII]+ versus 2.280(3) Å for [SrII(OH)FeIII]+, and M1–O7 = 1.826(2) Å for [SrII(OH)MnIII]+ versus 1.872(2) Å for [SrII(OH)FeIII]+). These structural observations are consistent with the differences in electron configurations between the high-spin FeIII (d5) and MnIII (d4) complexes. Because the MIII centers in the [SrII(OH)MIII]+ complexes have local C3 symmetry, the dz2 orbital is a σ-antibonding orbital and is highest in energy within the d-manifold. Thus, the d5 electron configuation for the FeIII center in [SrII(OH)FeIII]+ places a single electron in the σ* orbital while this orbital remains empty in the [SrII(OH)MnIII]+ analog. A similar trend was observed in the [CaII(OH)MIII]+ complexes of Mn and Fe as well as in other MIII–O(H) complexes (MIII = Mn, Fe) with local C3 symmetry.16
[(OTf)BaII(OH)FeIII] Complex
The [(OTf)BaII(OH)FeIII] complex, [(OTf)18-crown-6⊃BaII-(μ-OH)-FeIIIMST] (Fig. 5, Table 1), contained a similar heterobimetallic core structure with a bridging hydroxo ligand having an Fe1–O7 bond length of 1.859(2) Å and an O7-Fe1-N1 bond angle of 172.91(9)°. However, because [(OTf)BaII(OH)FeIII] needed 18-crown-6 to stabilize the complex, the BaII center was 10-coordinate in the solid state with a triflate ion occupying one of the coordination sites. In addition, the BaII ion sits further within the plane formed by the crown ether oxygen atoms (d[MII−Ocrown] = 0.776 Å) compared to the CaII and SrII analogs (1.156 Å and 1.334 Å, respectively). Despite these differences, some general structural trends were found within the series of [MII(OH)FeIII] complexes. The MII–OMST and MII–O7 bond distances increased proportionally with the size of the group 2 metal ion (Table 1) as expected based on their ionic radii. In addition, the MII···FeIII distances increased in a similar manner with the largest separation found in [(OTf)BaII(OH)FeIII]. However, the effects on the metrical parameters around the iron centers did not follow any clear trends. For instance, the Fe1–O7 bond lengths and O7···O5 distances in the three complexes were equivalent within experimental error, suggesting that these parameters were not sensitive to the choice of group 2 metal ion. Moreover, the index of trigonality parameter (τ)17 did not follow any specific trend, with [SrII(OH)FeIII]+ having the largest τ-value of 0.943 (Table 1). These structural findings do show that the [MST]3− ligands can serve as a platform for isolating a range of heterobimetallic complexes containing one transition metal ion and one group 2 metal ion.
Fig. 5.

Thermal ellipsoid diagram depicting the molecular structure of [(OTf)BaII(OH)FeIII]. Ellipsoids are drawn at the 50% probability level, and only the hydroxo hydrogen atom is shown for clarity.
[CaII(OH)GaIII]+ Complex
The paramagnetic nature of the FeIII and MnIII complexes described above limited the elucidation of their solution-state structures. Therefore, we synthesized the diamagnetic [CaII(OH)GaIII]+ complex to serve as a structural probe for the heterobimetallic complexes of iron. The ionic radii of 5-coordinate GaIII and FeIII ions are nearly identical (GaIII = 0.55 Å versus FeIII = 0.58 Å), which imparts similar Lewis acidities and coordination properties on the two ions.18 These similarities are apparent in the solid-state structures of [CaII(OH)GaIII]+ and [CaII(OH)FeIII]+. The complexes are isomorphous, with most deviations in the metrical parameters varying by the difference in the respective ionic radii (Fig. 6, Table 2). For example, the average MIII–Neq bond distances are 1.992 Å for the GaIII complex and 2.029(2) Å for the FeIII complex. In addition, the MIII–N1 and MIII–O7 distances (1.845 Å and 2.213 Å for [CaII(OH)GaIII]+ versus 1.865(2) Å and 2.230(2) Å for [CaII(OH)FeIII]+, respectively) differ by only 0.02 Å. Further indication of the structural similarities between the GaIII and FeIII complexes can be seen in the τ values, which are 0.793 and 0.798, respectively. The analogous [SrII(OH)GaIII]+ complex was also isolated, but single crystals suitable for X-ray diffraction studies could not be obtained.
Fig. 6.

Thermal ellipsoid diagram depicting the molecular structure of [CaII(OH)GaIII]+. Ellipsoids are drawn at the 50% probability level, and only the hydroxo hydrogen atom is shown for clarity.
Table 2.
Selected metrical parameters for the [CaII(OH)-GaIII]+ complex.
| Bond Distances (Å) | |
|---|---|
| Ga1—N1 | 2.209(2) |
| Ga1—N2 | 1.986(2) |
| Ga1—N3 | 1.986(2) |
| Ga1—N4 | 2.010(2) |
| Ga1—O7 | 1.847(2) |
| O7···O5 | 2.688(6) |
| Ca1—O7 | 2.296(2) |
| Ca1···O1 | 2.344(2) |
| Ca1···O3 | 2.385(2) |
| Ga1···Ca1 | 3.712(2) |
| Avg M1—Ocrown | 2.486(2) |
| d[Ga––Neq] | 0.335 |
| d[M1—Ocrown] | 1.156 |
|
| |
| Bond Angles (°) | |
| O7-Ga1-N1 | 173.31(8) |
| O7-Ga1-N2 | 96.82(8) |
| O7-Ga1-N3 | 105.22(8) |
| O7-Ga1-N4 | 97.62(8) |
| N1-Ga1-N2 | 79.93(8) |
| N1-Ga1-N3 | 81.44(8) |
| N1-Ga1-N4 | 79.81(9) |
| N2-Ga1-N3 | 111.47(9) |
| N3-Ga1-N4 | 114.51(9) |
| N2-Ga1-N4 | 125.65(10) |
| Ga1-O7-Ca1 | 126.94(9) |
| τ value | 0.794 |
Solution Studies on the [CaII(OH)GaIII]+ Complex
To probe the solution structure of [CaII(OH)GaIII]+, the complex was analyzed using nuclear magnetic resonance (NMR) spectroscopy. In CDCl3 at 298 K, the 1H NMR spectrum contained resonances at 2.75 and 2.95 ppm associated with the ethylene units in the [MST]3− backbone, and signals from the mesityl groups appeared at 2.30, 2.75, and 6.95 ppm (Fig. 7). The presence of single resonances for each proton signal in [MST]3− is consistent with the complex exhibiting C3 symmetry in solution. The hydroxo proton resonance was located at 4.73 ppm, and its identity was verified by D2O exchange experiments. A doublet of multiplets centered at 3.80 ppm was observed for the 15-crown-5 signal, which is normally an averaged singlet when free in solution (3.69 ppm). This suggested that the crown ether protons were diastereotopic, with half the macrocycle facing the cavity and half exposed to the solvent. Nuclear Overhauser Effect Spectroscopy (NOESY) showed through-space interactions with the ortho-methyl groups of [MST]3−, the hydroxo ligand, and the crown ether (Fig. 8), which is consistent with the spatial proximity of these groups as shown in the solid-state structure (~4 Å). Moreover, the NOE interactions were only observed with one half of the 15-crown-5 resonances, indicating that the more upfield multiplet resonance corresponded to the protons proximal to the complex cavity. These NMR studies unequivocally illustrate that the [15-crown-5⊃CaII(μ-OH)GaIIIMST]+ complex remains assembled in solution; this property should apply similarly to the Fe analogs.
Fig. 7.

1H NMR spectrum of [CaII(OH)GaIII]+ in CDCl3 at 298 K. Asterisks denote residual solvent peaks.
Fig. 8.

1H NOESY NMR spectrum of [CaII(OH)GaIII]+ in CDCl3 at 298 K. Asterisks denote residual solvent peaks, and the drawn lines indicate NOE interactions.
Comparison of the 1H NMR spectra of the [CaII(OH)GaIII]+ and [SrII(OH)GaIII]+ complexes yielded information about the effect of the group 2 metal ion on the hydroxo unit. For the [SrII(OH)GaIII]+ complex, the hydroxo proton resonance was at 4.49 ppm (Figs. S6 and S7), which is shifted to 4.73 ppm in [CaII(OH)GaIII]+. This difference in chemical shift of the hydroxo proton can be correlated to the difference in the Lewis acidities of the CaII and SrII ions. Binding of a CaII ion should remove more electron density from the OH unit compared to a SrII ion, yielding a more deshielded proton (i.e., a more acidic OH ligand) in [CaII(OH)GaIII]+.
Kinetics of Dioxygen Activation
We explored the effects of group 2 metal ions on the rate of O2 activation with the [FeIIMST]− complex (Fig. S8). In the absence of a group 2 metal ion, [NMe4][FeIIMST] showed sluggish reactivity with O2 at 20°C (Fig. 9, Table 3), having an initial rate of 2.2(1) × 10−4 s−1.19,20 The addition of group 2 metal ions increased the rate of O2 activation, as was expected from the previous studies on [MnIIMST]−.10 Treating a mixture of [NMe4][FeIIMST] and 3 equiv of Ba(OTf)2/18-crown-6 with excess dioxygen produced a modest increase in initial rate to 2.3(6) × 10−3 s−1.21 Significantly greater rate enhancements were found with SrII and CaII ions. The addition of M(OTf)2/15-crown-5 (M = CaII, SrII) gave the statistically identical initial rates of 7.9(4) × 10−3 (for CaII) and 7.4(1.6) × 10−3 s−1 (for SrII). These changes correspond to a greater than 30 fold increase in rate compared to that found for [NMe4][FeIIMST] (Fig. 9A).
Fig. 9.
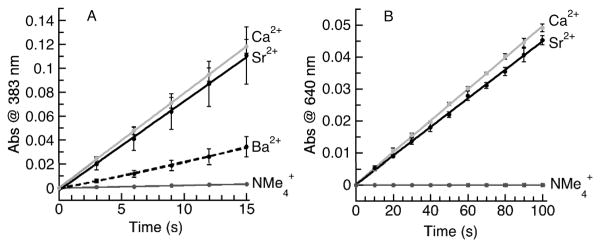
Initial rate data for (A) The reaction of [FeIIMST]− and O2 in the presence of [NMe4]+, 3 equiv of Ba(OTf)2/18-crown-6, 3 equiv of Sr(OTf)2/15-crown-5, and 3 equiv of Ca(OTf)2/15-crown-5. (B) The reaction of [MnIIMST]− and O2 in the presence of 1 equiv of Ca(OTf)2/15-crown-5Ca(OTf)2/15-crown-5 and Sr(OTf)2/15-crown-5.
Table 3.
Initial rate values for the reaction of [FeIIMST]− and O2 in the presence of [NMe4]+, 3 equiv of Ba(OTf)2/18-crown-6, 3 equiv of Sr(OTf)2/15-crown-5, and 3 equiv of Ca(OTf)2/15-crown-5.
| Cation | Initial Rate (s−1) | Rate relative to NMe4+ |
|---|---|---|
| NMe4+ | 2.2 ± 0.1 × 10−4 | 1 |
| Ba(OTf)2/18-crown-6 | 2.3 ± 0.6 × 10−3 | 10 |
| Sr(OTf)2/15-crown-5 | 7.4 ± 1.6 × 10−3 | 34 |
| Ca(OTf)2/15-crown-5 | 7.9 ± 0.4 × 10−3 | 36 |
We also compared the rate enhancements of O2 activation by the [MnIIMST]− complex in the presence of 1 equiv of SrII and CaII ions. Although the rates were not statistically identical as they were in the case of [FeIIMST] −, they differ by less than 10%, with the addition of CaII ions causing a greater rate enhancement than SrII ions (4.9(1) × 10−4 s−1 and 4.5(1) × 10−4 s−1 respectively, Fig. 9B).10,22 Non-redox active metal ions have been reported to 5 accelerate electron-transfer processes by transition metal complexes,1–3 yet the finding that CaII and SrII ions have nearly identical effects on the rates of dioxygen reduction by iron and manganese complexes has not been observed previously. Moreover, this discovery is counter to our initial predicted trend 10 for rate acceleration, which was based on the Lewis acidity scale of group 2 metal ions. The cause(s) for the observed similarities in rate enhancement for CaII and SrII ions are not yet understood.
Redox Properties of the [MII(OH)FeIII]+ and [MII(OH)MnIII]+ Complexes
The redox properties of the [MII(OH)MIII]+ complexes were further explored using cyclic voltammetry (Fig. 10). Each complex has a quasi-reversible one-electron redox process that is assigned to the MIII/MII couple. A striking outcome of this study is the similarity in reduction potential for the CaII and SrII analogs within the two series. In the [MII(OH)FeIII]+ complexes, the difference in potential between [CaII(OH)FeIII]+ (−1.13 V) and [SrII(OH)FeIII]+ (−1.12 V) is only 0.01 V, whereas the potential for [BaII(OH)FeIII]+ was found at the significantly more negative potential of −1.22 V (Fig. 10A).23 Similarly, in the [MII(OH)MnIII]+ series, the MnIII/MnII couples in [CaII(OH)MnIII]+ and [SrII(OH)MnIII]+ differ by only 0.02 V (−0.72 V and −0.70 V, respectively) while the [BaII(OH)MnIII]+ complex lies at a more negative potential (−0.76 V) and is much less reversible (Fig. 10B). The almost identical potentials for the [CaII(OH)MIII]+ and [SrII(OH)MIII]+ complexes again demonstrate the similar effects that these two group 2 ions can impart on the redox properties of transition metal complexes.
Fig. 10.
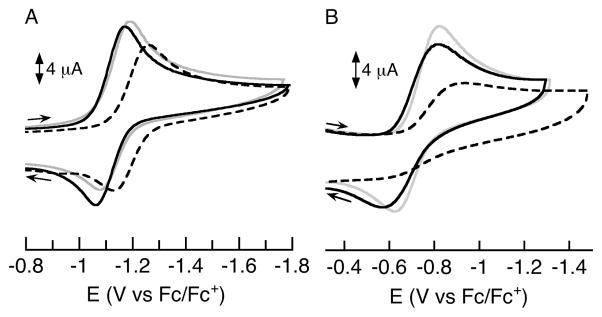
Cyclic voltammograms of the (A) [MII(OH)FeIII]+ and (B) [MII(OH)MnIII]+ complexes of [CaII(OH)MIII]+ (grey), [SrII(OH)MIII]+ (black), and [BaII(OH)MIII]+ (dashed black) showing a quasi-reversible FeII/FeIII or MnII/MnIII redox couple in CH2Cl2 (0.1 M TBAP, 100 mV/s) in the presence of ferrocene as an internal standard.
Summary
The observation that group 2 metal ions have functional consequences within the OEC in Photosystem II5 prompted us to investigate whether similar effects occur in the activation of dioxygen. The studies presented here involved a series of [MII(OH)FeIII]+ complexes formed with a sulfonamido tripod, which further establishes the utility of this ligand system in designing heterobimetallic complexes, especially those containing s-block and transition metal ions. The structural studies built upon our initial work on the related manganese systems and establish a series of heterobimetallic complexes containing MII(OH)FeIII cores. Introduced in this study are the SrII analogs, [SrII(OH)MIII]+, for both the iron(III) and manganese(III) series, which allowed us to compare the influence of these group 2 ions on transition metal-mediated processes. The structural studies were aided by the preparation and characterization of the diamagnetic [CaII(OH)GaIII]+ and [SrII(OH)GaIII]+ complexes. The solid-state structure determined for the [CaII(OH)GaIII]+ complex was found to be isomorphous with the iron analog, and the small differences in metrical parameters between the two complexes correlated with the difference in the ionic radii of the FeIII and GaIII ions (0.03 Å). NMR studies showed that the [MII(OH)GaIII]+ complexes maintain their heterobimetallic structures in solution, suggesting that similar structures are present in solution for the [MII(OH)FeIII]+ analogs. Moreover, the chemical shift for the hydroxo proton in [CaII(OH)GaIII]+ was downfield from that found for [SrII(OH)GaIII]+, which agrees with the trend predicted based on the Lewis acidities of CaII and SrII ions.
In the study of dioxygen activation, we found that CaII or SrII ions had the same initial rates for the formation of the [MII(OH)FeIII]+ complexes, which are significantly faster than the process involving BaII ions. In addition, only 0.01 V separate the one-electron reduction potentials for [CaII(OH)FeIII]+ and [SrII(OH)FeIII]+. Similar results with CaII and SrII ions were found in the manganese series, with the initial rates for O2 reduction differing by less than 10% and the one-electron reduction potentials differing by 0.02 V. The corresponding redox events for the [BaII(OH)MIII]+ were at lower redox potentials. These findings showed that the systems containing BaII ions follow trends based on Lewis acidities, whereas CaII and SrII ions do not follow this relationship, suggesting that electron-transfer processes do not always follow predictions based solely on Lewis acidity.8 Note that our findings for CaII and BaII ions differ from those reported by Fukuzumi, who showed that the conversion of dioxygen to superoxide by CoIITPP is 10 times faster in the presence of CaII ions than SrII ions.2e
In the OEC, CaII and SrII ions are the only the metal ions that combine with the Mn4O4 cluster to promote water oxidation.5a Brudvig has suggested that the similarities in coordination properties and Lewis acidity of CaII and SrII ions provide the correct balance for activity,7c with the CaII derivative reacting faster. This premise asserts that CaII and SrII ions bind to the Mn-cluster with similar coordination geometries and therefore maintain the intricate H-bonding network that surrounds the cluster. Moreover, the pKa of the coordinated water molecule(s) are comparable to ensure proper function. Within our heterobimetallic complexes, we also observed nearly identical molecular structures for the [CaII(OH)MIII]+ and [SrII(OH)MIII]+ complexes, with small metrical differences that can be attributed, in part, to their ionic radii (CaII, 1.12 Å; SrII, 1.26 Å).18a,24 In addition, we observed only small changes in the chemical shifts for the OH protons in [CaII(OH)GaIII]+ and [SrII(OH)GaIII]+ complexes, suggesting only minor differences in acidity. Furthermore, our work illustrates that CaII and SrII ions can have equivalent affects on multi-electron redox processes, which we suggest could also be operative within the OEC. If this were the case, the similar redox potentials for the Mn4O4Ca and Mn4O4Sr clusters should contribute to the comparable function observed for water oxidation.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health, USA (GM50781) for support of this work and Dr. P. Dennison for help in obtaining the NOESY experiments.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/b000000x/
See supporting information for experimental detail.
Notes and References
- 1.General reviews: Yamashita Y, Tsubogo T, Kobayashi S. Chem Sci. 2012;3:967–975.Kobayashi S, Yamashita Y. Acc Chem Res. 2011;44:58–71. doi: 10.1021/ar100101b.Harder S. Chem Rev. 2010;110:3852–3876. doi: 10.1021/cr9003659.Hanusa TP. Coord Chem Rev. 2000;210:329–367.Westerhausen M. Z Anorg Allg Chem. 2009;635:13–32.Westerhausen M. Coord Chem Rev. 2008;252:1516–1531.Westerhausen M, Gärtner M, Fischer R, Langer J, Yu L, Reiher M. Chem Eur J. 2007;13:6292–6306. doi: 10.1002/chem.200700558.Sarish SP, Nembenna S, Nagendran S, Roesky HW. Acc Chem Res. 2011;44:157–170. doi: 10.1021/ar100103h.Barrett AGM, Crimmin MR, Hill MS, Procopious PA. Proc R Soc, A. 2010;466:927–963.
- 2.(a) Kanady JS, Tsui EY, Day MW, Agapie T. Science. 2011;333:733–736. doi: 10.1126/science.1206036. [DOI] [PubMed] [Google Scholar]; (b) Morimoto Y, Kotani H, Park J, Lee YM, Nam W, Fukuzumi S. J Am Chem Soc. 2011;133:403–405. doi: 10.1021/ja109056x. [DOI] [PubMed] [Google Scholar]; (c) Fukuzumi S, Morimoto Y, Kotani H, Naumov P, Lee YM, Nam W. Nature Chem. 2010;2:756–759. doi: 10.1038/nchem.731. [DOI] [PubMed] [Google Scholar]; (d) Karlin KD. Nature Chem. 2010;2:711–712. doi: 10.1038/nchem.813. [DOI] [PubMed] [Google Scholar]; (e) Fukuzumi S, Ohkubo K. Chem Eur J. 2000;6:4532–4535. doi: 10.1002/1521-3765(20001215)6:24<4532::aid-chem4532>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]; (f) Miller CG, Gordon-Wylie SW, Horwitz CP, Strazisar SA, Peraino DK, Clark GR, Weintraub ST, Collins TJ. J Am Chem Soc. 1998;120:11540–11541. [Google Scholar]; (g) Boussac A, Rutherford AW. Biochemistry. 1988;27:3476–3483. [Google Scholar]
- 3.For other examples showing the importance of redox-inactive metal ions in the binding and activation of small molecules, see: Darensbourg MY, Darensbourg DJ, Burns D, Drew DA. J Am Chem Soc. 1976;98:3127–3136.Gambarotta S, Arena F, Floriani C, Zanazzi PF. J Am Chem Soc. 1982;104:5082–5092.Lee Y, Peters JC. J Am Chem Soc. 2011;133:4438–4446. doi: 10.1021/ja109678y.Lee Y, Mankad NP, Peters JC. Nat Chem. 2010;2:558–565. doi: 10.1038/nchem.660.Betley TA, Peters JC. J Am Chem Soc. 2003;125:10782–10783. doi: 10.1021/ja036687f.Ding K, Brennessel WW, Holland PL. J Am Chem Soc. 2009;131:10804–10805. doi: 10.1021/ja902812y.Ding K, Pierpont AW, Brennessel WW, Lukat-Rodgers G, Rodgers KR, Cundari TR, Bill E, Holland PL. J Am Chem Soc. 2009;131:9471–9472. doi: 10.1021/ja808783u.Smith JM, Sadique AR, Cundari TR, Rodgers KR, Lukat-Rodgers G, Lachicotte RJ, Flaschenriem CJ, Vela J, Holland PL. J Am Chem Soc. 2006;128:756–769. doi: 10.1021/ja052707x.Park J, Morimoto Y, Lee YM, Nam W, Fukuzumi S. J Am Chem Soc. 2011;133:5136–5141. doi: 10.1021/ja200901n.Park J, Morimoto Y, Lee YM, You Y, Nam W, Fukuzumi S. Inorg Chem. 2011;50:11612–11622. doi: 10.1021/ic201545a.Rodriguez MM, Bill E, Brennessel WW, Holland PL. Science. 2011;334:780–783. doi: 10.1126/science.1211906.
- 4.Umena Y, Kawakami K, Shen JR, Kamiya N. Nature. 2011;473:55–60. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 5.(a) Yocum CF. Coord Chem Rev. 2008;252:296–305. [Google Scholar]; (b) Ono TA, Inoue Y. FEBS Lett. 1988;227:147–152. [Google Scholar]; (c) Boussac A, Zimmermann JL, Rutherford AW. Biochemistry. 1989;28:8984–8989. doi: 10.1021/bi00449a005. [DOI] [PubMed] [Google Scholar]; (d) Boussac A, Zimmermann JL, Rutherford AW. FEBS Lett. 1990;277:69–74. doi: 10.1016/0014-5793(90)80811-v. [DOI] [PubMed] [Google Scholar]; (e) Ghanotakis DF, Babcock GT, Yocum CF. FEBS Lett. 1984;167:127–130. [Google Scholar]; (f) Sandusky PO, Yocum CF. Biochim Biophys Acta. 1984;766:603–611. [Google Scholar]; (g) Sandusky PO, Yocum CF. Biochim Biophys Acta. 1986;849:85–93. [Google Scholar]
- 6.(a) Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]; (b) Yano J, Kern J, Sauer K, Latimer MJ, Pushkar Y, Biesiadka J, Loll B, Saenger W, Messinger J, Zouni A, Yachandra VK. Science. 2006;314:821–825. doi: 10.1126/science.1128186. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Barber J. Inorg Chem. 2008;47:1700–1710. doi: 10.1021/ic701835r. [DOI] [PubMed] [Google Scholar]; (d) Sauer K, Yano J, Yachandra VK. Coord Chem Rev. 2008;252:318–335. doi: 10.1016/j.ccr.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Guskov A, Kern J, Gabdulkhakov A, Broser M, Zouni A, Saenger W. Nat Struct Mol Biol. 2009;16:334–342. doi: 10.1038/nsmb.1559. [DOI] [PubMed] [Google Scholar]
- 7.(a) Pecoraro VL, Baldwin MJ, Caudle MT, Hsieh WY, Law NA. Pure Appl Chem. 1998;70:925–929. [Google Scholar]; (b) Cady CW, Crabtree RH, Brudvig GW. Coord Chem Rev. 2008;252:444–455. doi: 10.1016/j.ccr.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brudvig GW. Philos Trans R Soc, B. 2008;363:1211–1219. doi: 10.1098/rstb.2007.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mullins CS, Pecoraro VL. Coord Chem Rev. 2008;252:416–443. doi: 10.1016/j.ccr.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Betley TA, Wu Q, VanVoorhis T, Nocera DG. Inorg Chem. 2008;47:1849–1861. doi: 10.1021/ic701972n. [DOI] [PubMed] [Google Scholar]
- 8.For the effect of CaII/SrII substitution on the electronic structure of the OEC, see: Cox N, Rapatskiy L, Su JH, Pantazis DA, Sugiura M, Kulik L, Dorlet P, Rutherford AW, Neese F, Boussac A, Lubitz W, Messinger J. J Am Chem Soc. 2011;133:3635–3648. doi: 10.1021/ja110145v.
- 9.(a) Mishra A, Wernsdorfer W, Abboud KA, Christou G. Chem Commun. 2005:54–56. doi: 10.1039/b413680b. [DOI] [PubMed] [Google Scholar]; (b) Hewitt IJ, Tang JK, Madhu NT, Clérac R, Buth G, Anson CE, Powell AK. Chem Commun. 2006:2650–2652. doi: 10.1039/b518026k. [DOI] [PubMed] [Google Scholar]; (c) Nayak S, Nayek HP, Dehnen S, Powell AK, Reedijk J. Dalton Trans. 2011;40:2699–2702. doi: 10.1039/c0dt01463j. [DOI] [PubMed] [Google Scholar]; (d) Sarish S, Nembenna S, Nagendran S, Roesky HW, Pal A, Herbst-Irmer R, Ringe A, Magull J. Inorg Chem. 2008;47:5971–5977. doi: 10.1021/ic800284u. [DOI] [PubMed] [Google Scholar]; (e) Blake MP, Kaltsoyannis N, Mountford P. J Am Chem Soc. 2011;133:15358–15361. doi: 10.1021/ja207487j. [DOI] [PubMed] [Google Scholar]
- 10.Park YJ, Ziller JW, Borovik AS. J Am Chem Soc. 2011;133:9258–9261. doi: 10.1021/ja203458d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.We incorrectly noted that complexes with tripodal ligands containing sulfonamido group have not been reported;10 in fact, this type of tripodal ligand has been used previously to prepare metal complexes: Schwarz AD, Chu Z, Mountford P. Organometallics. 2010;29:1246–1260.Schwarz AD, Herbert KR, Paniagua C, Mountford P. Organometallics. 2010;29:4171–4188.
- 12.Benetollo F, Bombieri G, Samaria KM, Vallarino LM, Williams JW. Polyhedron. 2001;20:3143–3148. [Google Scholar]
- 13.The solid-state structure of the [(OTf)BaII(OH)FeIII] complex is different from the others in this series because the triflate ion is bonded to the barium center, thus limiting the direct comparison of the vibrational properties.
- 14.We only explored the isotopic effects in [SrII(OH)FeIII]+: observing the isotopic shift in [CaII(OH)FeIII]+ was hindered by the broadness of the ν(OH) band and the relatively slow rate of formation of [BaII(OH)FeIII]+ limited its use in this study.
- 15.An error of 3σ was used in this analysis.
- 16.(a) MacBeth CE, Gupta R, Mitchell-Koch KR, Young VG, Jr, Lushington GH, Thompson WH, Hendrich MP, Borovik AS. J Am Chem Soc. 2004;126:2556–2567. doi: 10.1021/ja0305151. [DOI] [PubMed] [Google Scholar]; (b) MacBeth CE, Hammes BS, Young VG, Jr, Borovik AS. Inorg Chem. 2001;40:4733–4741. [PubMed] [Google Scholar]; (c) MacBeth CE, Golombek AP, Young VG, Jr, Yang C, Kuczera K, Hendrich MP, Borovik AS. Science. 2000;289:938–941. doi: 10.1126/science.289.5481.938. [DOI] [PubMed] [Google Scholar]
- 17.Addison AW, Rao TN, Reedijk J, van Rijn J, Verschoor GC. J Chem Soc Dalton Trans. 1984:1349–1356. [Google Scholar]
- 18.(a) Shannon RD. Acta Crystallogr, Sect A. 1976;32:751–767. [Google Scholar]; (b) Maelia LE, Koch SA. Inorg Chem. 1986;25:1896–1904. [Google Scholar]; (c) Ueno T, Nishikawa N, Moriyama S, Adachi S, Lee K, Okamura T, Ueyama N, Nakamura A. Inorg Chem. 1999;38:1199–1210. doi: 10.1021/ic980710u. [DOI] [PubMed] [Google Scholar]
- 19.All attempts to isolate the major product from this reaction as single crystals were unsuccessful.
- 20.The initial rate for [NMe4][FeIIMST] (2.5(1) × 10−4 s−1) was 40 times faster than that observed for [NMe4][MnIIMST] (6.2 × 10−6 s−1)10 at 25°C in CH2Cl2.
- 21.An excess of group 2 metal ion did not substantially affect the initial rate of the reaction:10 only a small decrease in the initial rates observed above 1 equiv. Three equiv of group 2 ions were used to avoid any fluctuations in the initial rates.
- 22.The initial rate for the reaction of [MnIIMST]− with O2 in the presence of 1 equiv Ca2+ ions has been reanalyzed and found to be 4.9(1) × 10−4 s−1 instead of the rate previously published (5.8(4) × 10−4 s−1);10 however, within experimental error, the rates differ by less than 10%.
- 23.We assume that in solution the triflate ion is not coordinated to the [BaII(μ-OH)MIII]+ complexes.
- 24.The stated ionic radii are for 8-coordinate complexes. The ionic radius for a 10-coordinate BaII ion is 1.61 Å.18a
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.