

Abstract
An abiotic formation of meso- and dl-tartrates in 80% yield via the cyanide-catalyzed dimerization of glyoxylate under alkaline conditions is demonstrated. A detailed mechanism for this conversion is proposed, supported by NMR evidence and 13C-labeled reactions. Simple dehydration of tartrates to oxaloacetate and an ensuing decarboxylation to form pyruvate are known processes that provide a ready feedstock for entry into the citric acid cycle. While glyoxylate and high hydroxide concentration are atypical in the prebiotic literature, there is evidence for natural, abiotic availability of each. It is proposed that this availability, coupled with the remarkable efficiency of tartrate production from glyoxylate, merits consideration of an alternative prebiotic pathway for providing constituents of the citric acid cycle.
Introduction
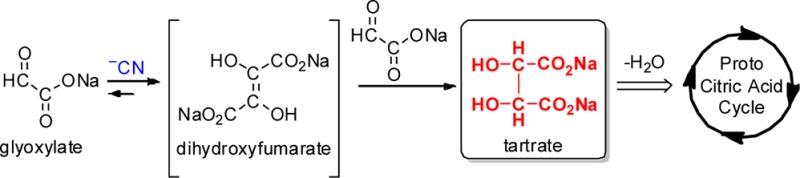
The emergence of primordial metabolism has been postulated to play a central role in the origins of life.1 Many of the investigations so far have centered on the reductive citric acid cycle.2,3 However, in his “glyoxylate scenario”,4 Eschenmoser theorized that glyoxylate 1 may have played an important role in the development of a primordial metabolism, acting as a primal source for biogenic molecules such as sugars, amino acids, and nucleobases. Crucial to the scenario was the hypothetical formation of dihydroxyfumarate (DHF) 5, by dimerization of glyoxylic acid under the influence of an umpolung catalyst such as cyanide.5 DHF was proposed to undergo reaction with glyoxylate and aldoses, ultimately yielding biologically relevant α-keto acids and sugars, respectively.4 Subsequently it was shown by Sagi et al.6 that the reaction of DHF with glyoxylate, glycolaldehyde, and glyceraldehyde led, via α-keto acid intermediates, to the formation of triose, tetrulose, and pentuloses, with remarkable efficiency. This success emphasized the need to demonstrate the formation of DHF from glyoxylate. To this end, we set out to investigate the cyanide-mediated dimerization of glyoxylate (“glyoxoin reaction”) with the expectation of chemistry similar to the benzoin reaction.7 Herein we report the results of this investigation: the unanticipated formation of meso- and dl-tartrates (the formal reduction products of DHF) rapidly and in high yield (Scheme 1). We also present evidence that the glyoxoin reaction proceeds via DHF as a putative intermediate, thus strengthening the proposals made in the “glyoxylate scenario”.4 This facile production of tartrate combined with the known dehydration of tartrate to give oxaloacetate,8 and its decarboxylation to give pyruvate,9 potentially, provides an alternate entry into the citric acid cycle based on the “glyoxylate scenario”.4
Scheme 1. Cyanide-Mediated Dimerization of Glyoxylate Leads Predominantly to Tartrates and Oxalate.
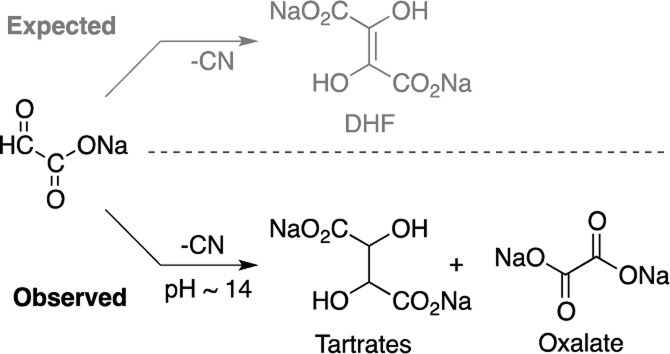
Cyanide-Mediated Dimerization of Glyoxylate
The (homogeneous) glyoxoin reaction of glyoxylate (1.0 M) with a catalytic amount of NaCN (0.1 M) in aqueous medium at room temperature in 2.0 M NaOH (pH ≈ 14) showed complete consumption of glyoxylate (by 13C NMR after 28 h); no signals corresponding to the expected DHF were observed. (High pH was required to ensure cyanide was present only as its anion; however pH 14 was obtained when 2.0 M NaOH was prepared rather than 0.5 M.) Rather, the 13C NMR spectrum of the crude reaction mixture (Figure 1) suggested the formation of meso- and dl-tartrates 8 in high yields (in some cases exceeding 80%, see Table S1 for example calculations) along with carbonate, oxalate 9, formate 11, tartronate 12, and glycolate 13. The identities of products were confirmed by comparison to and by spiking with authentic materials (Figure S1). In addition, the tartrates were isolated from the reaction mixture as calcium salts according to literature procedure10 and confirmed by 13C NMR spectroscopy and mass spectrometry (Figure S2). The meso-tartrate is produced in equal or greater quantity than the combined dl-tartrates (Table S1). No interconversion of meso-tartrate to and from dl-tartrates (nor conversion to any other reaction product) was observed under reaction conditions (Figure S3).
Figure 1.

Typical 13C NMR of the glyoxoin reaction. Reaction of 1.0 M sodium glyoxylate (182/94 ppm, completely consumed) with 0.1 M cyanide (167 ppm) in aqueous 2.0 M NaOH (room temperature, 1 h) produces meso-tartrate 8 (178.9/76.1 ppm) and dl-tartrates 8 (179.8/75.0 ppm). Signals for carbonate (169.1 ppm), oxalate 9 (173.6 ppm), formate 11 (172 ppm), tartronate 12 (179.7/76.2 ppm), and glycolate 13 (trace, 181.4/62.4 ppm) were observed. CD3OD was used as external standard (49.15 ppm).
The glyoxoin reaction also produced tartrates at lower concentrations (0.01 M of 1 and 0.002 M CN–) and at lower temperatures (4 °C). The reaction is, in fact, so resilient that even under heterogeneous conditions (when insoluble lithium glyoxylate was substituted in place of sodium glyoxylate), tartrates were still produced (Figure S4). However, when the pH was lowered (pH ≈ 9), tartrates were not observed, indicating that high pH was crucial. When a control reaction (at pH ≈ 14) was conducted by omitting the cyanide ion, no tartrates were formed; only the disproportionation of glyoxylate to glycolate 13 and oxalate 9 was observed, proceeding through the well-known Cannizzaro reaction (Figure S5C).11,12
Reaction of DHF with Glyxoylate
The production of tartrates (as opposed to DHF) from the glyoxoin reaction in basic media was unanticipated but did implicate DHF as an intermediate, since tartrate is formally the reduction product of DHF. Therefore, a reaction of DHF 5 (5 mmol, insoluble), NaCN (1 mmol/0.1 M), and glyoxylic acid (5 mmol/0.5 M) in aqueous NaOH (2.0 M/10 mL) at room temperature was investigated. A vigorous bubbling was observed in this heterogeneous reaction mixture for ∼2 h, at which point a 13C NMR spectrum of the supernatant showed the production of tartrates 8 along with carbonate, oxalate 9, formate 11, tartronate 12, and glycolate 13. More revealingly, when the DHF/glyoxylate reaction was repeated, omitting the cyanide, tartrates 8 were still formed. When the reaction of 0.25 M of DHF 5 with 0.25 M glyoxylic acid (in the absence of cyanide) was repeated in a mixture of 1.0 M NaOH and 1.0 M LiOH, the reaction mixture became homogeneous (similar to the glyoxoin reaction). 13C NMR spectrum, after 1 h, showed production of tartrates 8, (in 69% yield by quantitative 13C NMR) with complete glyoxylate conversion and little carbonate formation (Figure 2); the ratios of the accompanying product peaks were similar to the glyoxoin reaction (Figure S5A,B) with higher proportion of meso-tartrate (Table S1). Interestingly, in the heterogeneous case, the 13C NMR spectrum of the reaction was also nearly identical to the glyoxoin reaction with the exception of a much more intense carbonate peak (Figure S6B). In both heterogeneous and homogeneous cases, these results demonstrated that DHF could, by itself, mediate the transformation of glyoxylate to tartrates (Figure S6).
Figure 2.

Typical 13C NMR of the homogeneous DHF/glyoxylate (without cyanide) reaction. Reaction of 0.25 M glyoxylate with 0.25 M DHF in aqueous 1.0 M NaOH and 1M LiOH (room temperature, 1 h). For NMR details see caption of Figure 1.
Mechanistic Investigations
While these reactions showed that DHF might be an intermediate, it may not be the only path in the cyanide-mediated glyoxoin reaction. Tartrates could have been additionally produced from the base catalyzed condensation reaction of glycolate 13 (a product of the Cannizzaro reaction) with glyoxylate 1. This pathway was ruled out, since tartrates were observed neither in the glyoxylate control reaction discussed above nor when glyoxylate and glycolate were combined under identical reaction conditions. This leaves DHF formation and its reaction with glyoxylate as the most likely route to tartrates. Two possible mechanisms were considered for this formal reduction of DHF 5 (Scheme 2). The first is a simple cross-Cannizzaro reaction13 in which the hydroxide adduct of glyoxylate transfers a hydride to the carbonyl of the keto form of DHF leading directly to tartrates 8 and oxalate 9. The second pathway involves an aldol reaction between DHF and glyoxylate resulting in a six-carbon tricarboxylate 6. This six-carbon compound 6 can undergo a hydroxide promoted fragmentation to form tartrates 8 and oxalate 9 (Scheme 2).
Scheme 2. Two Possible Routes to Tartrate from Glyoxylate.
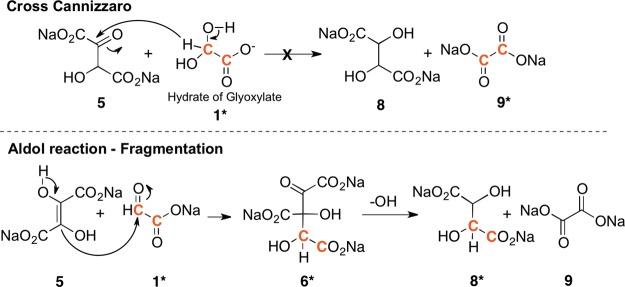
Cross-Cannizzaro reaction involving a hydride transfer (top) versus an aldol-reaction followed by hydroxide-mediated fragmentation (bottom).
To differentiate between these two possible pathways, a reaction of DHF and glyoxylate was conducted using 13C-dilabeled glyoxylate. If the cross-Cannizzaro hydride transfer was the sole pathway, only the formation of 13C labeled oxalate 9* would be expected. If the fragmentation of the six-carbon tricarboxylate 6* was the only pathway, only signals corresponding to labeled tartrates 8* would be observed. However, when 13C-dilabeled glyoxylic acid (0.1 M) was reacted with DHF (0.1 M) in 1.0 M NaOH and 1.0 M LiOH (homogeneous reaction), the13C NMR spectrum showed that both labeled tartrates 8* and oxalate 9* were formed.
While the presence of labeled tartrates 8* indicates that an aldol reaction–fragmentation reaction must have occurred, formation of labeled oxalate 9* could be explained via the competing self-Cannizzaro (as opposed to the cross-Cannizzaro) reaction of glyoxylate. However, integration of the 13C signals corresponding to the carboxylate peaks of oxalate 9* and glycolate 13* revealed that there is ∼50% less labeled oxalate than labeled glycolate in the reaction mixture (Figure S7C). This excess of glycolate indicated that (a) there is likely some side reaction that results in the production of labeled glycolate 13*; and (b) if the cross-Cannizzaro hydride transfer reaction is taking place, it is doing so to a lesser extent than this glycolate producing side reaction. Therefore, although the cross-Cannizzaro reaction cannot be ruled out entirely, this experiment supports the aldol reaction–fragmentation sequence as the primary pathway. A separate reaction using unlabeled glyoxylate (1.0 M) and 13C-labeled NaCN (0.01 M), clearly revealed the glyoxylate cyanohydrin 2 at ≈125 ppm an otherwise empty spectral region; as the reaction progressed, a second peak appeared at ≈126 ppm (attributed to the DHF cyanohydrin 4) persisting for ∼4 h before disappearing (Figure S8).
In the heterogeneous reaction (in 2.0 M aq NaOH) of labeled glyoxylate with DHF, a second pathway was suggested by the formation of significant amount (∼10%) of singly labeled tartrates 8† (Figure S7B), along with singly labeled tartronate 12†. Interestingly, the (single) labeling occurs only at the carboxylate moiety of tartrates 8† and tartronate 12† as evidenced by a lack of splitting of the carboxylate signal. Some of this could be explained by incomplete labeling of the starting material; however, the starting material contains <1% of singly labeled material (by 13C NMR, Figure S7A). This suggested that the carbon–carbon bond in glyoxylate is being broken during the course of the reaction.
Based on the above observations, we propose an overall mechanism (Scheme 3), which accounts for the bulk of the tartrates 8 and oxalate 9. In this mechanism, cyanide adds to glyoxylate 1 to form the glyoxylate cyanohydrin 2. The deprotonated glyoxylate cyanohydrin 3 then reacts with an additional molecule of glyoxylate to form the cyanohydrin adduct of DHF 4, which is then converted to the keto form of DHF 5 (which tautomerizes to the typical enol form). DHF, thus formed, can react via an aldol reaction with an additional molecule of glyoxylate (13C labeling shown in red) to yield a six-carbon tricarboxylate intermediate 6*, which can (under high pH) rearrange to 7* via a cyanide-mediated retro-aldol pathway.
Scheme 3. Major Pathway Leading to Tartrates and Oxalate from Glyoxylate.
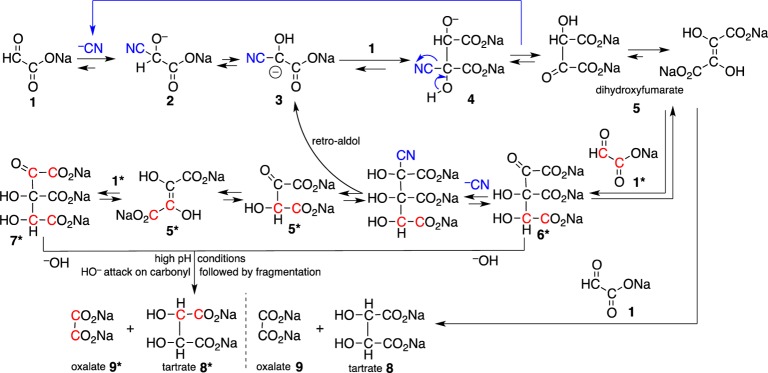
The catalytic role of cyanide is shown in blue. Results of labeled experiments are shown by the position of red carbons. This mechanism accounts for the primary pathway to tartrates, in which oxalate and tartrates are formed in a 1:1 ratio.
These intermediates 6*/7* can be attacked at the carbonyl by a free hydroxide anion (under high pH) and undergo fragmentation to yield tartrates 8/8* and oxalate 9/9*. This mechanism accounts for the primary pathway to tartrates, in which oxalate and tartrates are formed in a 1:1 ratio. Additional side products are formed by pathways (Scheme 4) that include a series of benzoin-type rearrangements,4 which are possible under these high alkaline conditions14,15 and which also account for the formation of singly labeled products. In these pathways the tricarboxylate intermediate 6(6*)/7(7*) can undergo benzoin-type rearrangements to a six-carbon aldehyde intermediate which fragments to yield singly labeled tartrates and formate.
Scheme 4. Potential Pathways Leading to Side Products.

The three pathways shown above account for all observed side products, including isotopically labeled side products.
Alternatively 6(6*)/7(7*) can undergo a retro-aldol reaction generating DHF 5/5*, which under these high-alkaline conditions undergoes a benzoin-type rearrangement to an aldehyde intermediate 10/10*. This intermediate can then react with hydroxide and fragment to form bicarbonate, formate 11/11*, tartronate 12/12*, and glycolate 13/13*. This second reaction pathway was identified from the experiments dealing with the heterogeneous decomposition of DHF alone to give formate and tartronate in 2.0 M NaOH. This also accounts for the presence of formate in these samples; glycolate and oxalate are also produced by the hydroxide promoted fragmentation reaction of the keto form of DHF.
In addition, there is a side reaction that occurs when glyoxylate 1 is in a far higher concentration than DHF 5, where tartrates 8 and tartronate 12 are formed in a 1:1 ratio. This side reaction (Scheme 4, bottom) begins at intermediate 6/7, which decarboxylates to intermediate 14, which then undergoes an aldol reaction with an additional molecule of glyoxylate 1 to form a seven carbon tricarboxylate 15. Further benzoin-type rearrangements followed by fragmentation gives rise to tartrates 8 and tartronate 12.
Discussion
The results of the DHF–glyoxylate reaction presented here are in sharp contrast to the work of Sagi et al.;6 there, DHF was reacted with glyoxylate in lithium hydroxide at pH 7–8 to produce dihydroxyacetone and pentulosonic acid by way of six- and seven-carbon tricarboxylate intermediates (6 and 15) via several decarboxylations. However, in our work, 6 and 15 undergo attack by free hydroxide at the carbonyl, yielding oxalate 9, tartronate 12, and tartrates 8. While the carbonate produced in the glyoxoin reaction is indicative of decarboxylations occurring, no evidence was seen for any of the intermediates or products observed in the work of Sagi et al. (6) This contrasting result demonstrates how even in consistently basic environments, pH may be used to alter the product suite of this reaction, from selective production of pentulosonic acid to selective production of tartrate. This dependence on pH underscores the need to investigate a wide range of conditions for potentially prebiotic reactions to fully explore the possibilities for producing these biologically relevant molecules.
The products of this simple and robust glyoxoin reaction are stable in the high-pH environment, illustrating their potential as feedstock for further reactions. However two questions must be raised to determine the relevance to prebiotic chemistry; specifically, what are the abiotic availabilities of glyoxylate and of a high-hydroxide concentration? For the former there are many possibilities: (a) Glyoxylate has been shown to be readily produced by photo-oxidation of acetylene under anoxic conditions;16 (b) reductive conversion of carbon dioxide and carbon monoxide to glyoxylate has been experimentally demonstrated;17−21 some of these could be reasonably extrapolated to early earth scenarios; and (c) glyoxylate was shown by Weber to form reliably in the reaction of glycolaldehyde and formaldehyde, catalyzed by various primary amines,22 though it was not the primary product. Overall these observations provide an array of options for production of glyoxylate. The results of this investigation underscore the need and the opportunity to further explore these options and fully investigate formation of glyoxylate under prebiotic conditions.
High-hydroxide concentrations are considered to be extreme (and unusual) in the conventional prebiotic line of thought. This usually renders any high-pH reaction problematic in this context. However, there are at least three plausible prebiotic scenarios one can consider. The first possible scenario is the widely investigated alkaline hydrothermal vent system;23−25 however, this system is not without drawbacks.26 Related reservoirs of natural hydroxide are lakes fed by alkali springs,27 which could act as a natural reactor for this type of chemistry. While these lakes may have the advantage of sidestepping the magnitude of both dilution and thermolysis faced by oceanic vents, they too have their limitations. Another possible high-alkaline environment can be found in the interlayer framework of double-layered hydroxides (e.g., hydrotalcites), which are especially conducive for uptake and concentration of anions, such as glyoxylate and cyanide, from dilute solutions.28,200 While each of these possibilities appear to be highly localized with their attendant weaknesses in a prebiotic context, they do offer potential for highly alkaline environments on early earth, that need to be validated from a prebiotic perspective, for the chemistry described here.
Notwithstanding the arguments presented above, it remains that the cyanide-mediated dimerization of glyoxylate at high pH to produce tartrates proceeds with remarkable consistency and speed, even at low temperature, low concentration, and low solubility. The fact that these reactions occur reliably and that the products are stable at high hydroxide concentrations demonstrates that a high pH environment need not be antithetical to production of such organics in a primordial setting. Moreover, the use of cyanide as a catalyst is to be contrasted with many other conventional prebiotic scenarios that start with cyanide as a source molecule.30,31
In the context of primordial metabolism, the robust production of tartrates from glyoxylate opens up new venues for the origins of biologically relevant small molecules. The simple and known dehydration of tartrates8 results in oxaloacetate which is known to decarboxylate to give pyruvate,9 thus providing an entry into a prebiotic citric acid cycle. In extant biology, oxaloacetate is the entry point to the citric acid cycle, reacting with pyruvate to form citrate.29 There are examples in extant biology where tartrate is utilized and metabolized for production of oxaloacetate to be used in the citric acid cycle.32−34 Thus, it is reasonable to posit that tartrates could have acted as a source of small molecules, which could become part of an emerging proto-metabolic process (e.g., reductive citric acid cycle).2,3 Thus, the glyoxoin reaction (and the “glyoxylate scenario”) could serve as a plausible alternative start for rudimentary chemical evolution (Scheme 5).4
Scheme 5. A Potential Abiotic Pathway to the Citric Acid Cycle.

Experimental Section
Reaction of Glyoxylate with Cyanide
Glyoxylic acid monohydrate (0.1–10 mmol, 10–100 equiv) and sodium cyanide (0.01–1 mmol, 1 equiv) were each weighed in individual 6 dram vials. Ten mL of 2.0 M NaOH or 2.0 M LiOH was then added to the vial containing the sodium cyanide. This vial was mixed until no solid remained. The solution was then transferred by syringe to the vial containing the glyoxylic acid. The cyanide solution was added down the side of the vial to the glyoxylic acid over the course of ∼20 s and then closed, and the vial was cooled (by running cool water over the vial) as necessary to avoid with excess heating. The pH measurements of the resulting solutions were ≈14.
Heterogeneous Reaction of DHF with Glyoxylate and Cyanide
Dihydroxyfumaric acid dihydrate (0.1–5 mmol, 5 equiv), glyoxylic acid monohydrate (0.1–5 mmol, 5 equiv), and sodium cyanide (0.01–1 mmol, 1 equiv) were each weighed in individual 6 dram vials. Five mL of 2.0 M NaOH was then added to the vial containing the sodium cyanide. This vial was mixed until no solid remained. In a separate vial an additional 5 mL of 2.0 M aq NaOH was then added to the vial containing the glyoxylic acid. The glyoxylate and sodium cyanide solutions were then added to the dihydroxyfumaric acid. The glyoxylate solution was added down the side of the vial to the dihydroxyfumaric acid over the course of ∼20 s and then closed, and the vial was cooled (by running cool water over the vial) as necessary to avoid with excess heating. The pH of the resulting solution was ≈14.
Homogeneous Reaction of DHF with Glyoxylate
Dihydroxyfumaric acid dihydrate (0.1–5 mmol, 1 equiv) and glyoxylic acid monohydrate (0.1–5 mmol, 1 equiv) were each weighed in individual 6 dram vials. Five mL of 2.0 M NaOH as then added to the vial containing the glyoxylic acid monohydrate. This vial was mixed until no solid remained. In a separate vial, 5 mL of 2.0 M aq LiOH was then added to the vial containing the dihydroxyfumaric acid. This vial was mixed until no solid remained. The glyoxylate solution was then transferred into the vial containing the DHF solution, and the vial was closed and shaken. Alternately, adding DHF to glyoxylate resulted in no noticeable differences. The pH of the resulting solutions was ≈14 by pH paper.
Acknowledgments
The work was supported jointly supported by NSF and the NASA Astrobiology program under the NSF Center for Chemical Evolution, Grant CHE-1004570.
Supporting Information Available
Description of materials and methods, authentic, and standard comparison NMR spectra, mass spectrometry data of isolated compounds, experimental comparison NMR spectra, quantitative NMR information, calculated yields, and chemical shift information. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Wächtershäuser G. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morowitz H. J.; Kostelnik J. D.; Yang J.; Cody G. D. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E.; Morowitz H. J. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 13168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eschenmoser A. Tetrahedron 2007, 63, 12821. [Google Scholar]
- Eschenmoser A. Chem. Biodiversity 2007, 4, 554. [DOI] [PubMed] [Google Scholar]
- Sagi V. N.; Punna V.; Hu F.; Meher G.; Krishnamurthy R. J. Am. Chem. Soc. 2012, 134, 3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapworth A. J. Chem. Soc., Trans. 1903, 83, 995. [Google Scholar]
- Chattaway F. D.; Ray F. E. J. Chem. Soc., Trans. 1921, 119, 34. [Google Scholar]
- Krebs H. Biochem. J. 1942, 36, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearon W. M.; Lo C. F.; Witte J. F. U.S. Patent 3998878 A; Boise Cascade Corp.: Portland, OR, 1976.
- Cannizzaro S. Liebigs Ann. 1853, 88, 129. [Google Scholar]
- Geissman T. A. In Organic Reactions, 2 ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 1944; p 94. [Google Scholar]
- Socha R.; Weiss A.; Sakharov M. J. Catal. 1981, 67, 207. [Google Scholar]
- Armstrong F. B.; Hedgecock C. J. R.; Reary J. B.; Whitehouse D.; Crout D. H. G. J. Chem. Soc., Chem. Commun. 1974, 351. [Google Scholar]
- Crout D. H. G.; Hedgecock C. J. R. J. Chem. Soc., Perkin Trans. 1 1979, 1982. [Google Scholar]
- Menor-Salván C.; Marín-Yaseli M. R. Chem.—Eur. J. 2013, 19, 6488. [DOI] [PubMed] [Google Scholar]
- Eggins B. R.; Irvine J. T. S.; Murphy E. P.; Grimshaw J. J. Chem. Soc., Chem. Commun. 1988, 1123. [Google Scholar]
- Pokhodenko V. D.; Koshechko V. G.; Titov V. E.; Lopushanskaja V. A. Tetrahedron Lett. 1995, 36, 3277. [Google Scholar]
- Tanaka K.; Matsui T.; Tanaka T. J. Am. Chem. Soc. 1989, 111, 3765. [Google Scholar]
- Kudo K.; Ikoma F.; Mori S.; Komatsu K.; Sugita N. J. Chem. Soc., Perkin Trans. 2 1997, 679. [Google Scholar]
- Eggins B. R.; Robertson P. K. J.; Murphy E. P.; Woods E.; Irvine J. T. S. J. Photochem. Photobiol., A 1998, 118, 31. [Google Scholar]
- Weber A. Origins Life Evol. Biospheres 2001, 31, 71. [DOI] [PubMed] [Google Scholar]
- Martin W.; Russell M. J. Philos. Trans. R. Soc., B 2007, 362, 1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin W.; Baross J.; Kelley D.; Russell M. J. Nat. Rev. Microbiol. 2008, 6, 805. [DOI] [PubMed] [Google Scholar]
- Glansdorff N.; Xu Y.; Labedan B. Res. Microbiol. 2009, 160, 522. [DOI] [PubMed] [Google Scholar]
- Miller S. L.; Bada J. L. Nature 1988, 334, 609. [DOI] [PubMed] [Google Scholar]
- Pedersen K.; Nilsson E.; Arlinger J.; Hallbeck L.; O’Neill A. Extremophiles 2004, 8, 151. [DOI] [PubMed] [Google Scholar]
- Boclair J.; Braterman P.; Brister B.; Jiang J.; Lou S.; Wang Z.; Yarberry F. Origins Life Evol. Biospheres 2001, 31, 53. [DOI] [PubMed] [Google Scholar]
- A successful example is the double-layered hydroxide-induced formation of sugar phosphates (starting from glycolaldehyde phosphate) at near-neutral pH, which otherwise needs a pH of ∼11 to proceed in solution.35,36 Experiments along these lines are underway.
- Pitsch S.; Eschenmoser A.; Gedulin B.; Hui S.; Arrhenius G. Origins Life Evol. Biospheres 1995, 25, 297. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy R.; Pitsch S.; Arrhenius G. Origins Life Evol. Biospheres 1999, 29, 139. [DOI] [PubMed] [Google Scholar]
- Oro J.; Lazcano-Araujo A.. The role of HCN and its derivatives in prebiotic evolution. In Cyanide in Biology; Vennesland B., Conn E. E., Knowles C. J., Westley J.; Wissing F., Eds.; Academic Press: London, 1981; pp 517–541. [Google Scholar]
- Huber C.; Kraus F.; Hanlik M.; Eisenreich W.; Wächtershäuser G. Chem.—Eur. J. 2012, 18, 2063. [DOI] [PubMed] [Google Scholar]
- Cody G. D.; Boctor N. Z.; Filley T. R.; Hazen R. M.; Scott J. H.; Sharma A.; Yoder H. S. Science 2000, 289, 1337. [DOI] [PubMed] [Google Scholar]
- Dagley S.; Trudgill P. Biochem. J. 1963, 89, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlbert R. E.; Jakoby W. B. J. Biol. Chem. 1965, 240, 2772. [PubMed] [Google Scholar]
- Kohn L. D. J. Biol. Chem. 1968, 243, 4426. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.