

Abstract
Informed consent is an ethical and legal requirement for research involving human participants. It is the process where a participant is informed about all aspects of the trial, which are important for the participant to make a decision and after studying all aspects of the trial the participant voluntarily confirms his or her willingness to participate in a particular clinical trial and significance of the research for advancement of medical knowledge and social welfare. The concept of informed consent is embedded in the principles of Nuremberg Code, The Declaration of Helsinki and The Belmont Report. Informed consent is an inevitable requirement prior to every research involving human being as subjects for study. Obtaining consent involves informing the subject about his or her rights, the purpose of the study, procedures to be undertaken, potential risks and benefits of participation, expected duration of study, extent of confidentiality of personal identification and demographic data, so that the participation of subjects in the study is entirely voluntary. This article provides an overview of issues in informed consent: The obligations of investigator, sponsor and Institutional Review Board to protect rights and welfare of human research subjects. It discusses about the basic elements of informed consent and the process to be followed while obtaining informed consent. Some of the circumstances under which informed consent can be waived and ethical challenges faced by physicians in obtaining informed consent from subjects are also highlighted in this article.
Keywords: Human subjects, informed consent, institutional review board
INTRODUCTION
For a drug to get approved and enter into the market it has to prove its safety and efficacy in clinical trials. Clinical trial is a term used to describe all research related activities, which use human being as subjects. As no individual has right to infract fundamental rights of another person for the sake of fulfilling his own purpose, so an important tool called “informed consent” came into existence.
The informed consent is described in ethical codes and regulations for human subject's research. The goal of the informed consent process is to provide sufficient information to a potential participant, in a language which is easily understood by him/her, so that he/she can make the voluntary decision regarding “to” or “not to” participate in the research study.
Conventionally informed consent is thought to be in terms of the documents signed and dated by participants, setting forth the purpose, benefits, risks and other study information necessary to allow the participants to make an informed and voluntary decision to participate in the clinical study. In reality, informed consent is the process that applies to each communication to participants, commencing with the subject recruitment material and the initial telephone screening of potential subjects through the conclusion of the study. It also describes the obligation of the investigator to inform the subject about personal benefits and risk, individual faces in study.
Informed consent is not only required for clinical trials but is an essential prerequisite before enrolling each and every participant in any type of research involving human subjects including; diagnostic, therapeutic, interventional, bioequivalence, social and behavioral studies and for all research conducted domestically or abroad. Obtaining consent involves informing the subject about his or her rights, the purpose of the study, the procedures to be undergone, the potential risks and/or benefits of participation and alternative treatments available if any. Subjects in the study must participate willingly only after consenting based on the information given.[1,2,3]
THE ORIGINS OF INFORMED CONSENT
Informed consent is a central tenet of research ethics involving human beings and has evolved into present shape over a period of time. The journey of informed consent is briefly described below [Tables 1-3].[4,5,6,7]
Table 1.
Evolution of informed consent

Table 3.
Informed consent and obligations

Table 2.
Classification of informed consent

BASIC ELEMENTS FOR WRITTEN INFORMED CONSENT DOCUMENTS
A statement that the study involves research;
An explanation of the purpose of research and the expected duration of the subject's participation;
A description of the procedures to be followed and identification of any procedures that are experimental;
A description of any foreseeable risks or discomforts to the subject, an estimate of their likelihood and a description of what steps will be taken to prevent or minimize them;
A description of any benefits to the subject or to others that may reasonably be expected from the research. Monetary compensation is not a benefit;
A disclosure of any appropriate alternative procedures or courses of treatment that might be advantageous to the subject;
A statement describing to what extent records will be kept confidential, including a description of who may have access to research records;
For research involving more than minimal risk, an explanation and description of any compensation and any medical treatments that are available if research subjects are injured; where further information may be obtained and whom to contact in the event of a research-related injury;
Information on the amount of remuneration/compensation, if any, that will be provided to subjects;
An explanation of whom to contact for answers to pertinent questions about the research and the research subject's rights (include the clinical center's patient representative and telephone number);
A statement that participation is voluntary and that refusal to participate or discontinuing participation at any time will involve no penalty or loss of benefits to which the subject is otherwise entitled.[13]
REQUIREMENTS FOR OBTAINING INFORMED CONSENT
The investigator or a person designated by investigator must obtain informed consent
Informed consent must be obtained before non-routine screening procedures are performed and/or before any change in the subject's current medical therapy is made for the purpose of the clinical trial
The subject/subject's legally acceptable representative should not be forced to sign on consent or participate/continue to participate in the trial
The subject/legally acceptable representative and individual obtaining consent must personally sign with date the form. The signature of the prospective subject/legally acceptable representative on informed consent document indicated that content of informed consent document has been adequately discussed and the subject/subject's legally acceptable representative freely gave the informed consent [Figure 1].[13]
Figure 1.
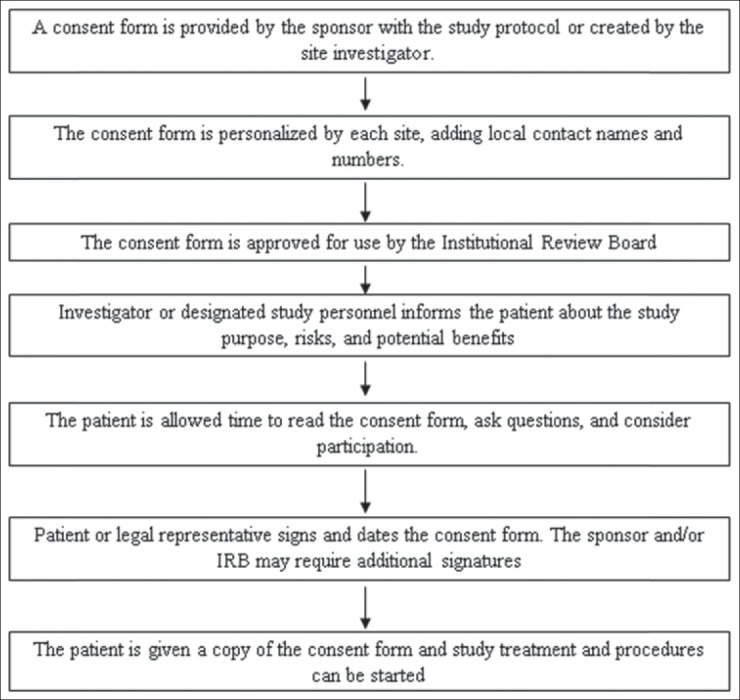
Flow chart regarding various informed consent processes
INFORMED CONSENT PROCESS FLOW
Notes to Flowchart
Source documents must reflect that consent was obtained before the start of study treatment and procedures
A copy of the signed consent form must be kept at the site
All versions of approved consent forms must be kept in the site study file; only the current Institutional Review Board (IRB) approved version may be used to consent new patients.[14]
WAIVERS TO INFORMED CONSENT
A waiver of informed consent under 45 Code of Federal Regulations (CFR) 46.116 (d)
-
An alteration of consent under 45 CFR 46.116 (d)
An IRB may waive the requirements to obtain informed consent provided the IRB finds and documents that:
- The research involves no more than minimal risk to the subjects;
- The waiver or alteration will not adversely affect the rights and welfare of the subjects;
- The research could not practicably be carried out without the waiver or alteration and
- Whenever appropriate, the subjects will be provided with additional pertinent information after participation.
A waiver of parent/guardian permission under 45CFR 46.408 (c)
A waiver of assent under 45 CFR 46.408 because the minors are not capable of assent
A waiver of assent under 45 CFR 46.408 because the research holds out a prospect of direct benefit that is available only in the context of the research.[15]
CHALLENGES IN INFORMED CONSENT PROCESS
Language Barriers
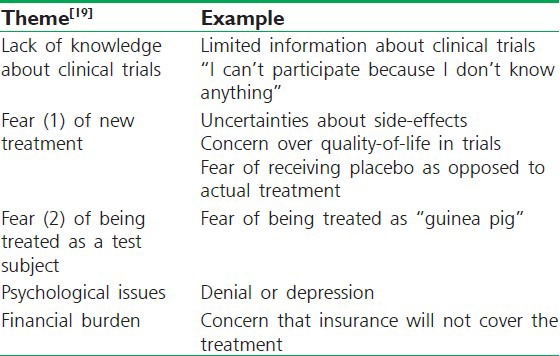
It is assumed that the individual who signs the consent form does so with full understanding of what is stated on the consent form. However, it is very difficult to evaluate their viewpoint about trial since there is no established method to measure the level of understanding that a participant has about the information given. Thus, it can be assumed that there is a degree of misunderstanding that occurs. Misunderstandings can occur because of incorrect or inadequate language translations. Many individuals sign the consent form without being fully aware of what they are signing, which results in withdrawal of subject at later stages of ongoing clinical studies. Hence, the responsibility of researcher enlarges when a study is performed in multilingual subjects [Table 4].[16]
Table 4.
Barriers to participation in clinical trials
Religious Influence
The informed consent process is designed to give every participant the liberty to decide whether to accept or refuse the recommended medical treatment. Sometimes their decision for participating in researcher projects is influenced by the religious beliefs. It is commonly observed that how the methodology of the experiment come into conflict with the rules of behavior set by a participant's religion.[17,18]
False Expectations
Even when there are no language barriers or religious impediments to hinder the communication relationship between researcher and participant, misunderstanding can still occur due to participants false expectations of the experiment outcome. Some patient fear of being treated as mere “experimental model” for the studies while others refuse to take part because of historical evidences of clinical trial fraud and misconducts known to them.[17,18]
Patient Perceptions
Most patients believe that, trials will put extra burden on them. They assume that the conventional treatment is best and they are afraid of the unknown side-effects of new treatment. Convincing and receiving an informed consent from such patient is most difficult. In some case disclosing too much information of the potential side-effects may unnecessarily scare the patient away from a potentially life-saving or life-enhancing surgery or procedure.[17,18]
Children
Where research involves children (under the age of 18) consent/permission has to be obtained from parents. If the child is above 7 years of age then “child assent” is also mandatory. It is arguable that children are capable of being partners in research and that they have rights to receive information, to be listened to, have their wishes and feelings taken into account and to give or withhold consent if judged competent to do so. Difficulty arises when parents give their consent while child refuses to assent.[20]
Vulnerable People and Groups
Vulnerable groups include the person who is absolutely or relatively incapable of protecting their interests. Obtaining informed consent is critical when working with them, specifically with some groups like people with learning disabilities. There may be potential problems of understanding what the research is about, what their role in the research will be and how the research will be used. Hence, obtaining informed consent can be difficult and special care needs to be taken to develop the appropriate strategies for communicating the implications of involvement in research.[21,22]
Indian Scenario
In countries like India, the clinical investigations are based on regional values and practices, the concept of disease as perceived through social values and power hierarchies in family of villages based on cultural systems. To get a meaningful and ethical informed consent in these settings become challenging due to differences in cultural values in western countries and local customs in developing countries including India. In a study by DeCosta et al.[23] that was carried out in a village of Haryana state of India, the majority of respondents interviewed by them could decide on clinical trial participation after discussing with community members. Another important factor emerged from this study, which showed an implicit trust by respondents in the medical system and ignorance about the information that should be known before consenting to be a part of the research study. These factors put a huge responsibility on the part of the investigator to get informed consent. The investigator must explain in most comprehensive and complete manner the risks involved in participating in the research study. Thus, investigator should have the patience to get informed consent from these subjects allowing them to discuss with other family and community members. The ethical principles of western countries require all adults to be the primary decision makers of their participation, which may not be applicable in Indian system, which is culturally and socially different from the western world.[23] Another important aspect of informed consent arises in psychiatric clinical studies. As large numbers of psychiatry studies are conducted in India, these studies present complex and unique challenges in Indian context. These issues include risk of worsening of illness, use of placebo and validity of informed consent. The informed consent procedure requires patient to be of sound mind and in understanding the information presented and make a sound judgment regarding participation. Assessment of consent capacity may be difficult due to fluctuation in illness, which requires continued assessment of consent capacity. Thus, conducting clinical trials and obtaining informed consent for psychiatry studies is difficult and raises a doubt on the conduct of clinical trials due to lack of trained researchers. The guidelines are prepared keeping in mind the western culture and may not replicate the same results due to cultural variability in non-western countries like India.[24] The dilemma in obtaining informed consent from subjects with cognitive impairment includes validity of informed consent by subject, implications and validity of third party consent, protection of human subjects. Regulations don’t provide information and guidance on ethical issues of psychiatry research.[25]
CONCLUSION
Though enveloped by challenges, informed consent is an important tool in clinical trials, which facilitates the entry of new therapeutic interventions into the market. No research activity involving human subjects can be conducted and proceed unless informed consent is completely sought. The responsibility of conducting trial ethically and genuinely lies in the hands of those involved in it. Everyone must understand their obligations and should not misuse their power for own benefit. Rights, safety and well-being of trial subjects should always prevail over the interest of science and society, so that a layman never feels being deceived off in name of a social cause. The issue of informed consent in India is a challenge on the part of investigator as a lot of complexities arise. Further, regulations are based on the western guidelines, which do not necessarily reflect the requirements of India. The guidelines on informed consent in India should be based on complex factors such as culture, level of education, demographics and risks involved during the study.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
REFERENCES
- 1.International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use. Guideline for good clinical practice E6(R1) June. 1996. [Last accessed on 2011 Apr 19]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6_R1/Step4/E6_R1__Guideline.pdf .
- 2.Berry IR. New York: Marcel Dekker; 2005. The Pharmaceutical Regulatory Process. [Google Scholar]
- 3.Lee C, Lee LH, Wu CL, Lee BR, Chen ML. Florida: CRC Press, Taylor and Francis Groups; 2006. Clinical Trials of Drugs and Biopharmaceuticals. [Google Scholar]
- 4.The Nuremberg code (1947) [Last accessed on 2011 Apr 19];Br Med J. 1996 313:1448. Available from: http://www.cirp.org/library/ethics/nuremberg . [Google Scholar]
- 5.World Medical Association declaration of Helsinki. Ethical principles for medical research involving human subjects. [Last accessed on 2011 Apr 20]. Available from: http://www.ohsr.od.nih.gov/guidelines/helsinki.html .
- 6.The Belmont Report ethical principles and guidelines for the protection of human subjects of research. The National Commission for the protection of human subjects of biomedical and behavioral research; April 18. 1979. [Last accessed on 2011 Apr 20]. Available from: http://www.ohsr.od.nih.gov/guidelines/belmont.html .
- 7.Geneva: Council for International Organizations of Medical Sciences (CIOMS) in collaboration with the World Health Organization (WHO); 2002. [Last accessed on 2011 Apr 20]. International ethical guidelines for biomedical research. Available from: http://www.nccr.gov.my/index.cfm?menuid=23 and parentid=17 . [Google Scholar]
- 8.Research involving children under the age of 18. [Last accessed on 2011 Apr 20]. Available from: http://www.belmont.edu/irb/instructions/Consent.html#Children .
- 9.CFR-Code of Federal Regulations Title 21; Part 50-Protection of Human Subjects. Short form written consent document. [Last accessed on 2011 Apr 21]. Available from: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=50.27 .
- 10.Investigator responsibilities-Protecting the rights safety, and welfare of study subjects 2009 October. [Last accessed on 2011 Apr 21]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ UCM187772.pdf .
- 11.Informed consent. [Last accessed on 2011 Apr 22]. Available from: http://www.hhs.gov/ohrp/archive/irb/irb_chapter 3.htm#e2 .
- 12.Niazi SK. USA: Informa Healthcare; 2007. Handbook of Bioequivalence Testing. [Google Scholar]
- 13.Guidelines for writing informed consent documents. OHSR Information Sheets/Forms 2006 December. [Last accessed on 2011 Apr 26]. Available from: http://www.ohsr.od.nih.gov/info/sheet6.html .
- 14.Informed Consent Process Flow. [Last accessed on 2011 Apr 27]. Available from: https://www.ctnbestpractices.org/resources/study-patient-management/informed-consent/Informed_Consent_Process.pdf/view .
- 15.Waiver of Informed Consent. [Last accessed on 2011 Apr 29]. Available from http://www.irb.purdue.edu/waiveconsent.shtml .
- 16.Escobedo C, Guerrero J, Lujan G, Ramirez A, Serrano D. Ethical Issues with Informed Consent. [Last accessed on 2011 Apr 29]. Available from: http://www.cstep.cs.utep.edu/research/ezine/EzineEthicalIssueswithInformedConsent.pdf .
- 17.Jehovah's Witness ethics 2009 September 29. [Last accessed on 2011 Apr 30]. Available from: http://www.bbc.co.uk/religion/religions/witnesses/witnessethics/ethics_1.shtml .
- 18.Jehovah's Witnesses and Blood Transfusions. [Last accessed on 2011 May 2]. Available from: http://www.jwfacts.com/watchtower/blood.transfusions.php .
- 19.Gonzalez LE, Quinn GP, McIntyre J. Main Barriers to Participation. [Last accessed on 2011 May 4]. Available from: http://www.kon.org/urc/v9/gonzalez.html .
- 20.Child Assent Requirement 2010 January. [Last accessed on 2011 May 6]. Available from: http://www.nhlbi.nih.gov/childrenandclinicalstudies/terms_assent.php .
- 21.Ethical Principles for Researching Vulnerable Groups 2003 May. [Last accessed on 2011 May 7]. Available from: http://www.ofmdfmni.gov.uk/ethicalprinciples.pdf .
- 22.Levine RJ. Michigan: Urban and Schwarzenberg; 1981. The Ethics and Regulation of Clinical Research. [Google Scholar]
- 23.DeCosta A, D’souza N, Krishnan S, Chhabra MS, Shihaam I, Goswami K. Community based trials and informed consent in rural north India. J Med Ethics. 2004;30:318–23. doi: 10.1136/jme.2002.001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kharawala S, Dalal J. Challenges in conducting psychiatry studies in India. Perspect Clin Res. 2011;2:8–12. doi: 10.4103/2229-3485.76284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta UC, Kharawala S. Informed consent in psychiatry clinical research: A conceptual review of issues, challenges, and recommendations. Perspect Clin Res. 2012;3:8–15. doi: 10.4103/2229-3485.92301. [DOI] [PMC free article] [PubMed] [Google Scholar]