

Abstract
Clobazam is a newer 1,5-benzodiazepine used for the treatment of epilepsy. It is better tolerated and less sedating than other benzodiazepines. Absorption of the drug can be impacted by oral fast dissolving dosage form; this may have implications for epilepsy in pediatrics and those having difficulty in swallowing tablets/capsules resulting in improved patient compliance. The purpose of the present investigation was to formulate and optimize clobazam oro-dissolving tablets by direct compression method using response surface methodology (RSM). Oro-dispersible tablets of clobazam were prepared by direct compression method using crospovidone (2-6%) as a superdisintegrant, microcrystalline cellulose (MCC) (20-40%) was used as diluents along with directly compressible mannitol to enhance mouth feel. A 32 full factorial design was applied to investigate the combined effect of two formulation variables: amount of crospovidone and MCC over the independent variables disintegration time, wetting time and percent drug release. Disintegration time showed by all formulations was found to be in the range of 24.3-193 s based on evaluation parameters the formulation containing 6% of crospovidone and 30% of MCC showed promising performance against all other formulations. The results demonstrated that the RSM could efficiently be applied for the formulation of clobazam oro-dispersible tablets; therefore, constitute an advance in the management of epileptic attacks.
Keywords: Antiepileptics, crospovidone, disintegration, microcrystalline cellulose, response surface methodology
INTRODUCTION
Recent advances in novel drug delivery systems aim to enhance safety and efficacy of drug molecule by formulating a convenient dosage form for administration and thus to achieve better patient compliance, i.e. one which rapidly disintegrate within the oral cavity without the need of water.[1] One such system is fast dissolving drug delivery system. These novel types of systems disintegrate/disperse in saliva within few seconds. According to European pharmacopoeia the orally disintegrating tablets (ODTs) should disintegrate/disperse in less than 3 min.[2] They are also known as oro-dissolving, rapid-dissolving, oro-dispersible, repi-melt, quick dissolving and melt in mouth drug delivery systems. ODTs offer many advantages over other solid oral dosage forms. They can be easily swallowed (in contrast to conventional tablets and hard gelatine capsules) and can be used with patients who have difficulty in swallowing such as stroke victims, psychiatric, pediatric and geriatric patients.[3] ODTs also need no preparatory step prior to administration and therefore preferable when compared with extemporaneous suspension or effervescent granules. They have pleasant mouth feel and acceptable taste and are preferred over the chewable tablets in which the bitter drug may leach during mastication. In addition, ODTs can be designed to provide fast onset of action by enhancing pregastric absorption through the buccal cavity, pharynx and oesophagus and to increase the bioavailability by incorporating emulsions within the tablets.[4] Clobazam is a 1,5-benzodiazepine derivative with antiepileptic and anti-psychotic properties. Clobazam is primarily indicated in conditions like adjunct in epilepsy, anxiety states in psychotic disorders.[5] Clobazam is a newer benzodiazepine used for the treatment of epilepsy. It is better tolerated and results in less sedation when compared to the other benzodiazepines. The half-life of clobazam is 18 h and up to 42 h for its active metabolite.[6] Several studies examined the efficacy of clobazam in partial and generalized seizures. Clobazam is a well-tolerated, safe and very effective antiepileptic drug. It has a broad spectrum of antiepileptic activity, minimal side-effects and is relatively inexpensive.[7] Wider use of this drug is recommended in children with intractable epilepsy.[8,9] Since epileptic patients have to strictly follow the dosage regimen for prevention of sub therapeutic concentration, Fast Dissolving Tablet (FDT) will avoid missing out of dose even during travelling or other situation where there is no access to water; offer a suitable and one of the practical approach in serving the desired objective of fast disintegration and reproducible dissolution characteristics within the oral cavity and hence increased bioavailability, also clobazam is primarily recommended in children's so it is a good candidate for the present investigation, in view of above discussion the current study aims at developing and optimizing the fast disintegrating tablets of clobazam using response surface methodology (RSM) as the technique requires minimum experimentation and time, thus proving to be far more effective and cost-effective than the conventional methods of formulating dosage form.
MATERIALS AND METHODS
Materials
Clobazam and crospovidone were received as a gift sample from Consern Pharma Pvt. Ltd., (Ludhiana, India) and Dr. Reddy's Lab (Baddi, India), respectively. Microcrystalline cellulose (MCC) and directly compressible mannitol were received from Lobe chemise Pvt. Ltd (Mumbai, India). A spartame was procured from Ipza Pharmaceutical (Patiala, India). All other chemical used were of analytical reagent grade.
Methods
Drug excipient compatibility studies
The pure drug clobazam and physical mixture of clobazam and crospovidon were mixed with IR grade potassium bromide pellets in the ratio of 100:1 and corresponding pellets were prepared in a hydraulic press. The pellets were scanned over a wave number range of 4000-500 cm−1 in using PerkinEmler spectrum 400 USA, Fourier transform infrared (FTIR) instrument.
Preparation of oro-dissolving tablets of clobazam
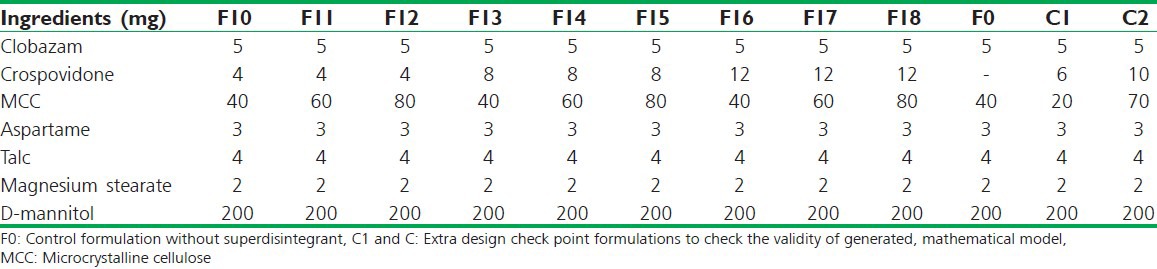
Preliminary screening on three different superdisintegrants croscarmellose sodium (CCS), sodium starch glycolate (SSG) and crospovidone was carried out. After preliminary trials the formulations were designed according to the 32 full factorial designs, allowing the simultaneous evaluation of the two formulation variables and their interaction. Oro-dispersible tablets of clobazam were prepared by direct compression method according to the formulae given in Table 1. All ingredients as per the formulae were weighed and passed through #60 mesh and mixed in geometrical order. Then lubricants and glidant were added and mixed for further 5 min. The blend thus obtained was evaluated for pre-compression parameters and was then directly compressed using 7 mm flat round punches into tablets of 200 mg on double compression tablet machine (AK Industries Jalandhar, India). A batch of 60 tablets was formulated for each designed formulation for preliminary trials and further for validation of data 200 tablets of each formulation batch was formulated. Formulation of all batches (full factorial design layout) and composition of factorial design batches are shown in Tables 1 and 2 respectively.[10]
Table 1.
32 factorial design layout
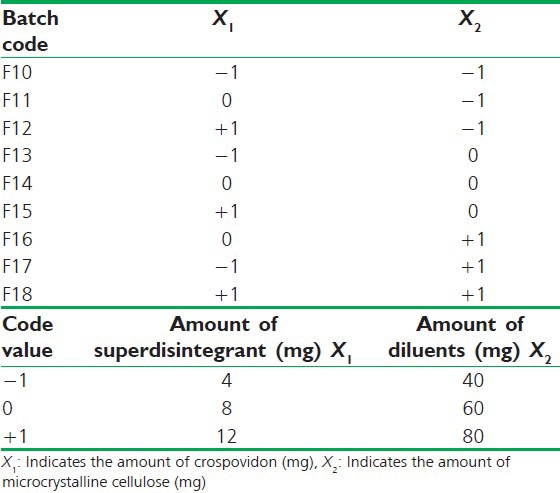
Table 2.
Composition of oro-dispersible tablets of clobazam using 32 factorial design
Full factorial design
A 32 randomized factorial design was adopted to optimize the formulation variables, in this design, two factors were evaluated each at three levels and experimental trials were performed at all nine possible combinations with two extra check point design formulations (C1 and C2) to check the validity of generated mathematical model. The amount of disintegrant (crospovidone) and the diluent (MCC) were chosen as independent variables. The disintegration time, wetting time and % drug released was selected as dependent variables. The software design expert from stat ease 8.0.7.1 trial version was used for generating the experimental design, modeling the response surface and calculating the statistical evaluation.
Evaluation of blend for pre-compression parameters
The formulation blend was evaluated for the following pre-compression parameters such as bulk density, tapped density, Carr's index, Hausner's ratio and angle of repose as per the official procedure.[11,12]
Evaluation of post-compression parameters of oro-dispersible tablets of clobazam
Tablet porosity
The tablet porosity was calculated using the equation:
€ (%) = 1 − M/ρtV
where € (%) is percent porosity, ρt is true density; M and V are the tablet weight and tablet volume respectively. The experiments were repeated 3 times.[13]
Weight variation
To determine weight variation, 20 tablets were selected randomly from each batch and weighed individually using an electronic precision balance (citizen, Mumbai). The individual weights were compared with the average weight for obtaining the weight variation.
Tablet hardness
Hardness of the tablets of each formulation was determined using the Monsanto hardness tester; an average of three readings was taken.
Tablet thickness
The thickness was determined using digital Vernier calipers (Mitutoyo Absolute Digimatic caliper Japan).[14,15]
Tablet friability
Tablet friability testing was carried out as per United States Pharmacopeio (USP) specification.[16] A sample of tablets corresponding to 6.5 g were placed in the Roch friability test apparatus, which was given 100 revolutions and the tablets were reweighed for the determination of percent friability.
Percent friability = initial weight − final weight/initial weight × 100
Wetting time
The tablet was placed in a petri dish (internal diameter of 7.5 cm) containing 10 ml of water at room temperature and the time for complete wetting was recorded. To check the reproducibility the measurements were carried out for 5 time and mean value was calculated.[17,18]
In vitro Disintegration Time
The in vitro disintegration time for all formulations was determined using USP tablet disintegration test apparatus.[19]
Drug Content
For the estimation of drug content five tablets were selected randomly and the average weight was calculated. The tablets were crushed in a mortar and an accurate weight equivalent to 5 mg of the drug was weighed. Then the sample were transferred to 100 ml volumetric flask and dissolved in methanol. The content was shaken periodically and 1 ml of this was withdrawn and diluted to 10 ml using methanol and drug content was estimated at 232 nm using double beam ultraviolet (UV)/visible spectrophotometer (Systronics AU-2701 Mumbai, India).[20]
In vitro Drug Release Study
In vitro dissolution test was performed according to the USP type II paddle apparatus (Labindia DS 8000 Mumbai, India). Test solution was 900 ml of phosphate buffer 6.8 at 37°C with a rotation speed of 50 rpm. Aliquots (5 ml) of samples were taken at time intervals from 1 to 30 min and the same volume of fresh phosphate buffer was replenished. The samples were filtered and suitably diluted and analyzed at 232 nm using double beam UV/visible spectrophotometer (Systronics AU-2701 Mumbai, India) the content of the drug was calculated.[21]
Scanning Electron Microscopy
Surface morphology of optimized tablet formulation was examined using Jeol JSM-6100 scanning electron microscope (Tokyo, Japan). Pictures were taken at an excitation voltage of 1.0 KV and a magnification of ×1000X.
Release Kinetics Study
To analyze the in vitro release data various kinetic models zero-order, first-order and Higuchi's, Hixson Crowell and Korsmeyer-Peppas models were used to describe the release kinetics.[22,23,24]
Regression Analysis
Clobazam oro-dispersible tablets were prepared by direct compression technique; formulation, optimization was done using 32 full factorial design employing crospovidone as superdisintegrant and MCC as diluent along with directly compressible mannitol to enhance the mouth feel. On the basis of preliminary trials a total of nine formulations, a control (without crospovidone) and two extra design check point formulation were designed. The general model (equation) was generated to fit the various data.
![]()
where Y is dependent variable, X1 the amount of crospovidone and X2 is the amount of MCC.
b0, is the arithmetic mean response of the nine runs and b1, b2, are estimated coefficients for the factors X1 and X2 respectively. The main effect (X1 and X2) represents the average effect of changing one factor at a time from its low to high value. The interaction term (X1 X2) shows the change in response when two factors are simultaneously changed. The polynomial term are included to investigate non-linearity.[25]
RESULTS AND DISCUSSION
Effect of Type and Concentration of Disintegrants
Initially tablets containing superdisintegrants in the concentrations 2, 4 and 6% w/w were tested for disintegration time. It was concluded that the disintegration time increases with the increase in concentration of SSG in the tablets, which was related to the disintegration mechanism of SSG, which act by swelling on contact with the aqueous medium. As swelling is reported to be accompanied by gelling this could possibly occlude the pore in the tablet preventing further penetration of water into tablet matrix hence delay in disintegration time as the concentration increases.[26,27] It indicates that increase in the concentration of SSG has a negative effect on the disintegration of tablets. It was observed that as the concentration of CCS was increased a significant variation was observed in disintegration time at different concentrations of CCS. In case of tablets containing crosspovidone, increasing concentration of crosspovidone from 2% to 6%, the disintegration times of tablets was not affected significantly, indicating positive effect on the disintegration time, which may be due to the higher capillary action and little tendency of the crosspovidone to form viscous gel.[28,29] Based on the disintegration results, the investigated superdisintegrants can be ranked according to their ability to swell in water as crosspovidone, CCS and SSG. On the basis of the results obtained in the preliminary screening studies, the batch containing crosspovidone showed the fastest disintegration. Hence, crosspovidon was selected for the formulation of fast dissolving tablets of clobazam.
Effect of Diluents on Disintegration Time
When MCC is combined with water soluble mannitol, it shows the shorter disintegration time than other diluents. This may be attributed to the high water solubility of mannitol, which may leave pores in the tablet matrix; afterward capillary action may be responsible for penetration of the surrounding fluid in the tablet matrix and thereafter rapid disintegration.
Effect of Tablet Hardness on Disintegration Time
The data shows that there is no remarkable effect of hardness on the disintegration time. As the concentration of crospovidone increases the disintegration time decrease, but increase in the concentration of MCC found to increase the disintegration time.[30] The basic reason of this that MCC has good property of compaction thus the tablet containing 30% of MCC and 6% of crospovidone (F17) has maximum hardness of 3.58 kg/cm2 with minimum disintegration time of 24.3 s, which may be attributed due to porous particle morphology of crospovidone, which rapidly absorb water through capillary action, which help in the faster disintegration.
Porosity Effect
The first step in the disintegration process is the penetration of water into the tablet.[31,32] Consequently; the tablet porosity has an important influence on the disintegration rate.[33] Porosity depends upon the pressure at which tablets are compressed as well as on the nature of the material being tablet. At low compression force levels many cavities remain in the tablet. This porous structure leads to reduced effectiveness of crospovidone in water because the swelling force of the disintegrant gets lost in the cavities, resulting in a slower dispersion of the tablet. On the other hand, a high compression force leads to tablets with a lower porosity. The water is hindered in penetrating the tablet and therefore the disintegration time is increased (Caramella et al. 1984) reported that there is a strong relationship between porosity and disintegrant concentration. Increasing the crospovidone percentage, porosity tends to increase, which is accompanied by a reduction in disintegrating force transmission. Obviously an optimum exists at a porosity of 13%. Since the formulation with 6% crospovidone covers this region best.
Drug Excipient Compatibility Studies
FTIR studies of pure drug and drug with crospovidone indicated that there is no interaction between drug and superdisintegrant used [Figure 1]. Pure clobazam displays a peak characteristic of C = O stretching vibration at 1694.7 cm−1, aromatic CH stretching at 3075.4 cm−1, C-C stretching at 1493.5 cm−1, C-N stretching at 1100-1200 cm−1, CH bending at 600-800 cm−1, CH3 bending at 1371 cm−1, The spectra of drug with crospovidone showed all characteristics peaks of drug indicating that the drug is compatible with crospovidone.
Figure 1.
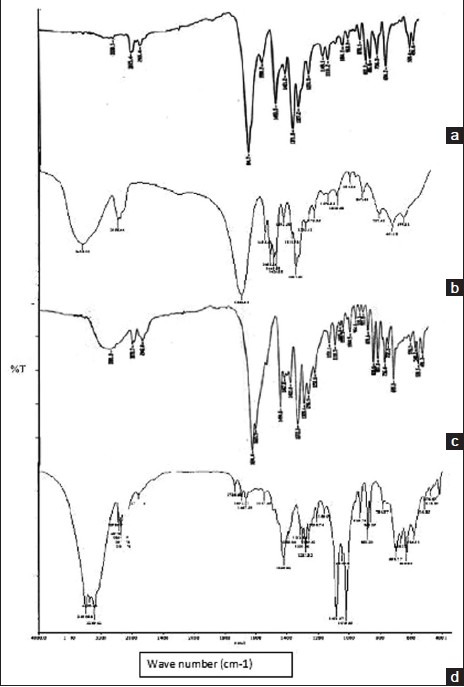
Fourier transform infrared spectra of (a) clobazam, (b) crospovidon, (c) physical mixture of clobazam and crospovidon, (d) optimized formulation (F17)
Pre-compression Parameters of Powder Blend
Powder blends were evaluated for pre-compression parameters such as the angle of repose, bulk density, tapped density and Carr's consolidation index as per official requirement. Bulk density was found to be in the range of 0.377-0.341 g/cm3, tapped density was in the range of 0.482-0.366 g/cm3, Carr's index was found to be in the range of 16.08-10.41%, Hausner ratio was found to varying between 2.103 and 1.012 and angle of repose was found to varying between 28.27° and 22.27°. All parameters were found to be in acceptable limits indicating fair to good flow properties.
Post-compression Parameters of Clobazam Oro-dispersible Tablets
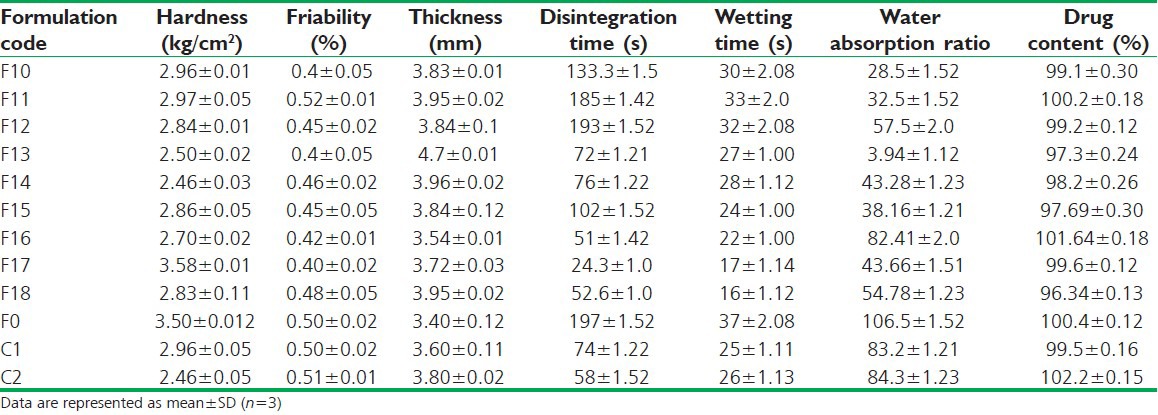
The tablets were evaluated for post-compression parameters – tablet porosity, weight variation, uniformity of drug content, hardness, friability, in vitro disintegration time, in-vitro dissolution studies, wetting time and water absorption ratio. Table 3 presents the results of physicochemical evaluation of all batches of clobazam oro-dispersible tablets. All prepared tablets were located within the acceptable weight variation varying between 200 mg and 210 mg, with acceptable limit as per IP specifications (±7.5%). Drug content was found to be in the range of 102.2-96.34%, hardness of the tablets was found to be in the range of 2.46-2.96 kg/cm2. Friability was found to be below 1% (0.4-0.52%); thickness was in the range of 3.40-4.7 mm.
Table 3.
Evaluation of factorial design formulations of clobazam tablets
In vitro Disintegration Time
In vitro disintegration time was found to be in the range of 24.3-193 s. Formulation F17 containing 6% of crospovidone and 30% of MCC was found to give minimum disintegration time against all other formulations. Wetting time is an important criterion for understanding the capacity of disintegrant to swell in the presence of water, which was in close agreement with the disintegration time and was found to be in the range of 16-37 s with F17 17 s. short wetting time is indicative of the highly porous nature of the tablet matrix. Further, it was found that as the concentration of crospovidone increases the disintegration time decrease, but increase in concentration of diluent (MCC) found to increase the disintegration time. Such delay in disintegration time may be due to tight binding between the molecules, which ultimately slow down the water up take by the tablets and thus superdisintegrant used do not get sufficient water to swell. Further decreased disintegration time of F17 formulation is attributed to increased porosity, which may be due to addition of water soluble filler which affect the internal structure of tablet matrix.[34] It was found that there is a positive correlation between wetting time, disintegration time and porosity.
In vitro Drug Release Study
In vitro dissolution testing in phosphate buffer pH 6.8 reveals that more than 80% of the drug gets released within 20 min for all the formulations with F17 showing maximum release of 98% [Figure 2]. Further formulation F17 was compared with marketed formulation for its in vitro release profile and it was found to be comparable to the marketed formulation of clobazam [Figure 3]. This result correlates well with disintegration time and wetting time. Addition of water soluble filler mannitol greatly affected the inner structure of the tablet with subsequent impact on the wetting time, disintegration time and drug dissolution profile.
Figure 2.
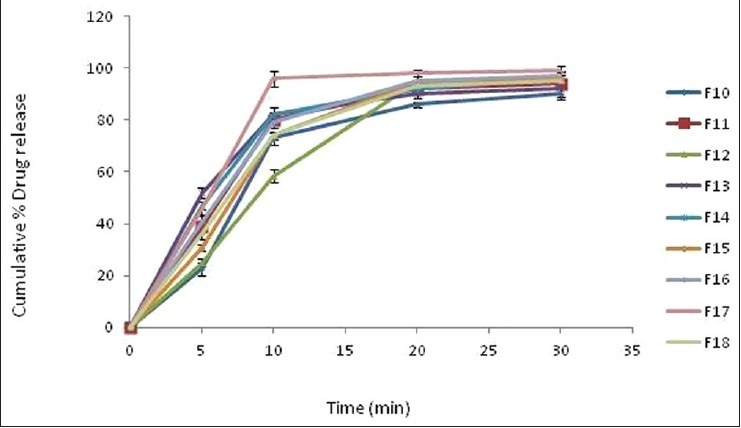
Comparative in vitro drug release profile of factorial design formulation batches F10-F18 of clobazam tablet. Data are represented as mean ± SD (n= 3)
Figure 3.
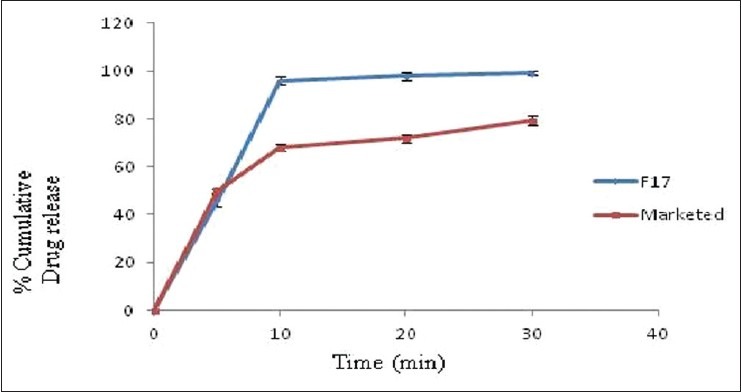
Comparative in vitro drug release profile of optimized formulation (F17) and marketed formulation of clobazam. Data are represented as mean ± SD (n= 3)
Scanning Electron Microscopy
Scanning electron micrographs of the surface view, cross-sectional surface of optimized (F17) and control formulation without superdisintegrant shows the highly porous nature of the prepared oro-dispersible tablet (F17) as compared with the control formulation [Figure 4], which suggests the rapid penetration of water, which results in rapid wetting, disintegration and dissolution in the oral cavity.
Figure 4.

Scanning electron microscopy of (a) cross-sectional surface of optimized formulation (F17) of clobazam, (b) cross-sectional surface of formulation F17, (c) sectional surface of formulation F17 (without superdisintegrant)
Release Kinetic Study
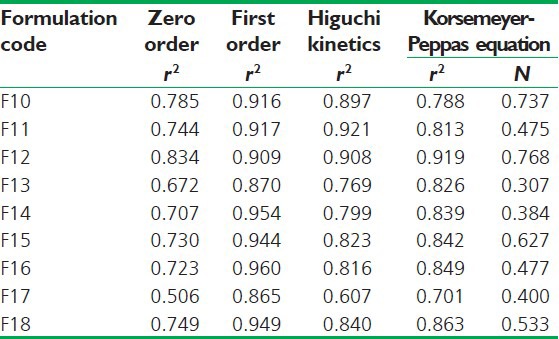
The mechanism and kinetics of drug release of clobazam were determined by the application of Korsmeyer-Peppas model, Higuchi's model, zero order and first order kinetics as shown in Table 4. From the release kinetics of formulations F10 to F18, it was found that the first order release kinetics was best fitted for clobazam oro-dispersible tablets. The correlation coefficient (r) was used as an indicator of the best fitting for each of the models considered. The release pattern of all developed formulations followed first order release model. The mechanisms of drug release are non-Fickian diffusion (anomalous transport) with “n” value less than 1. This indicates the drug release depends on swelling and diffusion mechanism of release.
Table 4.
Regression coefficient (r2) values of drug release data obtained from various kinetic models and ‘n’ value (diffusional exponent) according to Korsmeyer-Peppas model
Regression Analysis
Mathematical relationship generated using multiple linear regression analysis for the studied variables are expressed as follows:
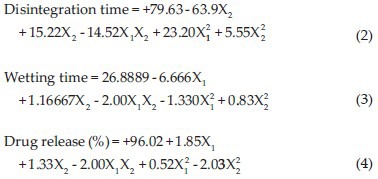
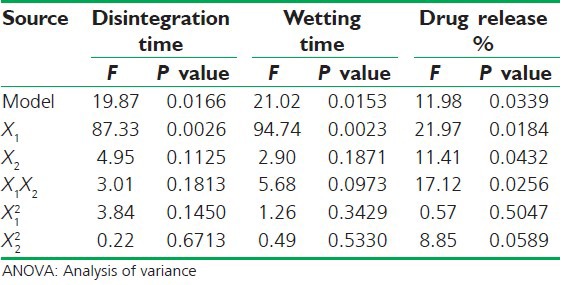
All polynomial equations were found to be statistically significant (P < 0.01) as determined using analysis of variance (ANOVA). The summary of ANOVA for all three responses is shown in Table 5.
Table 5.
ANOVA for all three responses
The results of multiple linear regression analysis as depicted in Table 6 shows that for all responses, i.e. disintegration time, wetting time and % drug release the amount of crospovidon (X1) had a negative effect while the concentration of MCC (X2) had a positive effect it means as the amount of crospovidone increases the wetting time and disintegration time decreases and there are an increase in % drug release while as the amount of MCC is increased both the wetting time and disintegration time increases. Therefore, high level of crospovidone and medium level of MCC should be selected for the rapid disintegration of the tablets. The relationship between the dependent and independent variable was further elucidated using surface response plots. The data of the response surface plots [Figure 5] demonstrated that both X1 and X2 effect the disintegration time, wetting time and % release of drug.
Table 6.
Summary of regression analysis results
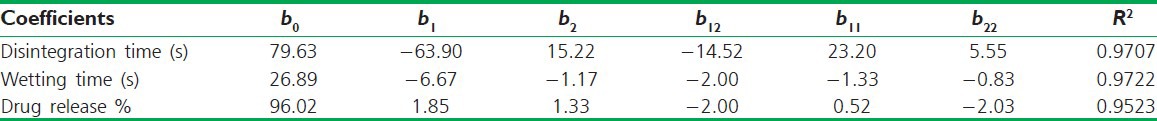
Figure 5.
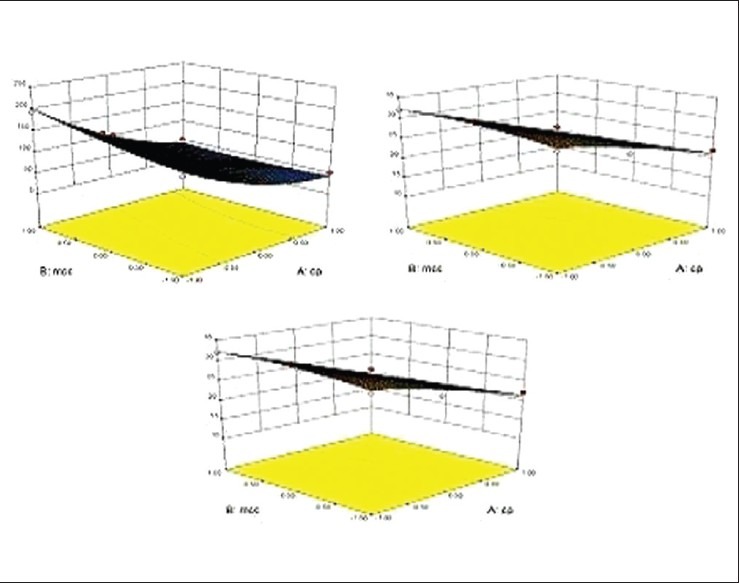
Response surface plot showing the influence of diluent microcrystalline cellulose and superdisintegrant (cp) over (a) disintegrantion time, (b) wetting time, (c) % drug release
Comparison between Conventional Marketed Product and Selected Formulation
A marketed conventional clobazam tablet was compared with the selected formulation F17 and results are reported in Table 7. From the comparison, it was found that formulation F17 is more effective than the conventional formulation of clobazam.
Table 7.
Comparison between one marketed product and selected formulation (F17)

CONCLUSION
The objective of the present investigations has been achieved by preparing fast dissolving drug delivery system for clobazam for the management of epileptic attacks with faster and quick onset of action particularly in children's using an optimum amount of superdisintegrant crospovidone and diluents MCC using direct compression technique. The present study has also revealed that RSM could efficiently be applied for the optimization and developing clobazam oro-dispersible tablets. Finally, it can be concluded that with a limited number of experiments an optimum formulation with required drug release and disintegration time can be designed with appropriate statistical experimental design and optimization technique.
ACKNOWLEDGMENT
We are thankful to Consern Pharma Pvt. Limited; Ludhiana; India, for providing us free sample of clobazam and Chitkara College of Pharmacy for providing all the facilities for performing experimental work.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
REFERENCES
- 1.Seager H. Drug-delivery products and the Zydis fast-dissolving dosage form. J Pharm Pharmacol. 1998;50:375–82. doi: 10.1111/j.2042-7158.1998.tb06876.x. [DOI] [PubMed] [Google Scholar]
- 2.Parkash V, Maan S, Deepika, Yadav SK, Hemlata, Jogpal V. Fast disintegrating tablets: Opportunity in drug delivery system. J Adv Pharm Technol Res. 2011;2:223–35. doi: 10.4103/2231-4040.90877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Solanki SS, Dahima R. Formulation and evaluation of aceclofenac mouth-dissolving tablet. J Adv Pharm Technol Res. 2011;2:128–31. doi: 10.4103/2231-4040.82951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradoo R, Shahani S, Poojary S, Deewan B, Sudarshan S. Fast dissolving drug delivery systems. J Am Med Assoc India. 2001;4:27–31. [Google Scholar]
- 5.Ashton H. Guidelines for the rational use of benzodiazepines. When and what to use. Drugs. 1994;48:25–40. doi: 10.2165/00003495-199448010-00004. [DOI] [PubMed] [Google Scholar]
- 6.Remy C. Clobazam in the treatment of epilepsy: A review of the literature. Epilepsia. 1994;35(Suppl 5):S88–91. doi: 10.1111/j.1528-1157.1994.tb05978.x. [DOI] [PubMed] [Google Scholar]
- 7.Buchanan N. Clobazam in the treatment of epilepsy: Prospective follow-up to 8 years. J R Soc Med. 1993;86:378–80. [PMC free article] [PubMed] [Google Scholar]
- 8.Keene DL, Whiting S, Humphreys P. Clobazam as an add-on drug in the treatment of refractory epilepsy of childhood. Can J Neurol Sci. 1990;17:317–9. doi: 10.1017/s0317167100030651. [DOI] [PubMed] [Google Scholar]
- 9.Jan MM, Shaabat AO. Clobazam for the treatment of intractable childhood epilepsy. Saudi Med J. 2000;21:622–4. [PubMed] [Google Scholar]
- 10.Kalia A, Khurana S, Bedi N. Formulation and evaluation of mouth dissolving tablets of oxcarbazepine. Int J Pharm Pharm Sci. 2009;1:12–23. [Google Scholar]
- 11.USP. <1174>flow USP 30 NF 25. 2007 [Google Scholar]
- 12.Carr RL. Evaluating flow properties of solids. Chem Eng. 1965;72:69–72. [Google Scholar]
- 13.Goel H, Kaur G, Tiwary AK, Rana V. Formulation development of stronger and quick disintegrating tablets: A crucial effect of chitin. Yakugaku Zasshi. 2010;130:729–35. doi: 10.1248/yakushi.130.729. [DOI] [PubMed] [Google Scholar]
- 14.Mishra D, Bindal M, Singh S, Kumar S. Rapidly disintegrating oral tablets of meloxicam. Indian Drugs. 2005;42:685–7. [Google Scholar]
- 15.Marshall K, Lachman L, Liberman H, Kanig J. 3rd ed. Mumbai: Varghese Publishing House; 1987. The Theory and Practice of Industrial Pharmacy; pp. 66–99. [Google Scholar]
- 16.Fu Y, Yang S, Jeong SH, Kimura S, Park K. Orally fast disintegrating tablets: Development, technologies, taste-masking and clinical studies. Crit Rev Ther Drug Carrier Syst. 2004;21:433–75. doi: 10.1615/critrevtherdrugcarriersyst.v21.i6.10. [DOI] [PubMed] [Google Scholar]
- 17.Bi Y, Sunada H, Yonezawa Y, Danjo K, Otsuka A, Iida K. Preparation and evaluation of a compressed tablet rapidly disintegrating in the oral cavity. Chem Pharm Bull (Tokyo) 1996;44:2121–7. doi: 10.1248/cpb.44.2121. [DOI] [PubMed] [Google Scholar]
- 18.Patil S, Sawant K. Formulation and evaluation of orodispersible tablets of ondensteron hydrochloride by direct compression using super disintegrant. Int J Pharma Sci Nanotechnol. 2008;1:106–11. [Google Scholar]
- 19.Mishra DN, Bindal M, Singh SK, Vijaya Kumar SG. Spray dried excipient base: A novel technique for the formulation of orally disintegrating tablets. Chem Pharm Bull (Tokyo) 2006;54:99–102. doi: 10.1248/cpb.54.99. [DOI] [PubMed] [Google Scholar]
- 20.Mohammad R, Yalda H, Majid S, Lida H, Geise B, Maryam M. Pharmacokinetics and bioequivalence study of clobazam 10 mg tablet. Int J Pharmacol. 2006;2:481–4. [Google Scholar]
- 21.James K. Dissolution testing of orally disintegrating tablets. Dissolution Technol. 2003;6:1–8. [Google Scholar]
- 22.Korsmeyer RW, Gurny R, Doelker EM, Buri P, Peppas NA. Mechanism of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15:25–35. doi: 10.1002/jps.2600721021. [DOI] [PubMed] [Google Scholar]
- 23.Peppas NA. Analysis of Fickian and non-Fickian drug release from polymers. Pharm Acta Helv. 1985;60:110–1. [PubMed] [Google Scholar]
- 24.Shoaib MH, Tazeen J, Merchant HA, Yousuf RI. Evaluation of drug release kinetics from ibuprofen matrix tablets using HPMC. Pak J Pharm Sci. 2006;19:119–24. [PubMed] [Google Scholar]
- 25.Jeevana B, Suneela G. Development of fast dissolving tablets of glibenclamide using crospovidon and its kneading mixture. Indian J Pharm Edu Res. 2010;44:334–40. [Google Scholar]
- 26.Remya K, Beena P, Bijesh P, Sheeba A. Formulation development, evaluation and comparative study of effects of super disintegrants in cefixime oral disintegrating tablets. J Young Pharm. 2010;2:234–9. doi: 10.4103/0975-1483.66794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao N, Augsburger LL. The influence of swelling capacity of superdisintegrants in different pH media on the dissolution of hydrochlorothiazide from directly compressed tablets. AAPS Pharm Sci Tech. 2005;6:E120–6. doi: 10.1208/pt060119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shu T, Suzuki H, Hironaka K, Ito K. Studies of rapidly disintegrating tablets in the oral cavity using co-ground mixtures of mannitol with crospovidone. Chem Pharm Bull (Tokyo) 2002;50:193–8. doi: 10.1248/cpb.50.193. [DOI] [PubMed] [Google Scholar]
- 29.Madan J, Sharma AK, Singh R. Fast dissolving tablets of aloe vera gel. Trop J Pharm Res. 2009;8:63–70. [Google Scholar]
- 30.Caramella C, Colombo P, Conte U, Gazzaniga A, La Manna A. The role of swelling in the disintegration process. Int J Pharm Techn Prod Manuf. 1984;2:1–5. [Google Scholar]
- 31.Van Kamp HV, Bolhuis GK, de Boer AH, Lerk CF, Lie-A-Huen L. The role of water uptake on tablet disintegration. Design of an improved method for penetration measurements. Pharm Acta Helv. 1986;61:22–9. [PubMed] [Google Scholar]
- 32.Shangraw R, Mitrevej A, Shah M. A new era of tablet disintegrants. Pharm Technol. 1980;4:49–57. [Google Scholar]
- 33.Patil BS, Rao DK, Kulkarni U, Hariprasanna RC, Mahesh MG. Formulation and evaluation of fast dissolving tablets of granistron hydrochloride by direct compression technique. Int J Curr Pharm Res. 2011;3:124–8. [Google Scholar]
- 34.Jacob S, Shirwarkar AA, Joseph A, Srinivasan KK. Novel co-processed excipients of mannitol and microcrystalline cellulose for preparing fast dissolving tablets of glipizide. Indian J Pharm Sci. 2007;69:633–9. [Google Scholar]