

Abstract
In addition to insulin sensitization, the thiazolidenedione drug pioglitazone exhibits favorable circulatory effects. Here, we hypothesized that pioglitazone protects against the hypertension and related vascular derangements caused by the immunosuppressant drug cyclosporine (CSA). Compared with vehicle (olive oil)-treated rats, chronic treatment with CSA (20 mg/kg/day s.c., for 14 days) increased blood pressure (BP), reduced the aortic protein expression of phosphorylated eNOS (p-eNOS), and impaired responsiveness of isolated aortas to endothelium-dependent vasorelaxations induced by carbachol. The effects of CSA on BP, aortic p-eNOS, and carbachol relaxations were abolished upon concurrent administration of pioglitazone (2.5 mg/kg/day). Serum levels of adiponectin, an adipose tissue-derived adipokine, were not altered by CSA but showed significant elevations in rats treated with pioglitazone or pioglitazone plus CSA. The possibility that alterations in the antioxidant and/or lipid profile contributed to the CSA-pioglitazone BP interaction was investigated. Pioglitazone abrogated the oxidative (aortic superoxide dismutase), lipid peroxidation (aortic malondialdyde), and dyslipidemic (serum LDL levels and LDL/HDL ratio) effects of CSA. Histologically, CSA caused focal disruption in the endothelial lining of the aorta and this effect disappeared in rats co-treated with pioglitazone. Collectively, pioglitazone abrogates the hypertensive effect of CSA via ameliorating detrimental changes in vascular endothelial NOS/NO pathway and oxidative and lipid profiles caused by CSA.
Keywords: Cyclosporine, pioglitazone, blood pressure, endothelium-dependent relaxations, oxidative stress
1. Introduction
Hypertension is one of the most troublesome effects often associated with the use of the immunosuppressant drug cyclosporine. Approximately 80% of patients treated with CSA after kidney transplantation develop hypertension [1]. Also, CSA causes hypertension in 70% of liver transplant patients and almost 100% of cardiac transplant recipients [2,3]. This clinical problem has been replicated in experimental animals in reported studies including ours [4,5]. Possible contributing factors include increased sympathetic [4], endothelin [6] and angiotensin activities [5], vascular endothelium dysfunction [7], interference with the testosterone-mediated vascular control [8,9], and deterioration of the cellular antioxidant profile [10,11].
Pioglitazone, like other thiazolidenedione drugs (TZD), reduces blood glucose level through improving insulin resistance. TZD produce their effect via binding to peroxisome proliferator-activated gamma receptors (PPARγ), a nuclear receptor that modulates among several other functions the transcription of genes essential for glucose transport, lipid homeostasis and metabolism, and arterial inflammation and atherogenesis [12]. In addition to their insulin sensitizing action, TZD have beneficial circulatory effects such as: (i) facilitation of endothelium-dependent vasodilation [13], (ii) inhibition of voltage-dependent calcium channels [14], (iii) inhibition of cell proliferation and migration, which precedes the development of vascular lesions [15], (iv) improving the antioxidant profile [13], and (v) increasing the expression and plasma levels of adiponectin [16]. The latter is a hormone derived from adipose tissue with protective effects against hypertension [17], endothelium dysfunction [18], and oxidative stress [19].
Despite their favorable circulatory effects, it is not known whether TZDs s can abrogate the hypertensive and vasculotoxic effects of CSA. This interesting possibility was investigated in the current study at the integrative (blood pressure) and in vitro (vascular reactivity) levels in rats that received chronic treatment of CSA, pioglitazone or their combination. The effect of either drug or their combination on endogenous mediators and ameliorators of endothelial dysfunction was investigated to gain insight into the molecular mechanisms that underlie their functional interaction.
2. Materials and Methods
Male Wistar rats (240–240 g, Faculty of Pharmacy, University of Alexandria, Egypt or Charles River, Raleigh, NC, USA) were used in the present study. Experiments were performed in strict accordance with institutional guidelines.
2.1. Intravascular cannulation
The method described in our previous studies [4,8] for intravascular cannulation and BP measurement in rats was adopted. Briefly, rats were anesthetized with thiopental (50 mg/kg i.p.). Catheters were placed into the abdominal aorta and vena cava via the left femoral vessels for measurement of arterial pressure and intravenous injections, respectively.
2.2. Rat isolated aortic ring preparations
Isolation of the rat aorta and recording of isometric contraction were performed as described in our previous studies [20,21]. Rats were euthanized with an overdose of thiopental sodium (100 mg/kg) and thoracic aortas were removed, trimmed free of connective tissues and cut into ring segments 3 mm in length. Aortic rings were mounted in 10 ml organ baths containing physiological solution at 37°C and aerated with 95% O2 and 5% CO2. The physiological solution was composed of the following (in mM): NaCl 118, KCl 4.7, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, and glucose 11.1. Aortic rings were mounted in organ baths by means of two stainless steel wire hooks inserted through the lumen of the ring. One of the hooks was anchored to a stationary pin at the bottom of the organ bath and the other was connected to an isometric force-displacement transducer (Grass FT-03C) which was connected to a Grass polygraph (Model 7D) for recording isometric contractions of the aorta. An optimum resting tension of 1.5 g was placed on the tissue and an equilibration period of 1 hr was allowed before the start of the experiment, with the bath fluid being replaced every 15 min. To study aortic responsiveness to vasorelaxants, aortas were precontracted with the α1-adrenoceptor agonist phenylephrine. To acclimatize the preparation, phenylephrine (1 μM) was added to the organ bath on two separate occasions during the 1 hr equilibration [20].
2.3. Western blotting
For the determination of total and phosphorylated aortic eNOS protein levels, the rat aorta was homogenized on ice in a homogenization buffer [50 mM Tris (pH 7.5), 0.1 mM EGTA, 0.1 mM EDTA, 2 μM leupeptin, 1 mM phenylmethylsulfonyl fluoride, 0.1% (vol/vol) Nonidet P-40, 0.1% SDS, and 0.1% deoxycholate]. After centrifugation (12,000 g for 10 min), protein in the supernatant was quantified (Bio-Rad protein assay system; Bio-Rad, Hercules, CA). Protein extracts (50 μg per lane) were run on a 4–12% SDS-polyacrylamide gel electrophoresis gel (Invitrogen, Carlsbad, CA) and electroblotted to nitrocellulose membranes. Blots were blocked for 120 min at room temperature in tris-buffered saline/tween20 (TBS/T) buffer (100 mM Tris pH 7.5, 150 mM NaCl, 0.1% Tween 20) containing 5% non-fat milk. They were then incubated overnight at 4°C with mouse antibody to total eNOS (1:2,500; BD Biosciences, San Jose, CA) or rabbit antibody to phospho-eNOS(Ser1177) (1:500, Cell Signaling Technology®, Inc. Danvers, MA) in TBS/T buffer containing 5% BSA. After 3 washes with TBS/T buffer, the blots were incubated for 60 min at room temperature with the appropriate horseradish peroxidase-linked species-specific anti-IgG (1:2,000, GE Healthcare BIO-Sciences Corp, Piscataway, NJ). After 3 washes with the TBS/T buffer, the blots were detected by enhanced chemiluminescence system and exposed to an X-ray film. Equivalent sample loading was confirmed by stripping membranes with blot restore membrane rejuvenation solution (SignaGen Laboratories, Gaithersburg, MD) and reprobing with anti-actin antibody (Sigma). Proteins bands were quantified by measuring the integrated density (mean density × area) using the NIH Image software (version 1.62); data was normalized in relation to actin, and expressed as a percent of control (olive oil) as in our previous studies [22,23].
2.4. Protocols and experimental groups
2.4.1. Effect of pioglitazone on CSA-evoked hypertension
Four groups of conscious rats (n=5–6 each) pre-instrumented for BP measurement were employed in this study to determine the effect of CSA on BP in absence or presence of pioglitazone. Rats were allocated to receive one of the following chronic treatments: (i) olive oil, (ii) CSA, (iii) pioglitazone, or (iv) CSA plus pioglitazone. Cyclosporine (20 mg/kg, dissolved in olive oil) or equal volume of the vehicle was injected s.c. in single daily doses for 14 consecutive days. This dose of cyclosporine has been shown to elevate BP and impair vasorelaxant activity [4,9,24]. Pioglitazone was mixed with food and served to rats in a dose of 2.5 mg/kg/day for 14 days. Rats were instrumented for BP measurement 2 days before starting drug treatments. BP was measured right before starting drug/vehicle treatment (day 0) and 3, 5, 8, 10, 12, and 14 days after the commencement of drug/vehicle administration. For BP measurement on any given day, the arterial catheter was connected to a Gould-Statham pressure transducer (Oxnard, CA, U.S.A.) and BP was displayed on a PowerLab system (ADInstruments, Inc., Colorado, USA). A period of at least 30 min was allowed at the beginning of each experiment for BP and heart rate stabilization. The blood and vasculature collected from these animals was used for the biochemical (serum adiponectin) and Western blot studies.
Two additional groups of rats (n=6–7 each) were employed to investigate the effect of captopril, an angiotensin converting enzyme inhibitor, on the hypertensive effect of CSA. Rats were treated with captopril alone (10 mg/kg/day i.p.) or combined with CSA (20 mg/kg/day) for 14 consecutive days.
2.4.2. CSA-pioglitazone interaction on aortic vasorelaxations
Another four groups of rats (n=7–8) were used to evaluate the effects of pioglitazone on the CSA-induced alterations in aortic responsiveness to endothelium-dependent and independent relaxations and aortic eNOS protein expression. As described in the preceding experiment, rats received olive oil, CSA, pioglitazone, or CSA plus pioglitazone. On the day of the experiment, rats were sacrificed, thoracic aortic rings were prepared as described earlier. Two cumulative concentration-response curves to stepwise cumulative addition of carbachol (3×10−8–3×10−4 M) or SNP (1×10−10–1×10−5 M) were established in aortic rings precontracted with phenylephrine (1 μM) as described in our previous studies [20,25]. Each new addition was made after the response to the previous addition had attained a steady state. A wash period of 60 min was allowed after the first curve to help the muscle relax to baseline tension. The aorta was then re-contracted with phenylephrine (1 μM) to permit construction of the SNP concentration-response curve.
2.4.3. CSA-pioglitazone redox, adiponectin and lipid interactions
Four groups of rats (n=6–7 each) were utilized and received similar treatments as described under section 2.4.2. After 14 days of chronic treatment, overnight-fasted rats were sacrificed and aortas were quickly removed, washed in ice-cold saline, blotted dry, weighed and homogenized in ice-cold saline (Kinematica Gmbh PT 45180, Lucerne, Switzerland). Tissues homogenates were divided into portions and stored at −20 °C till used for the determination of TBARs [26] and SOD [27]. Blood samples were collected and spun at 1200 g for 10 min and serum was aspirated, divided into aliquots and stored at −20 °C till analyzed. Serum samples were also prepared and used for the determination of triglycerides (TG), total cholesterol (TC), high density lipoprotein cholesterol (HDL-C), and glucose using the Randox® assay kit (Randox Laboratories Ltd., United Kingdom). The total lipids, low density lipoprotein cholesterol (LDL-C), LDL/HDL ratio were computed. Serm adiponectin was measured by Adiponectin (rat) ELISA (ALPCO Immunoassays, Salem, NH, USA). For histopathological examination, cross sections of the aorta were fixed in 10% formaldehyde, stained by Haematoxylin and Eosin stain, and examined under a light microscope.
2.5. Drugs
Carbachol (carbamoylcholine chloride), sodium nitroprusside (Sigma Chemical Co., St. Louis, MO, U.S.A.), thiopental (Thiopental, Biochemie GmbH, Vienna, Austria), povidone-iodine solution (Betadine, Nile Pharmaceutical Co., Cairo, Egypt) and Penicid (Cid Pharmaceutical Co., Cairo, Egypt) were purchased from commercial vendors. Cyclosporine, pioglitazone hydrochloride, and captopril were gifts from Novartis Pharma, AG (Basel, Switzerland), Takeda Pharmaceutical Company Ltd (Osaka, Japan), and PHARCO Pharmaceutical Co. (Alexandria, Egypt), respectively.
2.6. Data analysis and statistics
Values are expressed as means±S.E.M. Mean arterial pressure (MAP) was calculated as diastolic pressure + one third pulse pressure (systolic-diastolic pressures). Nonlinear regression was used to fit sigmoidal curves to individual concentration-response curves to determine agonist potency (EC50), the dose of carbachol or SNP giving half the maximum vasodilation, and maximum effect (Emax). The analysis of variance (ANOVA) followed by a Newman-Keuls post-hoc analysis was used for multiple comparisons with the level of significance set at P<0.05.
3. Results
3.1. Effect of pioglitazone on CSA-evoked hypertension
Baseline MAP values in rats subsequently receiving vehicle, pioglitazone, captopril, CSA, pioglitazone plus CSA, or captopril plus CSA were not statistically different (107±6, 106±5, 102±4, 103±4, 105±4, and 102±3 mmHg, respectively). Chronic treatment of rats with CSA (20 mg/kg/day for 14 days) resulted in significant increases in MAP compared with vehicle-treated animals (Fig. 1). The hypertensive effect of CSA appeared 5 days after starting CSA regimen and continued through the remaining period of the study (Fig. 1). Concurrent treatment with pioglitazone (2.5 mg/kg/day) or captopril (10 mg/kg/day) for 14 days virtually abolished CSA-induced hypertension (Fig. 1). While treatment with pioglitazone alone had no effect on MAP, captopril caused reductions in MAP that were statistically significant compared with control values during the first 10 days of the study (Fig. 1).
Figure 1.

Changes in mean arterial pressure (MAP) of conscious rats treated with cyclosporine (CSA, 20 mg/kg/day), pioglitazone (PIO, 2.5 mg/kg/day), captopril (Capto, 10 mg/kg/day), or their combination for 14 days. Values are means±S.E.M. of 5–7 observations. *P<0.05 compared with vehicle (olive oil) values, +P<0.05 compared with CSA values.
3.2. Effect of CSA-pioglitazone combination on aortic vasorelaxation and p-eNOS content
Changes evoked by chronic treatment with CSA, pioglitazone, or their combination on vasorelaxant responses of the aorta to carbachol or SNP are shown in figures 2 and 3. The cumulative addition of carbachol (3×10−8–3×10−4 M) or SNP (1×10−10–1×10−5 M) resulted in concentration-related relaxations of phenylephrine (1 μM) precontracted aortic rings (Fig. 2). Compared with corresponding vehicle (olive oil) values, chronic treatment with CSA (20 mg/kg/day for 14 days) caused significant reductions in the vasorelaxant responses to carbachol (Fig. 2). The EC50 (41×10−7±16×10−7 vs. 9×10−7±2×10−7 M) and Emax (58±6 vs. 86±5%) of carbachol in aortic preparations of CSA-treated rats were significantly higher and lower, respectively, than those of control values. The CSA-evoked attenuation of carbachol responses disappeared in preparations treated simultaneously with pioglitazone (2.5 mg/kg/day, Fig. 2). Also, the concurrent exposure to CSA and pioglitazone restored the EC50 and Emax values of carbachol to near-control values (Fig. 3). In preparations treated with pioglitazone alone, the relaxant responses (Fig. 2) and EC50 and Emax values (Fig. 3) of carbachol were not statistically different from control values. Unlike carbachol, the relaxant responses elicited by SNP were not affected by CSA, pioglitazone, or their combination (Figs. 2 and 3).
Figure 2.

Vasorelaxant effects of carbachol or sodium nitroprusside in phenylephrine (1 μM)-precontracted aortic rings obtained from rats treated with cyclosporine (CSA, 20 mg/kg/day), pioglitazone (2.5 mg/kg/day) or their combination for 14 days. Values are means±S.E.M. of 7–8 observations. *P<0.05 compared with vehicle (olive oil) values, +P<0.05 compared with CSA values.
Figure 3.

The vasorelaxant potency (EC50) and maximum effect (Emax) of carbachol or sodium nitroprusside in phenylephrine (1 μM)-precontracted aortic rings obtained from rats treated with cyclosporine (CSA, 20 mg/kg/day), pioglitazone (2.5 mg/kg/day) or their combination for 14 days. Values are means±S.E.M. of 7–8 observations. *P<0.05 compared with vehicle (olive oil) values, +P<0.05 compared with CSA values.
Western blotting showed that compared with vehicle values, 2-week CSA treatment significantly decreased phosphorylated eNOS expression in aortic tissues whereas total eNOS remained unchanged (Figs. 4 and 5). The decrease in p-eNOS expression caused by CSA was abrogated in rats treated concurrently with pioglitazone (Figs. 4 and 5).
Figure 4.

The protein expression of total or phosphorylated eNOS (p-eNOS) in aortic tissues of Wistar rats treated with cyclosporine (CSA, 20 mg/kg/day), pioglitazone (2.5 mg/kg/day) or their combination for 14 days. Values are means±S.E.M of 6–7 observations. *P<0.05 compared with vehicle (olive oil) values, +P<0.05 compared with CSA values.
Figure 5.
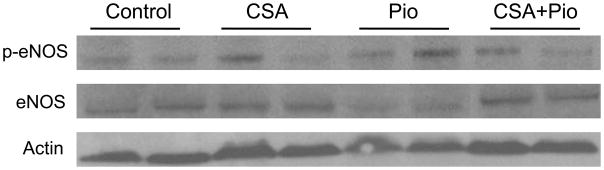
Illustrative gels depicting total or phosphorylated eNOS (p-eNOS) expression in homogenates of aortic tissues of Wistar rats treated with cyclosporine (CSA, 20 mg/kg/day), pioglitazone (2.5 mg/kg/day) or their combination for 14 days.
3.3. CSA-pioglitazone interaction on adiponectin, SOD and lipid profiles
Figures 6 and 7 illustrate changes caused by different drug treatments in serum adiponectin, aortic SOD and lipid profiles. Compared with vehicle-treated rats, serum adiponectin was not altered in rats treated with CSA but showed significant increases in rats treated with pioglitazone or pioglitazone plus CSA (Fig. 6A). CSA caused significant reduction in aortic SOD activity (Fig. 6B) and increase in MDA (Fig. 6C). Also, serum LDL (Fig. 7E) and LDL/HDL ratio (Fig. 7G) were increased by CSA treatment. On the other hand, serum total lipids, total cholesterol, triglycerides, HDL, VLDL, and glucose were not affected by CSA (Fig. 7). The deleterious effects of CSA on the antioxidant and lipid profiles disappeared upon concurrent administration of pioglitazone (Figs. 6 and 7). Histological examination of the aorta revealed that chronic exposure to CSA was associated with focal disruption in the endothelial lining of the aorta (Fig. 8). This effect of CSA was virtually abolished in rats treated simultaneously with pioglitazone (Fig. 8). No apparent histological changes were demonstrated in smooth muscle and connective tissue layers in the aortas of different rat preparations (Fig. 8).
Figure 6.

Plasma adiponectin (panel A) and aortic superoxide dismutase (SOD, panel B) and malondialdyde (MDA, panel C) levels in rats treated with cyclosporine (CSA, 20 mg/kg/day), pioglitazone (2.5 mg/kg/day) or their combination for 14 days. Values are means±S.E.M. of 6–7 observations. *P<0.05 compared with vehicle (olive oil) values, +P<0.05 compared with CSA values.
Figure 7.

Serum glucose and lipid levels in rats treated with cyclosporine (CSA, 20 mg/kg/day), pioglitazone (2.5 mg/kg/day) or their combination for 14 days. Values are means±S.E.M. of 6–7 observations. *P<0.05 compared with vehicle (olive oil) values, +P<0.05 compared with CSA values.
Figure 8.
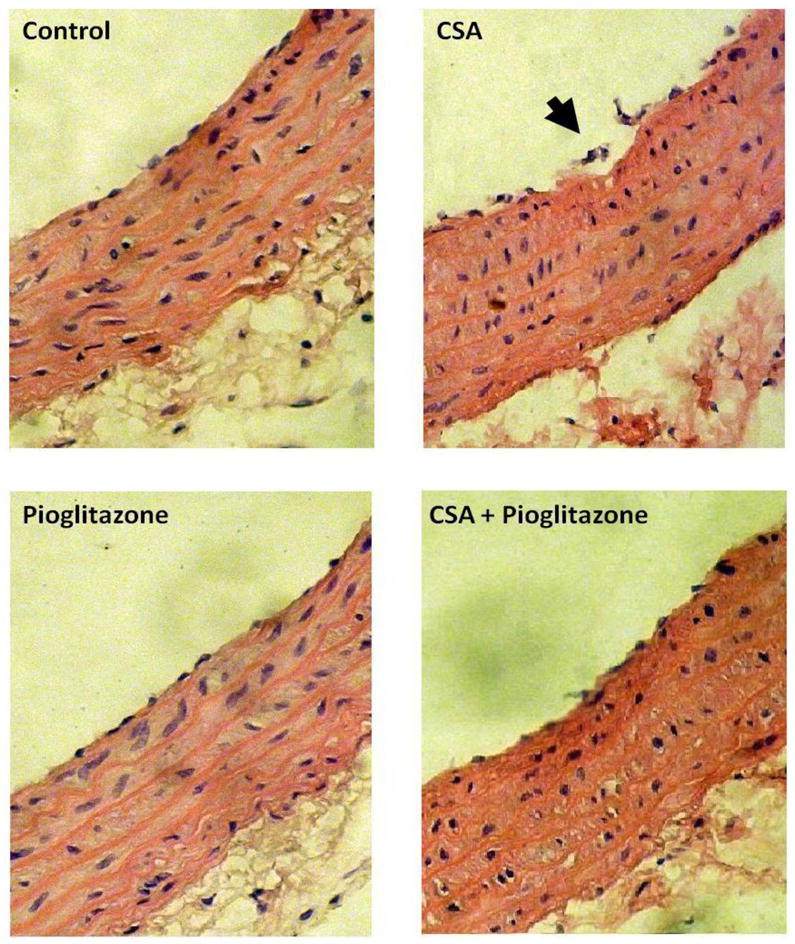
Hematoxylin and Eosin stained photomicrographs (× 100) of aortas obtained from rats treated with cyclosporine (CSA, 20 mg/kg/day), pioglitazone (2.5 mg/kg/day) or their combination for 14 days. In contrast to the intact endothelial cell layer seen in vehicle (olive oil) preparations, the aorta of CSA-treated rats shows focal disruption of the endothelial lining (arrow). This effect of CSA disappears in preparations treated concurrently with pioglitazone.
4. Discussion
The current study is the first to report on the potential protective effect of the insulin sensitizing drug pioglitazone against the hypertensive and deleterious vascular effects of chronic CSA. Concurrent pioglitazone administration virtually abolished the CSA-induced hypertensive response. The following findings explain, at least partly, pioglitazone counteraction of CSA-induced hypertension: (i) pioglitazone ameliorated the functional (impairment of endothelium-dependent relaxations), protein expression (reduced p-eNOS), and structural (disruption of vascular endothelial lining) abnormalities caused by CSA in the rat aorta, (ii) the CSA-induced lipid peroxidation, oxidative stress, and dyslipidemia were also abrogated by concurrent treatment with pioglitazone, and (iii) pioglitazone increased circulating adiponectin levels. These findings established convincing evidence that pioglitazone offsets the hypertensive effect of CSA via ameliorating significant vascular derangements caused by CSA.
We reasoned that concurrent exposure to pioglitazone would mitigate or at least minimize the hypertensive action of CSA and potentially related vasculotoxic manifestations. This assumption was ascertained in the current study because the hypertensive effect of CSA disappeared when rats were treated simultaneously with pioglitazone. We focused on vascular NOS/NO signaling because of its importance in CSA-evoked endothelial dysfunction and hypertension [28] and in the favorable vascular effects of pioglitazone [13]. Several lines of evidence are presented here that highlight a pivotal role for endothelial NOS/NO signaling in the pioglitazone amelioration of CSA-evoked endothelial dysfunction and hypertension. Histopathological and functional studies showed that pioglitazone circumvented the CSA-induced disruption in the aortic endothelial lining and impairment in endothelium-dependent vasorelaxant responses elicited by carbachol. This favorable effect of pioglitazone on endothelium-dependent vasorelaxations appears to be directly related to its ability to counterbalance the decline in p-eNOS expression caused by CSA in vascular smooth muscle. Notably, our finding that eNOS protein was expressed in the rat aorta is consistent with previous reports, which detected appreciable amounts of eNOS in aortic tissue homogenates [29]. Because treatment with CSA, pioglitazone, or their combination failed to alter aortic responsiveness to sodium nitroprusside, it is unlikely that alterations in aortic smooth muscle reactivity to NO or downstream effectors, e.g. GC/cGMP pathway, contributed to the vascular interaction between CSA and pioglitazone.
The role of oxidative stress in the hypertensive and vasculotoxic actions of CSA and the abrogation of these effects by pioglitazone was evaluated by measuring the activity of SOD in aortic tissues. SOD constitutes a biologically important group of metalloprotein enzymes that protect cells against the cytotoxic effect of the superoxide anion [30]. The lipid peroxidation product MDA, which reflects oxidative destruction of polyunsaturated fatty acids of biological membranes [31], was also assessed. Our observations that CSA caused significant decrease and increase in aortic SOD and MDA contents, respectively, are in line with the established role of oxidative stress caused by reactive oxygen species and lipid peroxides in the pathophysiology of CSA-induced hypertension, vascular injury, and endothelium dysfunction [10,11,19]. The subsequent increases in vascular reactive oxygen species, e.g. O2−. and H2O2, interfere with endothelial cell integrity and attenuate endothelium-dependant relaxations and elevates BP [19,32]. By the same token, the capability of concurrently administered pioglitazone to abrogate the CSA-induced alterations in SOD and MDA points to a key role for improved redox and lipid peroxidation states in the protective effect of pioglitazone against hypertension and vascular toxicity evoked by CSA. These findings are in harmony with reported favorable effects of pioglitazone in other pathological models of oxidative stress. For example, pioglitazone normalizes the activity of CAT, GPx and SOD in spontaneously hypertensive hyperlipidemic rats [33]. Similar protective effects for pioglitazone are demonstrated in cell cultures stimulated by high concentrations of glucose, where pioglitazone reduces MDA and restores SOD activity [34].
One more evidence that implicates oxidative stress in the vascular interaction between pioglitazone and CSA emerged from the assessment of the lipid profile. Our findings showed that the increases caused by CSA in serum LDL and LDL/HDL ratio were abrogated in rats treated concurrently with pioglitazone. Notably, clinical studies highlighted that long-term administration of CSA increases total cholesterol, triglycerides, LDL and/or VLDL, which correlate with increased risk of atherosclerosis [35,36]. The dyslipidemic effect of CSA has been attributed to the facilitation of cholesterol synthesis via increasing 3-hydroxy-3- methylglutaryl coenzyme A (HMG-CoA) reductase activity [36] and inhibition of cholesterol-26-hydroxylase [37]. Indeed, the CSA-induced dyslipidemia and elevated LDL together with the increased levels of reactive oxygen radicals promote the oxidation of lipoproteins and formation of proatherogenic ox-LDL [38]. The latter is believed to inactivate NO and induce endothelial dysfunction [11]. Our finding that pioglitazone favorably affects the dyslipidemic effect of CSA is consistent with reports that activation of PPARγ induces monocyte/macrophages differentiation and uptake of ox-LDL [39] and inhibits the production of inflammatory enzymes such as iNOS and matrix metalloproteinase-9 [40], and pro-inflammatory cytokines such as TNF-α, IL-1β, IL-6 [41].
The possibility that alterations in circulating adiponectin contributed to the CSA-pioglitazone hemodynamic interaction was investigated. We are not aware of any study that investigated the effect of CSA on adiponectin production. Although serum adiponectin was found to be decreased in renal transplant recipients on CSA therapy [10], it is not clear whether the reduction in adiponectin level is a direct effect of CSA or it occurs secondary to the after-transplantation improvement in kidney and vascular functions. In the present study, serum adiponectin remained unaltered in CSA-treated rats, thereby arguing against a possible role for adiponectin in the hypertensive and vasculotoxicity induced by CSA. Nonetheless, because serum adiponectin was increased by pioglitazone, the probability that adiponectin was responsible, at least partly, for the favorable effect of pioglitazone on the hypertensive and vascular effects of CSA cannot be ruled out. Indeed, the beneficial circulatory effects of pioglitazone have been attributed, among other things, to increases in the expression and plasma level of adiponectin [16]. As mentioned earlier, adiponectin acts to counterbalance rises in BP [17] and endothelium dysfunction [18]. More studies are clearly needed to identify more precisely the role of adiponectin, and possibly other adipokines, in the CSA-pioglitazone hemodynamic interaction.
The current study establishes that pioglitazone could be an effective therapeutic regimen for combating the hypertensive action of CSA. In this regard, pioglitazone appears to be as effective as the standard antihypertensive drug captopril. The latter prevented CSA-evoked hypertension in this experimental study as well as in reported clinical studies [42]. The questions remain, however, whether (i) the additional endothelial/antioxidant benefits of pioglitazone makes it a better drug than others currently used in clinical practice, and (ii) other glitazones, e.g. rosiglitazone and troglitazone, can similarly abrogate the hemodynamic consequences of CSA. These important issues will be addressed in future studies from our laboratory.
In summary, our functional, biochemical, molecular, and histological findings suggest a protective effect for pioglitazone against the hypertensive and vasculotoxic effects of CSA. Apparently, the amelioration of the CSA-evoked lipid peroxidation, oxidative stress, and dyslipidemia contributes to the inhibitory effect of pioglitazone on the CSA-evoked hypertension. The clinical importance of this research is warranted particularly in view of recent information that the activation of PPARγ boosts the immunosuppressant effect of CSA via the inhibition of T cell proliferation and IL-2 release [43]. Interestingly, the combined use of PPARγ ligands and low-dose CSA might represent a rationale therapeutic approach for the prevention of CSA nephrotoxicity while maintaining adequate immunosuppression [43,44].
Acknowledgments
Supported by the Faculty of Pharmacy, University of Alexandria, Egypt, and by Grant [AA07839] from the National Institute on Alcohol Abuse and Alcoholism. The authors thank Dr. Neveen Eldeeb, Assistant Professor of Pathology, Faculty of Medicine, Alexandria University, Egypt, for assistance with the histopathological examination, and Ms. Kui Sun, East Carolina Univ., USA, for technical assistance. Cyclosporine, pioglitazone hydrochloride, and captopril were generously supplied by Novartis Pharma, AG (Basel, Switzerland), Takeda Pharmaceutical Company Ltd (Osaka, Japan), and PHARCO Pharmaceuticals (Alexandria, Egypt), respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ponticelli C, Montagnino G, Aroldi A, Angelini C, Braga M, Tarantino A. Hypertension after renal transplantation. Am J Kidney Dis. 1993;21:73–8. doi: 10.1016/0272-6386(93)70098-j. [DOI] [PubMed] [Google Scholar]
- 2.Diaz MA, Cisneros C, Montejo JC, Garcia C, Cantalapiedra JA, Perez F. Systemic arterial hypertension in the immediate postoperative period of liver transplantation. Transplant Proc. 1989;21:3547–8. [PubMed] [Google Scholar]
- 3.Eid A, Perkins JD, Rakela J, Krom RA. Conversion from standard cyclosporine to low-dose cyclosporine in liver transplant recipients: effect on nephrotoxicity and hypertension beyond one year. Transplant Proc. 1989;21:2238–9. [PubMed] [Google Scholar]
- 4.El-Mas MM, Omar AG, Helmy MM, Mohy El-Din MM. Interruption of central neuronal pathway of imidazoline I1 receptor mediates the hypertensive effect of cyclosporine in rats. Brain Res. 2009;1248:96–106. doi: 10.1016/j.brainres.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 5.Nishiyama A, Kobori H, Fukui T, Zhang GX, Yao L, Rahman M, Hitomi H, Kiyomoto H, Shokoji T, Kimura S, Kohno M, Abe Y. Role of angiotensin II and reactive oxygen species in cyclosporine A-dependent hypertension. Hypertension. 2003;42:754–60. doi: 10.1161/01.HYP.0000085195.38870.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gardiner SM, March JE, Kemp PA, Fallgren B, Bennett T. Regional haemodynamic effects of cyclosporine A, tacrolimus and sirolimus in conscious rats. Br J Pharmacol. 2004;141:634–43. doi: 10.1038/sj.bjp.0705659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.El-Mas MM, Mohy El-Din MM, El-gowilly SM, Sharabi FM. Relative roles of endothelial relaxing factors in cyclosporine-induced impairment of cholinergic and β-adrenergic renal vasodilations. Eur J Pharmacol. 2004;487:149–58. doi: 10.1016/j.ejphar.2004.01.025. [DOI] [PubMed] [Google Scholar]
- 8.El-Mas MM, Afify EA, Omar AG, Sharabi FM. Cyclosporine adversely affects baroreflexes via inhibition of testosterone modulation of cardiac vagal control. J Pharmacol Exp Ther. 2002;301:346–54. doi: 10.1124/jpet.301.1.346. [DOI] [PubMed] [Google Scholar]
- 9.El-Mas MM, Afify EA, Omar AG, Mohy El-Din MM, Sharabi FM. Testosterone depletion contributes to cyclosporine-induced chronic impairment of acetylcholine renovascular relaxations. Eur J Pharmacol. 2003;468:217–24. doi: 10.1016/s0014-2999(03)01720-5. [DOI] [PubMed] [Google Scholar]
- 10.Navarro-Antolín J, Redondo-Horcajo M, Zaragoza C, Alvarez-Barrientos A, Fernández AP, León-Gómez E, Rodrigo J, Lamas S. Role of peroxynitrite in endothelial damage mediated by Cyclosporine A. Free Radic Biol Med. 2007;42:394–403. doi: 10.1016/j.freeradbiomed.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 11.Galle J, Lehmann-Bodem C, Hubner U, Heinloth A, Wanner C. CyA and OxLDL cause endothelial dysfunction in isolated arteries through endothelin-mediated stimulation of O(2)(−) formation. Nephrol Dial Transplant. 2000;15:339–46. doi: 10.1093/ndt/15.3.339. [DOI] [PubMed] [Google Scholar]
- 12.Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, Grimaldi PA, Kadowaki T, Lazar MA, O’Rahilly S, Palmer CN, Plutzky J, Reddy JK, Spiegelman BM, Staels B, Wahli W International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev. 2006;58:726–41. doi: 10.1124/pr.58.4.5. [DOI] [PubMed] [Google Scholar]
- 13.Majithiya JB, Paramar AN, Balaraman R. Pioglitazone, a PPARgamma agonist, restores endothelial function in aorta of streptozotocin-induced diabetic rats. Cardiovasc Res. 2005;66:150–61. doi: 10.1016/j.cardiores.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 14.Heppner TJ, Bonev AD, Eckman DM, Gomez MF, Petkov GV, Nelson MT. Novel PPARgamma agonists GI 262570, GW 7845, GW 1929, and pioglitazone decrease calcium channel function and myogenic tone in rat mesenteric arteries. Pharmacology. 2005;73:15–22. doi: 10.1159/000081070. [DOI] [PubMed] [Google Scholar]
- 15.Dubey RK, Zhang HY, Reddy SR, Boegehold MA, Kotchen TA. Pioglitazone attenuates hypertension and inhibits growth of renal arteriolar smooth muscle in rats. Am J Physiol. 1993;265:R726–32. doi: 10.1152/ajpregu.1993.265.4.R726. [DOI] [PubMed] [Google Scholar]
- 16.Maeda N, Takahashi M, Funahashi T, Kihara S, Nishizawa H, Kishida K, Nagaretani H, Matsuda M, Komuro R, Ouchi N, Kuriyama H, Hotta K, Nakamura T, Shimomura I, Matsuzawa Y. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes. 2001;50:2094–9. doi: 10.2337/diabetes.50.9.2094. [DOI] [PubMed] [Google Scholar]
- 17.Iwashima Y, Katsuya T, Ishikawa K, Ouchi N, Ohishi M, Sugimoto K, Fu Y, Motone M, Yamamoto K, Matsuo A, Ohashi K, Kihara S, Funahashi T, Rakugi H, Matsuzawa Y, Ogihara T. Hypoadiponectinemia is an independent risk factor for hypertension. Hypertension. 2004;43:1318–23. doi: 10.1161/01.HYP.0000129281.03801.4b. [DOI] [PubMed] [Google Scholar]
- 18.Barac A, Campia U, Matuskey LA, Lu L, Panza JA. Effects of peroxisome proliferator-activated receptor-gamma activation with pioglitazone on plasma adipokines in nondiabetic patients with either hypercholesterolemia or hypertension. Am J Cardiol. 2008;101:980–5. doi: 10.1016/j.amjcard.2007.11.058. [DOI] [PubMed] [Google Scholar]
- 19.Matsunami T, Sato Y, Ariga S, Sato T, Kashimura H, Hasegawa Y, Yukawa M. Regulation of oxidative stress and inflammation by hepatic adiponectin receptor 2 in an animal model of nonalcoholic steatohepatitis. Int J Clin Exp Pathol. 2010;3:472–4. [PMC free article] [PubMed] [Google Scholar]
- 20.El-Mas MM, Abdel-Galil AA-G, El-Gowelli HM, Daabees TT. Short-term aortic barodenervation diminishes α1-adrenoceptor reactivity in rat aortic smooth muscle. Eur J Pharmacol. 1997;322:201–10. doi: 10.1016/s0014-2999(97)00010-1. [DOI] [PubMed] [Google Scholar]
- 21.El-Mas MM, Sharabi FM, El-gowilly SM, Mohy El-Din MM. Inhibition of nitric oxide-guanylate cyclase-dependent and -independent signaling contributes to impairment of β-adrenergic vasorelaxations by cyclosporine. Biochem Pharmacol. 2007;73:359–67. doi: 10.1016/j.bcp.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 22.El-Mas MM, Zhang J, Abdel-Rahman AA. Upregulation of vascular inducible nitric oxide synthase mediates the hypotensive effect of ethanol in conscious female rats. J Appl Physiol. 2006;100:1011–8. doi: 10.1152/japplphysiol.01058.2005. [DOI] [PubMed] [Google Scholar]
- 23.El-Mas MM, Fan M, Abdel-Rahman AA. Endotoxemia-mediated induction of cardiac inducible nitric-oxide synthase expression accounts for the hypotensive effect of ethanol in female rats. J Pharmacol Exp Ther. 2008;324:368–75. doi: 10.1124/jpet.107.127498. [DOI] [PubMed] [Google Scholar]
- 24.Gerkens JF. Cyclosporine treatment of normal rats produces a rise in blood pressure and decreased renal vascular responses to nerve stimulation, vasoconstrictors and endothelium-dependent dilators. J Pharmacol Exp Ther. 1989;250:1105–12. [PubMed] [Google Scholar]
- 25.Fahim M, El-Mas MM, Abdel-Rahman AA, Mustafa SJ. Influence of aortic baroreceptor denervation on adenosine receptor-mediated relaxation of isolated rat aorta. Eur J Pharmacol. 1994;254:183–91. doi: 10.1016/0014-2999(94)90386-7. [DOI] [PubMed] [Google Scholar]
- 26.Ohkawa H, Ohishi N, Yagi K. Assay of lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–8. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- 27.Marklund S, Marklund G. Involvement of superoxide anion radical in autooxidation of pyrogallol and a convenient assay for SOD. Eur J Biochem. 1974;47:469–74. doi: 10.1111/j.1432-1033.1974.tb03714.x. [DOI] [PubMed] [Google Scholar]
- 28.Oriji GK. Role of metoprolol, β1-adrenoceptor antagonist, thromboxane A2 and nitric oxide in CsA-induced hypertension. Prostaglandins Leukot Essent Fatty Acids. 2003;68:233–8. doi: 10.1016/s0952-3278(02)00276-4. [DOI] [PubMed] [Google Scholar]
- 29.Olukman M, Orhan CE, Celenk FG, Ulker S. Apocynin restores endothelial dysfunction in streptozotocin diabetic rats through regulation of nitric oxide synthase and NADPH oxidase expressions. J Diabetes Complications. 2010;24:415–23. doi: 10.1016/j.jdiacomp.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 30.Bertuglia S, Giusti A. Microvascular oxygenation and oxidative stress during postischemic reperfusion. PO2, ROS, and NO during reperfusion. Adv Exp Med Biol. 2005;566:23–9. doi: 10.1007/0-387-26206-7_4. [DOI] [PubMed] [Google Scholar]
- 31.Jung T, Engels M, Kaiser B, Poppek D, Grune T. Intracellular distribution of oxidized proteins and proteasome in HT22 cells during oxidative stress. Free Radic Biol Med. 2006;40:1303–13. doi: 10.1016/j.freeradbiomed.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 32.Ülker S, Mckeown PP, Bayraktutan U. Vitamins reverse endothelial dysfunction through regulation of eNOS and NAD(P)H oxidase activities. J Hypertens. 2003;41:534–9. doi: 10.1161/01.HYP.0000057421.28533.37. [DOI] [PubMed] [Google Scholar]
- 33.Saiki R, Okazaki M, Iwai S, Kumai T, Kobayashi S, Oguchi K. Effects of pioglitazone on increases in visceral fat accumulation and oxidative stress in spontaneously hypertensive hyperlipidemic rats fed a high-fat diet and sucrose solution. J Pharmacol Sci. 2007;105:157–67. doi: 10.1254/jphs.fp0070619. [DOI] [PubMed] [Google Scholar]
- 34.Zhang M, Zhou SH, Zhao S, Li XP, Liu LP, Shen XQ. Pioglitazone can downregulate bone morphogenetic protein-2 expression induced by high glucose in human umbilical vein endothelial cells. Pharmacology. 2008;81:312–6. doi: 10.1159/000119118. [DOI] [PubMed] [Google Scholar]
- 35.Quaschning T, Mainka T, Nauck M, Rump LC, Wanner C, Kramer-Guth A. Immunosuppression enhances atherogenicity of lipid profile after transplantation. Kidney Int Suppl. 1999;71:S235–S7. doi: 10.1046/j.1523-1755.1999.07162.x. [DOI] [PubMed] [Google Scholar]
- 36.Superko H, Haskell W, DiRiccio C. Lipoprotein and hepatic lipase activity and high-density lipoprotein sub-classes after cardiac transplantation. Am J Cardiol. 1990;66:1131–4. doi: 10.1016/0002-9149(90)90517-5. [DOI] [PubMed] [Google Scholar]
- 37.de Groen PC. Cyclosporine, low density lipoprotein, and cholesterol. Mayo Clin Proc. 1988;63:1012–21. doi: 10.1016/s0025-6196(12)64916-7. [DOI] [PubMed] [Google Scholar]
- 38.López-Miranda J, Vilella E, Perez-Jimenez F, Espino A, Jimenez-Pereperez JA, Masana L, Turner PP. Low-density lipoprotein metabolism in rats treated with cyclosporine. Metabolism. 1993;42:678–83. doi: 10.1016/0026-0495(93)90232-d. [DOI] [PubMed] [Google Scholar]
- 39.Moore KJ, Rosen ED, Fitzgerald ML, Randow F, Andersson LP, Altshuler D, Milstone DS, Mortensen RM, Spiegelman BM, Freeman MW. The role of PPAR-gamma in macrophage differentiation and cholesterol uptake. Nat Med. 2001;7:41–47. doi: 10.1038/83328. [DOI] [PubMed] [Google Scholar]
- 40.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 41.Bruemmer D, Blaschke F, Law RE. New targets for PPARgamma in the vessel wall: implications for restenosis. Int J Obes. 2005;29:S26–S30. doi: 10.1038/sj.ijo.0802910. [DOI] [PubMed] [Google Scholar]
- 42.Curtis JJ, Laskow DA, Jones PA, Julian BA, Gaston RS, Luke RG. Captopril-induced fall in glomerular filtration rate in cyclosporine-treated hypertensive patients. J Am Soc Nephrol. 1993;3:1570–4. doi: 10.1681/ASN.V391570. [DOI] [PubMed] [Google Scholar]
- 43.Rampino T, Ranghino A, Guidetti C, Gregorini M, Soccio G, Marasà M, Libetta C, Guida G, De Amici M, Dal Canton A. Activation of PPARgamma enhances in vitro the immunosuppressive effect of cyclosporine on T lymphocytes. Transpl Immunol. 2007;18:32–6. doi: 10.1016/j.trim.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 44.Tanaka Y, Hasegawa T, Chen Z, Okita Y, Okada K. Renoprotective immunosuppression by pioglitazone with low-dose cyclosporine in rat heart transplantation. J Thorac Cardiovasc Surg. 2009;138:744–51. doi: 10.1016/j.jtcvs.2009.04.019. [DOI] [PubMed] [Google Scholar]