

Abstract
Earlier studies showed that 2-methoxyestradiol (2ME2), an endogenous nonpolar metabolite of estradiol-17β, is a strong inducer of G2/M cell cycle arrest (based on analysis of cellular DNA content) in human cancer cell lines. The present study sought to investigate the molecular mechanism underlying 2ME2-induced cell cycle arrest. We found that 2ME2 can selectively induce mitotic prometaphase arrest, but not G2 phase arrest, in cultured MDA-MB-435s and MCF-7 human breast cancer cells. During the induction of prometaphase arrest, there is a time-dependent initial up-regulation of cyclin B1 and Cdc2 proteins, occurring around 12-24 h. The strong initial up-regulation of cyclin B1 and Cdc2 matches in timing the 2ME2-induced prometaphase arrest. The 2ME2-induced prometaphase arrest is abrogated by selective knockdown of cyclin B1 and Cdc2, or by pre-treatment of cells with roscovitine, an inhibitor of cyclin-dependent kinases, or by co-treatment of cells with cycloheximide, a protein synthesis inhibitor that was found to suppress the early up-regulation of cyclin B1 and Cdc2. In addition, we provided evidence showing that MAD2 and JNK1 are important upstream mediators of 2ME2-induced up-regulation of cyclin B1 and Cdc2 as well as the subsequent induction of mitotic prometaphase arrest. In conclusion, treatment of human cancer cells with 2ME2 causes up-regulation of cyclin B1 and Cdc2, which then mediate the induction of mitotic prometaphase arrest.
Keywords: 2-Methoxyestradiol, Anti-microtubule Agents, Mitotic Prometaphase Arrest, Cyclin-Dependent Kinases, MAD2
1. Introduction
2-Methoxyestradiol (2ME2), a well-known endogenous derivative of estradiol-17β (E2), is biosynthesized by catechol-O-methyltransferase using 2-hydroxy-E2 as substrate and S-adenosyl-L-methionine as methyl donor [1,2]. Studies have shown that 2ME2 at pharmacological concentrations has a strong growth-inhibitory effect in a variety of human cancer cell lines in culture [3–12]. This estrogen metabolite has also been tested in a number of preclinical as well as clinical studies for its potential usefulness in the treatment of solid tumors [17–22]. Since the anti-cancer activity of 2ME2 is not shared by E2, it is unlikely that such actions occur through its metabolism to estrogenic derivatives.
To understand its cellular mechanism of actions, a number of earlier studies have investigated its effect on cell cycle changes and cell death in cultured human cancer cells [6,10–16,23–25]. It has been consistently observed that the induction of G2/M cell cycle arrest (based on flow cytometric analysis of the cellular DNA content) is a predominant initial cellular change in 2ME2-treated cells [11,12,14–16,23,24], and this change is subsequently followed by increased apoptotic cell death [13–16,23,24]. It has been suggested that the ability of 2ME2 to induce G2/M cell cycle arrest is associated with its ability to interact with microtubules, thereby disrupting normal microtubule functions [26,27]. In line with this suggestion, the G2/M cell cycle arrest induced by 2ME2 can also be induced by other well-known anti-microtubule agents, such as nocodazole, paclitaxel, and vinblastine. These microtubule inhibitors can bind to tubulins or microtubules and suppress microtubule dynamics and functions, subsequently inducing G2/M cell cycle arrest as well as apoptotic cell death [28–32].
In a normal cell cycle, the activation of the cyclin B1/Cdc2 function plays a critical role in regulating the transition of cells from the G2 phase to mitotic M phase [33]. Generally, cells with a suppressed cyclin B1/Cdc2 activity would tend to be arrested in the G2 phase, whereas cells with an up-regulated cyclin B1/Cdc2 activity would be favored to enter and proceed through mitosis [33]. Notably, some of the earlier studies have probed the changes of some of the cell cycle regulatory proteins (such as cyclin B1 and Cdc2) following treatment of human cancer cells with 2ME2. While one study reported that the cellular level of cyclin B1 is down-regulated in MCF-7 human breast cancer cells treated with 2ME2[34], other studies using different human cancer cell lines showed an increased level of cyclin B1 and/or Cdc2 [11,35,36]. The observation of an increased cyclin B1/Cdc2 level during the occurrence of G2/M arrest in 2ME2-treated cells is rather intriguing, because this phenomenon is different from the known regulatory functions of cyclin B1 and Cdc2 during a normal cell cycle.
In the present study, therefore, we sought to further study the mechanism underlying 2ME2-induced G2/M cell cycle arrest in human breast cancer cells by focusing on the cell cycle regulatory proteins at the G2/M boundary. We found that 2ME2 only selectively induces mitotic prometaphase arrest, but not G2 phase arrest. During the induction of prometaphase arrest by 2ME2, there is a time-dependent early up-regulation of cyclin B1 and Cdc2 proteins. A series of experiments were then conducted to demonstrate that the cyclin B1/Cdc2 up-regulation in 2ME2-treated breast cancer cells contributes critically to the selective development of prometaphase arrest and some of the cellular morphological characteristics observed. In addition, we also showed that MAD2 and JNK1 are important upstream mediators responsible for the up-regulation of cyclin B1 and Cdc2 and the induction of prometaphase arrest.
2. MATERIALS AND METHODS
2.1. Chemicals and reagents
2ME2, nocodazole, roscovitine, cycloheximide, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were obtained from Sigma (St. Louis, MO). The JNK1/2 inhibitor SP600125 was obtained from Calbiochem (La Jolla, CA). The anti-c-Jun NH2-terminal kinase 1/2 (JNK1/2) antibody was obtained from Biosource (Camarillo, CA). Anti-p-c-jun, anti-Cdc2 (Cdk1), anti-Cdc2 (Try), anti-Cdc2 (Thr161), anti-cyclin B1, anti-Cdk2, anti-Cdc25C, anti-Cdc25A, and anti-GAPDH antibodies were purchased from Cell Signaling Technology (Beverly, MA). Anti-MAD2 antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The sources of other materials are described elsewhere [16].
2.2. Cell culture, MTT assay, and flow cytometric analysis
The culture conditions of MCF-7 and MDA-MB-435s human breast cancer cells (obtained from ATCC, Manassas, VA) were described earlier [16]. For treatment of cells with 2ME2, a stock solution of 2ME2 (5 mM in 200-proof ethanol) was diluted in the culture medium immediately before addition. Following the treatment, the MTT assay was performed to determine cell viability according to methods described earlier [16]. For flow cytometric analysis, cells were harvested and treated as described previously [16]. The analysis was performed on a model BD LSR II flow cytometer (BD Bioscience, San Jose, CA).
2.3. Nuclear and cytosolic extracts
The nuclear and cytosolic fractions were prepared using the subcellular fractionation kit obtained from Biovision (Mountain View, CA), by following the instructions of the manufacturer. Briefly, cells were suspended in hypotonic buffer and lysed with the proprietary detergent included in the kit. Samples were centrifuged at 800 g for 10 min at 4°C. The supernatant was collected, centrifuged 5 min at 16,000 g to remove any remaining nuclei, and then transferred to a new microtube (cytosolic protein fraction). The original pellet was re-suspended in the nuclear extraction buffer and then incubated on ice for 40 min with occasional vortexing. After salt extraction, the nuclear suspension was centrifuged at 16,000 g for 10 min, and the supernatant was collected and stored at −80°C as nuclear extract.
2.4. Western blotting
For Western blotting, cells were washed and then suspended in 100 mL lysis buffer, and the amount of proteins was determined. Proteins were separated by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and electrically transferred to a polyvinylidene difluoride membrane (Bio-Rad). After blocking the membrane using 5% skim milk, target proteins were immunodetected using specific antibodies. Thereafter, the horseradish peroxidase (HRP)-conjugated anti-rabbit IgG was applied as the secondary antibody, and the positive bands were detected using the Amersham ECL Plus Western blotting detection reagents (GE Healthcare, Piscataway, NJ).
2.5. Small interfering RNA (siRNA) treatment
The role of JNK1 in mediating 2ME2 actions was examined using the JNK1-siRNA (siJNK1) to silence its gene. The siJNK1 (catalog no. AM16704) and the negative control siRNA (siCon; catalog no. AM4611) were obtained from Ambion (Austin, TX). Similarly, the role of cyclin B1, Cdc2, and MAD2 in mediating 2ME2 actions was examined using the following specific siRNAs (obtained from Santa Cruz), namely, cyclin B1-siRNA (sicyclin B1; catalog no. sc-29284), Cdc2-siRNA (siCdc2; catalog no. sc-29252), and MAD2-siRNA (siMAD2; catalog no. sc-35837), to selectively silence their expression. MDA-MB-435s and MCF-7 cells were seeded 24 h earlier and reached a density of 30–50% confluency at the time of transfection. Sixty pmols of siJNK1 or forty nmols of sicyclin B1, siCdc2, siMAD2, or siCon were used for transfection with Lipofectamine 2000 (Invitrogen; Carlsbad, CA). Transfected cells were maintained in culture for 2 days before harvesting and further analyses. The efficiency of the siRNA knockdown for each gene was determined by Western blot analysis of its protein product.
2.6. Statistical analysis
Many of the quantitative data were expressed as mean ± S.D. Statistical significance was determined using the analysis of variance (ANOVA) followed by a multiple comparison test with a Bonferroni adjustment. The P value of less than 0.05 is considered statistically significant.
3. RESULTS
3.1. 2ME2 induces mitotic prometaphase arrest in human breast cancer cells
First, we examined the effect of 2ME2 on cell cycle changes in two representative human breast cancer cell lines (i.e., the ER-negative MDA-MB-435s cells and the ER-positive MCF-7 cells) in culture. As shown in Fig. 1A, 2ME2 at 1 and 2 μM increased the combined G2/M cell populations (based on flow cytometric analysis) in a dose-dependent manner. Time-course experiments showed that the 2ME2-induced G2/M arrest peaked around 12 h after treatment (Fig. 1B). Nocodazole, a prototypical microtubule inhibitor [31], was tested as a positive control for comparison. Treatment with 250 nM nocodazole for 12 h induced a similar G2/M cell cycle arrest in these two cell lines (Fig. 1C).
Fig. 1. Effect of 2ME2 on cell cycle arrest.

(A and B) Changes in the combined G2 and M cell populations following treatment with 2ME2. MDA-MB-435s and MCF-7 cells were treated with 1 or 2 μM 2ME2 for 24 h (A), or with 2 μM 2ME2 for 12, 24, 48, and 72 h (B). Treatment with 250 nM nocodazole for 12 h was also tested for comparison. Cells were harvested and analyzed by flow cytometry. Each bar is the mean ± S.D. (N = 6 replicates). * P < 0.05 versus the corresponding control. (C) Morphology of cells arrested in prometaphase (stained with Hoechst-33342) following treatment with 2 μM 2ME2 or 250 nM nocodazole at indicated time points. The morphology was viewed using both phase contrast (PC) and fluorescence microscopes (× 200 magnification). The arrow points the cell shown in the enlarged inset. (D) Quantitative data of the relative population of prometaphase-arrested MDA-MB-435s and MCF-7 cells following treatment with 2ME2 (2 μM). Each bar is the mean ± S.D. value from triplicate determinations, and each value (percentage) was based on counting ≥ 200 nuclei in each treatment group. * P < 0.05 versus the control.
Next, we examined the effect of 2ME2 on the nuclear morphological changes after the cells were stained with Hoechst-33342. Typical morphological features of untreated MCF-7 cells at different stages of mitosis are shown in Fig. S1A. As shown in Fig. 1C, most of the cells treated with 2ME2 at 12 and 24 h had a round shape, and there was no concurrent blebbing of the cell membranes, an indicator of cell death. However, many of the treated cells exhibited gross chromosomal condensation and segregation at 12 and 24 h, which are characteristic morphological changes in cells blocked in prometaphase (Fig. S1B)[37]. There was a close correlation between the time-dependent changes in 2ME2-induced prometaphase arrest (Fig. 1D) and the combined G2/M cell population (Fig. 1B). Notably, treatment of cells with nocodazole also induced similar morphological changes associated with prometaphase arrest (Fig. 1C, 1D). These data showed, for the first time, that 2ME2 selectively induces mitotic prometaphase arrest, but not G2 phase arrest, in human breast cancer cells in a time- and concentration-dependent manner.
3.2. Effect of 2ME2 on cell cycle regulatory proteins
To understand the mechanism by which 2ME2 induces mitotic prometaphase arrest, we first determined the expression of several cell cycle regulatory proteins at the G2/M boundary, including cyclin B1, cyclin A, Cdc2, Cdk2, Cdc25A, and Cdc25C. As shown in Fig. 2A and 2B, the levels of cyclin B1 and Cdc2 expression in MDA-MB-435s and MCF-7 cells were markedly increased at 3 h after 2ME2 treatment and peaked between 12 and 24 h. After the initial 48 h (i.e., between 48 and 72 h), their levels decreased in a time-dependent manner. In comparison, the initial increase in the first 24 h was not observed with cyclin A, Cdk2, and Cdc25A (data not shown), although similar time-dependent decreases from 24 to 72 h were seen for these regulatory proteins (Fig. 2B). The Cdc25’s mitotic migration band was detected in MCF-7 cells at 6 and 12 h after 2ME2 treatment, and was decreased after 24 h (Fig. 2A). Similarly, the levels of cyclin B1 and Cdc2 in MCF-7 cells treated with nocodazole were also changed in a comparable time-dependent manner (Fig. 2C).
Fig. 2. Effect of 2ME2on the cellular levels of cyclin B1, Cdc2, Cdk2 and cdc25C proteins.

(A) MDA-MB-435s and MCF-7 cells were treated with 2 μM 2ME2 for different lengths of time as indicated. Total cell lysates were prepared, and an equal amount of protein lysates was electrophoretically separated on the 10% SDS-polyacrylamide gel, and then transferred to a nitrocellulose membrane. The protein level was determined using specific antibodies on an enhanced chemiluminescence (ECL) apparatus. (B and C) Cells were treated with 2 μM 2ME2 or 250 nM nocodazole for different lengths of time as indicated. Total cell lysates were analyzed for the protein levels of cyclin B1, Cdc2, Cdk2, cyclin A, Cdc25C, and Cdc25A by Western blotting. D. MDA-MB-435s and MCF-7 cells were treated with 2 μM 2ME2 for 12 h, and the total cell lysates were analyzed for the levels of p-Cdc2(Try15) and p-Cdc2(Thr161) proteins by Western blotting using antibodies specific for each of the phosphorylated proteins.
It is known that the function of Cdc2 is regulated by its phosphorylation at Try15 (which results in inactivation) and Thr161 (which results in activation). We found that whereas the phosphorylation at Thr161 (activation) was markedly increased by 2ME2 treatment, the phosphorylation at Try15 (inactivation) was decreased (Fig. 2D). Together, these data show that 2ME2-induced Cdc2 activation includes both an increase in its protein level and a favorable change in its phosphorylation patterns.
3.3. Functional role of cyclin B1/Cdc2 up-regulation in 2ME2-induced prometaphase arrest
It is known that aberrant accumulation of cyclin B1 and Cdc2 in the nucleus of a cell will trigger chromosomal condensation and segregation [30]. Therefore, it is hypothesized that the up-regulation of cyclin B1/Cdc2 may be associated with the development of prometaphase arrest in 2ME2-treated cancer cells. To test this hypothesis, we first probed whether the early up-regulation of cyclin B1 and Cdc2 protein levels contributes to the observed nuclear morphological changes in 2ME2-treated cells, by examining the subcellular localization of these two proteins in control and 2ME2-treated cells using immunofluorescence staining and Western immunoblotting. As shown in Fig. 3A, while the levels of these two proteins were very low in the cytoplasm and nuclei of vehicle-treated control cells, their levels were dramatically increased in the nuclei of 2ME2-induced prometaphase cells (at 12 h) (Fig. 3A). The levels of cyclin B1 and Cdc2 were also increased in the nuclear fraction in a time-dependent manner (Fig. 3B).
Fig. 3. Effect of cyclin B1/Cdc2 knockdown on the development of 2ME2-induced prometaphase arrest.

(A) MCF-7 cells were treated with 2 μM 2ME2 for 12 h and then immunostained with anti-cyclin B1 or anti-Cdc2 antibodies. Nuclear accumulation of cyclin B1 (left panel) and Cdc2 (right panel) in 2ME2-treated cells that have developed prometaphase arrest. (B) Subcellular localization of cyclin B1 and Cdc2 in MCF-7 cells during 2ME2-induced prometaphase arrest (at 6, 12, and 24 h after treatment). Cytosolic and nuclear extracts were prepared from 2ME2-treated cells, and cyclin B1 and Cdc2 proteins were analyzed by Western blotting. (C, D, and E) MCF-7 cells were transfected with sicyclin B1 and siCdc2 or siCon, and then treated with 2ME2 for 12 h. Total cell lysates were analyzed by Western blotting for cyclin B1 and Cdc2. The DNA content of the cells was analyzed using flow cytometry (D), and their gross nuclear morphology was examined under fluorescence microscopy (original magnification, × 200) after the cells were stained with Hoechst-33342 (E, left panel). The quantitative data for the prometaphase-arrested cells (based on counting ≥ 200 nuclei in each treatment group) are shown in E(right panel). Each bar is the mean ± S.D. from triplicate measurements. * P < 0.05 versus vehicle control; #P < 0.05 versus 2ME2 treatment.
To provide more definitive evidence for the involvement of cyclin B1 and Cdc2, next we employed the siRNA approach to selectively knock down their expression in breast cancer cells. Twenty-four h after joint transfection with sicyclin B1 and siCdc2, cells were treated with 2ME2. Double knockdown of cyclin B1 and Cdc2 abrogated 2ME2-induced up-regulation of cyclin B1 and Cdc2 (Fig. 3C), and this change in cyclin B1 and Cdc2 levels was accompanied by significant reductions in G2/M arrest (based on flow cytometric analysis; Fig. 3D) and prometaphase arrest (based on morphological analysis; Fig. 3E) when the cells were analyzed at 12 h after 2ME2 treatment. At 24 h after 2ME2 treatment of these knockdown cells, the degree of cell death (based on PI and annexin V double staining) was also markedly reduced (data not shown).
The functional relationship between 2ME2-induced cyclin B1 and Cdc2 up-regulation and prometaphase arrest was also probed by using roscovitine (a known inhibitor of cyclin-dependent kinases) and cycloheximide (a protein synthesis inhibitor). As shown in Fig. 4A, treatment with 20 μM roscovitine suppressed cyclin B1/Cdc2 up-regulation induced by 2 μM 2ME2 or 250 nM nocodazole. While treatment of cells with roscovitine (20 μM) alone slightly increased the combined G2/M cell populations, co-treatment of roscovitine with of 2ME2 strongly suppressed the accumulation of the combined G2/M cell populations (Fig. 4B). Further quantification of the population of prometaphase-arrested cells showed that treatment with roscovitine alone did not appreciably enhance prometaphase arrest, but it dose-dependently suppressed 2ME2-induced accumulation of prometaphase-arrested cells (Fig. 4C).
Fig. 4. Effect of roscorvitine on 2ME2-induced prometaphase arrest.

(A and B) MCF-7 cells were pre-treated for 2 h with roscovitine (20 μM) and then further treated with 2 μM 2ME2 or 250 nM nocodazole for additional 12 h. Total cell lysates were analyzed by Western blotting for cyclin B1 and Cdc2 protein levels (A, upper panel). The relative protein levels of cyclin B1 and Cdc2 are calculated according to their densitometry readings, which are normalized to GAPDH protein level (A, lower panel). (B) Cells were pre-treated for 2 h with roscovitine (20 μM) and then treated in combination with 2 μM 2ME2 for additional 12 or 72 h. Cells were harvested and analyzed using flow cytometry. Each bar is the mean ± S.D. from triplicate measurements. (C, upper panel) Gross nuclear morphological changes in cells treated with a representative concentration (20 μM) of roscovitine. The nuclear morphology was examined under fluorescence microscopy (× 200 magnification) after the cells were stained with Hoechst-33342. The arrow points to a prometaphase-arrested cell. (C, lower panel) Quantitation of prometaphase cells based on counting 200 nuclei in each treatment group. * P < 0.05 versus vehicle control; #P < 0.05 versus 2ME2 or nocodazole treatment.
Pre-treatment of breast cancer cells with cycloheximide also strongly prevented 2ME2-induced increases in cyclin B1 and Cdc2 levels (Fig. 5A). Similarly, cycloheximide reduced 2ME2-induced mitotic prometaphase arrest (52% to 3.3%; Fig. 5B). Notably, treatment of cells with cycloheximide alone did not appreciably increase the population of prometaphase-arrested cells (Fig. 5B), nor of cell populations in the G1 or G2 or sub-G1 phases (data not shown).
Fig. 5. Effect of cycloheximide (CHX) on 2ME2-induced promataphase arrest.

Cells were pre-treated for 2 h with cycloheximide (1 μg/mL) and then treated for additional 12 h with 2 μM 2ME2. Total cell lysates were analyzed by Western blotting for cyclin B1 and Cdc2 protein levels (A, upper panel). The relative protein levels of cyclin B1 and Cdc2 were calculated according to their densitometry readings, which were normalized to GAPDH protein level (A, lower panel). Each value is mean ± S.D. from triplicate measurements. * P < 0.05 versus vehicle control; #P < 0.05 versus 2ME2 treatment. The gross nuclear morphology was examined under fluorescence microscopy (original magnification, × 200) after the cells were stained with Hoechst-33342 (B, upper panel). The arrow points to a prometaphase-arrested cell. The quantitative data (B, lower panel) was based on counting 200 nuclei in each treatment group. Each bar is the mean ± S.D. from triplicate determinations. * P < 0.05 versus vehicle control; #P < 0.05 versus 2ME2 treatment.
3.4. Role of JNK1 in 2ME2-induced prometaphase arrest
Earlier studies in human prostate cancer cells [11] and human breast cancer cells [16] have shown that treatment with 2ME2 results in increased phosphorylation of JNK. Here we sought to evaluate whether JNK is involved in 2ME2-induced prometaphase arrest in MDA-MB-435s and MCF-7 cells. As shown in Fig. 6A and 6B, treatment of these cells with 2 μM 2ME2 induced, in a time-dependent manner, the phosphorylation of JNK1, but not JNK2. The total JNK1 and JNK2 protein levels were not significantly altered by 2ME2 treatment. A marked increase in JNK1 phosphorylation was seen as early as 30 min in MCF-7 cells after 2ME2 treatment, and peaked at 60 min (Fig. 6B).
Fig. 6. 2ME2-induced activation of the JNK signaling pathway during the induction of prometaphase arrest.

(A and B) MDA-MB-435s and MCF-7 cells were first incubated with 2 μM 2ME2 for the indicated lengths of time, and then the cellular extracts were subjected to Western blotting of the levels of p-JNK1/2 and JNK1/2. The level of GAPDH protein was determined as a loading control. (C and D) MDA-MB-435s and MCF-7 cells were first transfected with siJNK1 or the negative control siRNAs, and they were then treated with 2ME2 for additional 24 h (C) or 12 h (D), respectively. p-JNK, c-jun, p21, cyclin B1, Cdc2, and p-Cdc2(Thr161) levels were determined by Western blotting. (E and F) MDA-MB-435s and MCF-7 cells were transfected with siJNK1 and the negative control siRNAs, and then treated with 2ME2 for additional 12 h. The DNA content of the cells was analyzed by flow cytometry (E). The gross nuclear morphology was examined under fluorescence microscopy after the cells were stained with Hoechst-33342, and the quantitative data (F) was based on counting 200 nuclei in each treatment group. Each bar is a mean ± S.D. from triplicate determinations. * P < 0.05 versus vehicle control; #P < 0.05 versus 2ME2 treatment.
To verify the role of JNK1 in 2ME2-induced G2/M arrest, we used the siRNA approach to suppress JNK1 expression in these cells. Twenty-four h after transfection with the siJNK1 or the negative control siRNAs, cells were treated with 2ME2 for additional 24 h. As shown Fig. 6C, knockdown of JNK1 abrogated 2ME2-induced increase in JNK1 and c-jun phosphorylation compared to cells transfected with the control siRNAs. Similarly, knockdown of JNK1 abrogated the 2ME2-induced up-regulation of cylin B1 and Cdc2 as well as p-Cdc2 (Thr161) at 12 h (Fig. 6D), suggesting that JNK1 phosphorylation contributes to 2ME2-induced up-regulation of cyclin B1/Cdc2.
In addition, as shown in Fig. 6E, JNK1 knockdown in MCF-7 and MDA-MB-435s cells reduced the degree of 2ME2-induced G2/M cell cycle arrest, from 61.5% to 36.8% and from 67.9% to 40.4%, respectively, compared to control siRNA-transfected cells. Notably, the reduction in the population of prometaphase cells was more pronounced in cells with JNK1 knockdown (from 48.0% to 11.0% and 54.0% to 9.8%, respectively) (Fig. 6F), compared to the reductions in the combined G2/M cell populations.
3.5. MAD2 mediates 2ME2-induced cyclin B1/Cdc2 accumulation and prometaphase arrest
Next, we examined whether MAD2 is involved in 2ME2-induced cyclin B1/Cdc2 accumulation and JNK1 phosphorylation in mitotically-arrested MCF-7 cells. We found that the MAD2 level in cells treated with 2ME2 was not increased by the treatment (Fig. 7A). Next, siRNA approach was used to selectively knock down the expression of MAD2. Following transfection of cells with siMAD2, the cells were then treated with 2ME2 and then harvested for Western blotting of p-JNK1/2, p-c-jun, p-Cdc2 (Thr161), cyclin B1, and MAD2 protein levels. Surprisingly, knockdown of MAD2 abrogated 2ME2-induced increases in JNK1 activation as well as the up-regulation of cyclin B1 and p-Cdc2 (Thr161) when compared to control siRNA-transfected cells (Fig. 7A). Moreover, MAD2 knockdown also abrogated 2ME2-induced prometaphase arrest at 12 and 24 h (52% to 2% and 43% to 6%, respectively) (Fig. 7B). Together, these observations show that the function of MAD2 is critically required for these changes seen in 2ME2-treated breast cancer cells.
Fig. 7. Effect of MAD2 knockdown on 2ME2-induced cyclin B1/Cdc2 up-regulation.
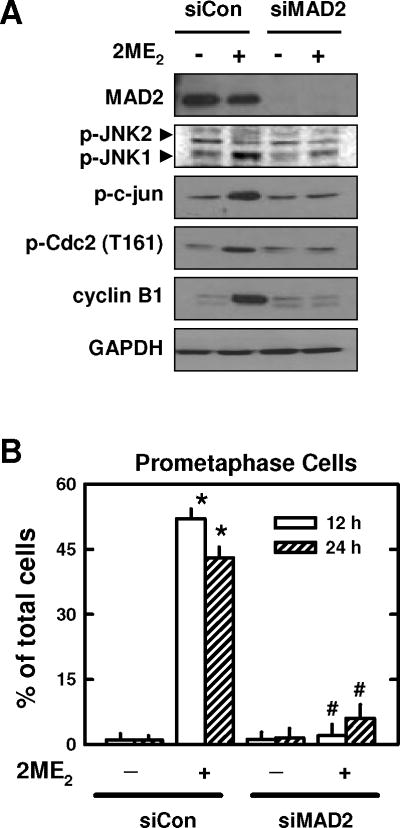
(A) MCF-7 cells were transfected with siMAD2 or the negative control siRNAs, and then treated with 2ME2 for additional 24 h. The protein levels of p-JNK, c-jun, cyclin B1, MAD2, and p-Cdc2(Thr161) were determined by Western blotting. (B) The degree of 2ME2-induced prometaphase arrest in cells with or without MAD2 knockdown. The quantitative value (percentage) was based on counting ≥ 200 nuclei in each treatment group. Each bar is the mean ± S.D. from three separate experiments. * P < 0.05 versus vehicle control; #P < 0.05 versus 2ME2 treatment.
4. DISCUSSION
In this study, we first confirmed earlier observations that 2ME2 can predominantly induce a combined G2/M arrest (based on flow cytometric analysis) in cultured human breast cancer cells. Further analysis of the nuclear morphology showed that 2ME2 mainly induces mitotic prometaphase arrest, but not G2 phase arrest, in these cells. The predominant induction of prometaphase arrest by 2ME2 in cancer cells is not surprising given that this estrogen metabolite was known to have an anti-microtubule activity [26]. Apparently, the ability of 2ME2 to induce prometaphase arrest is not related to its residual estrogenic activity or the presence of the estrogen receptors because it induces a similar level of prometaphase arrest regardless of the estrogen receptor status of the cell lines tested.
In a normal cell cycle, the transition from G2 phase to mitotic phase is triggered by the activation of the cyclin B1/Cdc2 complex. Cells with a suppressed cyclin B1/Cdc2 activity would tend to be arrested in the G2 phase, whereas cells with an elevated cyclin B1/Cdc2 activity would be favored to enter and then proceed through mitosis [33]. The results of our present study show that treatment of human breast cancer cells with 2ME2 (a microtubule inhibitor) not only causes a strong up-regulation of cyclin B1 and Cdc2 protein levels, but it also alters its phosphorylation patterns. Collectively, these changes are expected to result in marked increases in the functionality of the cyclin B1/Cdc2 complex. However, under these conditions, a higher percentage of cells is actually found to be selectively arrested in mitotic prometaphase; by contrast, the control cells that are not treated with 2ME2 and have lower cyclin B1/Cdc2 levels actually have far fewer cells arrested in the prometaphase. Evidently, these seemingly paradoxical changes are due to the presence of 2ME2, which disrupts microtubule functions and, in turn, creates a false signal in prometaphase cells that they do not have an adequate cyclin B1/Cdc2 activity to proceed through mitosis. Consequently, cells arrested in prometaphase would increase their cyclin B1 and Cdc2 levels and also change the phosphorylation pattern of Cdc2, as cellular compensatory responses to 2ME2 treatment. As discussed below, the results of this study provide strong support for the proposed novel concept that a strong, early up-regulation of cyclin B1 and Cdc2 following 2ME2 treatment contributes critically to the development of prometaphase arrest as well as some of the characteristic morphological changes.
Earlier studies have shown that a rapid, excessive activation of the cyclin B1-dependent Cdc2 in G2 phase cells can result in aberrant entry into mitotic phase [38,39]. Moreover, premature nuclear accumulation of cyclin B1 and Cdc2 can trigger chromosomal condensation and segregation. The results of our present study show that the marked early increase in cyclin B1 and Cdc2 protein levels is accompanied by rapid nuclear accumulation of these two proteins, in conjunction with the development of nuclear chromosomal condensation and segregation, which are characteristic features of cells blocked in prometaphase. Moreover, we showed that selective knockdown of cyclin B1 and Cdc2 strongly reduces the severity of nuclear chromosomal condensation and segregation as well as prometaphase arrest in 2ME2-treated cells. Similar reductions in nuclear chromosomal condensation and segregation as well as prometaphase arrest are also observed in 2ME2-treated cells upon co-treatment with roscorvitine (an inhibitor of the cyclin-dependent kinases) or with cycloheximide (a protein biosynthesis inhibitor that was found to reduce the levels of both cyclin B and Cdc2 in 2ME2-treated cells). Based on these observations, it is suggested that a stronger initial compensatory up-regulation of the cyclin B1/Cdc2 level following 2ME2 treatment would result in a more severe form of prometaphase arrest because higher cyclin B/Cdc2 levels likely would bring about a more severe degree of chromosomal condensation and segregation. In line with this study, we observed that when higher concentrations of 2ME2 are used, higher levels of cyclin B1 and Cdc2 are induced (data not shown), which is accompanied by a more severe form of prometaphase arrest.
Earlier studies with human breast cancer cells [16] or human prostate cancer cells [11] have shown that treatment with 2ME2 results in increased phosphorylation of JNK. In addition, it was also reported that JNK inhibition can attenuate 2ME2-induced apoptosis in human breast cancer cells [16]. In this study, we evaluated whether JNK is also involved in 2ME2-induced prometaphase arrest in MDA-MB-435s and MCF-7 cells. We showed that 2ME2 can induce, in a time-dependent manner, the phosphorylation of JNK1, but not JNK2, while the total JNK1/2 protein levels are not significantly changed. Knockdown of JNK1 abrogates 2ME2-induced up-regulation of cylin B1 and Cdc2 as well as p-Cdc2 (Thr161), suggesting that JNK1 phosphorylation contributes to 2ME2-induced up-regulation of cyclin B1 and Cdc2. As expected, JNK1 knockdown also results in a reduction in 2ME2-induced prometaphase arrest.
It is known that MAD2 is an essential member of the spindle checkpoint system, which restrains the metaphase-to-anaphase transition. This protein is present at unattached kinetochores and is essential to execute a block in the metaphase-to-anaphase transition when a microtubule inhibitor is present [40]. In this study, we found that although the MAD2 protein level is not increased by treatment with 2ME2, its knockdown drastically diminishes 2ME2-induced JNK1 activation as well as the subsequent up-regulation of cyclin B1 and Cdc2 and the development of prometaphase arrest. Since we also showed that JNK1 knockdown abrogates 2ME2-induced up-regulation of cyclin B1/Cdc2 and prometaphase arrest, it is evident that JNK1 is a downstream target of MAD2.
Notably, we observed that MAD2 knockdown produces a stronger reduction in prometaphase-arrested cells than does the joint knockdown of cyclin B1 and Cdc2. This observation is actually in agreement with the mechanistic explanation developed here. First, we showed in this study that MAD2 knockdown can reduce the levels of cyclin B1 and Cdc2. Second, it is known that MDA2 knockdown can also directly facilitate the procession of prometaphase cells through metaphase and anaphase [40]. Therefore, combination of these two effects would jointly help reduce the population of prometaphase-arrested cells, producing a stronger reduction in prometaphase arrest compared to cyclin B1 and Cdc2 knockdown alone.
In this study, roscovitine was found to suppress 2ME2-induced increases in cyclin B1 and Cdc2 levels. Flow cytometric analysis showed that the combined G2/M cell population is increased following treatment with roscorvitine alone, but the population of prometaphase cells is only slightly increased. Our observation is in agreement with an earlier study showing that roscorvitine blocks MCF-7 cells in the G2 phase [41]. However, when roscovitine is used in combination with 2ME2, it strongly suppresses 2ME2-induced accumulation of the G2/M population as well as prometaphase cells. The dose-dependent effect of roscorvitine in suppressing 2ME2-induced accumulation of both G2/M and prometaphase populations is closely correlated with its ability to suppress cyclin B1/Cdc2 up-regulation. Collectively, these data suggest that roscorvitine, when used in combination with 2ME2, can keep cells in the G2 phase and suppress cells from entering prometaphase through inhibition of the cyclin B1/Cdc2 complex.
We found that co-treatment of cancer cells with cycloheximide and 2ME2 completely suppresses the development of mitotic arrest. This effect is readily explained on the basis of our observation that cycloheximide can suppress 2ME2-induced up-regulation of cyclin B1 and Cdc2. Notably, the combination treatment of cells with cycloheximide and 2ME2 does not produce a stronger degree of cell death; instead, these two agents antagonize the cytotoxicity of each another. This observation is intriguing, and offers a mechanistic basis that these two classes of agents should not be used together as a combination anticancer regimen.
It is of interest to note that although flow cytometric analysis shows that the cells treated with 2ME2 or roscorvitine exhibit a similar G2/M cell cycle arrest pattern, the true nature of their cell cycle arrests is strikingly different, based on the comparisons made in this study. While roscorvitine predominantly induces G2 phase arrest (with minimal accumulation of prometaphase cells), 2ME2 predominantly induces prometaphase arrest, along with a reduction in the G2 phase cell population. Similar to the observation made with roscorvitine, selective knockdown of cyclin B1 and Cdc2 also decreases the population of prometaphase cells, while it does not markedly affect G2 phase cells. Based on the mechanistic explanation developed in this study, these results are readily understood because chemical inhibition or down-regulation of cyclin B1/Cdc2 would only block those cells that have already completed the DNA replication in the G2 phase and prevent them from entering mitosis. Consequently, they would not produce significant prometaphase arrest.
Based on the observations made in the present study, a novel mechanistic explanation (schematically depicted in Fig. 8) is proposed for the role of cyclin B1 and Cdc2 up-regulation in the induction of mitotic prometaphase arrest in 2ME2-treated cells. The presence of 2ME2 will cause disruption of microtubule formation in prometaphase cells, which subsequently results in failure of the microtubules to attach to kinetochores on the chromosomes. The unattached kinetochores will then be bound by the spindle checkpoint protein MAD2. The binding of MAD2 at the kinetochores prevents the progression from prometaphase to metaphase and further to anaphase. The occurrence of prometaphase arrest creates a feedback up-regulation of the cyclin B1 and Cdc2 protein levels, partly mediated by the actions of the kinetochore-bound MAD2 proteins. The rapid rise in these two proteins in prometaphase cells followed by rapid nuclear accumulation leads to the induction of the characteristic nuclear phenotypes. Following a prolonged prometaphase arrest, it is speculated that the 2ME2-treated cells will undergo cell death via activation of the intrinsic apoptotic pathways.
Fig. 8. Schematic explanation of the role of the early cyclin B1/Cdc2 up-regulation in the development of prometaphase arrest in cancer cells treated with 2ME2.

Supplementary Material
Acknowledgments
This study was supported, in part, by a grant from the National Institutes of Health (CA-97109). Some of the analytical and imaging instruments employed in this study are part of the COBRE core facility that is supported by the NIH Grant P20RR021940 from the National Center for Research Resources.
A list of nonstandard abbreviations used in the paper
- 2ME2
2-methoxyestradiol
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- Rosc
roscovitine
- CHX
cycloheximide
- Cdc2
cell division control 2
- CDK
cyclin-dependent kinase
- MAD2
mitotic arrest deficient 2
Footnotes
STATEMENT OF CONFLICTS OF INTEREST
The authors have no conflict of interest to declare.
References
- 1.Zhu BT, Conney AH. Functional role of estrogen metabolism in target cells: review and perspectives. Carcinogenesis. 1998a;19:1–27. doi: 10.1093/carcin/19.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Zhu BT, Conney AH. Is 2-methoxyestradiol an endogenous estrogen metabolite that inhibits mammary carcinogenesis? Cancer Res. 1998b;58:2269–2277. [PubMed] [Google Scholar]
- 3.Fotsis T, Zhang Y, Pepper MS, Adlercreutz H, Montesano R, Nawroth PP, Schweigerer L. The endogenous oestrogen metabolite 2-methoxyoestradiol inhibits angiogenesis and suppresses tumour growth. Nature. 1994;368:237–239. doi: 10.1038/368237a0. [DOI] [PubMed] [Google Scholar]
- 4.Klauber N, Parang S, Flynn E, Hamel E, D’Amato RJ. Inhibition of angiogenesis and breast cancer in mice by the microtubule inhibitors 2-methoxyestradiol and taxol. Cancer Res. 1997;57:81–86. [PubMed] [Google Scholar]
- 5.Pribluda VS, Gubish ER, Jr, Lavallee TM, Treston A, Swartz GM, Green SJ. 2-Methoxyestradiol: an endogenous antiangiogenic and antiproliferative drug candidate. Cancer Metastasis Rev. 2000;19:173–179. doi: 10.1023/a:1026543018478. [DOI] [PubMed] [Google Scholar]
- 6.Lin HL, Liu TY, Wu CW, Chi CW. 2-Methoxyestradiol-induced caspase-3 activation and apoptosis occurs through G(2)/M arrest dependent and independent pathways in gastric carcinoma cells. Cancer. 2001;92:500–509. doi: 10.1002/1097-0142(20010801)92:3<500::aid-cncr1348>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 7.Tinley TL, Leal RM, Randall-Hlubek DA, Cessac JW, Rao PN, Mooberry SL. Novel 2-methoxyestradiol analogues with antitumor activity. Cancer Res. 2003;63:1538–1549. [PubMed] [Google Scholar]
- 8.Liu ZJ, Zhu BT. Concentration-dependent mitogenic and antiproliferative actions of 2-methoxyestradiol in estrogen receptor-positive human breast cancer cells. J Steroid Biochem Mol Biol. 2004;88:265–275. doi: 10.1016/j.jsbmb.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Han GZ, Liu ZJ, Shimoi K, Zhu BT. Synergism between the anticancer actions of 2-methoxyestradiol and microtubule-disrupting agents in human breast cancer. Cancer Res. 2005;65:387–393. [PubMed] [Google Scholar]
- 10.Davoodpour P, Landström M M. 2-Methoxyestradiol-induced apoptosis in prostate cancer cells requires Smad7. J Biol Chem. 2005;280:14773–14779. doi: 10.1074/jbc.M414470200. [DOI] [PubMed] [Google Scholar]
- 11.Ray G, Dhar G, Van Veldhuizen PJ, Banerjee S, Saxena NK, Sengupta K, Banerjee SK. Modulation of cell-cycle regulatory signaling network by 2-methoxyestradiol in prostate cancer cells is mediated through multiple signal transduction pathways. Biochemistry. 2006;45:3703–3713. doi: 10.1021/bi051570k. [DOI] [PubMed] [Google Scholar]
- 12.Stander BA, Marais S, Vorster CJ, Joubert AM. In vitro effects of 2-methoxyestradiol on morphology, cell cycle progression, cell death and gene expression changes in the tumorigenic MCF-7 breast epithelial cell line. J Steroid Biochem Mol Biol. 2010;119:149–160. doi: 10.1016/j.jsbmb.2010.02.019. [DOI] [PubMed] [Google Scholar]
- 13.Lin HL, Liu TY, Chau GY, Lui WY, Chi CW. Comparison of 2-methoxyestradiol-induced, docetaxel-induced, and paclitaxel-induced apoptosis in hepatoma cells and its correlation with reactive oxygen species. Cancer. 2000;89:983–994. [PubMed] [Google Scholar]
- 14.Qadan LR, Perez-Stable CM, Anderson C, D’Ippolito G, Herron A, Howard GA, Roos BA BA. 2-Methoxyestradiol induces G2/M arrest and apoptosis in prostate cancer. Biochem Biophys Res Commun. 2001;285:1259–1266. doi: 10.1006/bbrc.2001.5320. [DOI] [PubMed] [Google Scholar]
- 15.Li L, Bu S, Bäckström T, Landström M, Ulmsten U, Fu X. Induction of apoptosis and G2/M arrest by 2-methoxyestradiol in human cervical cancer HeLaS3 cells. Anticancer Res. 2004;24:873–880. [PubMed] [Google Scholar]
- 16.Fukui M, Zhu BT. Mechanism of 2-methoxyestradiol-induced apoptosis and growth arrest in human breast cancer cells. Mol Carcinog. 2009;48:66–78. doi: 10.1002/mc.20458. [DOI] [PubMed] [Google Scholar]
- 17.Sweeney C, Liu G, Yiannoutsos C, Kolesar J, Horvath D, Staab MJ, Fife K, Armstrong V, Treston A, Sidor C, Wilding G. A phase II multicenter, randomized, double-blind, safety trial assessing the pharmacokinetics, pharmacodynamics, and efficacy of oral 2-methoxyestradiol capsules in hormone-refractory prostate cancer. Clin Cancer Res. 2005a;11:6625–6633. doi: 10.1158/1078-0432.CCR-05-0440. [DOI] [PubMed] [Google Scholar]
- 18.Sweeney C, Porter J, Selbe K, Treston A, Quon C, Sidor C. A single-center, safety and pharmacokinetic study of 2-methoxyestradiol Nanocrystal® Colloidal Dispersion (Panzem®NCD), administered orally to patients with advanced cancer. Clin Cancer Res. 2005b;11:B121. [Google Scholar]
- 19.Dahut WL, Lakhani NJ, Gulley JL, Arlen PM, Kohn EC, Kotz H, McNally D, Parr A, Nguyen D, Yang SX, Steinberg SM, Venitz J, Sparreboom A, Figg WD. Phase I clinical trial of oral 2-methoxyestradiol, an antiangiogenic and apoptotic agent, in patients with solid tumors. Cancer Biol Ther. 2006;5:22–27. doi: 10.4161/cbt.5.1.2349. [DOI] [PubMed] [Google Scholar]
- 20.Rajkumar SV, Richardson PG, Lacy MQ, Dispenzieri A, Greipp PR, Witzig TE, Schlossman R, Sidor CF, Anderson KC, Gertz MA. Novel therapy with 2-methoxyestradiol for the treatment of relapsed and plateau phase multiple myeloma. Clin Cancer Res. 2007;13:6162–6167. doi: 10.1158/1078-0432.CCR-07-0807. [DOI] [PubMed] [Google Scholar]
- 21.Tevaarwerk AJ, Holen KD, Alberti DB, Sidor C, Arnott J, Quon C, Wilding G, Liu G. Phase I trial of 2-methoxyestradiol NanoCrystal dispersion in advanced solid malignancies. Clin Cancer Res. 2009;15:1460–1465. doi: 10.1158/1078-0432.CCR-08-1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harrison MR, Hahn NM, Pili R, Oh WK, Hammers H, Sweeney C, Kim K, Perlman S, Arnott J, Sidor C, Wilding G, Liu G. A phase II study of 2-methoxyestradiol (2ME2) NanoCrystal(R) dispersion (NCD) in patients with taxane-refractory, metastatic castrate-resistant prostate cancer (CRPC) Invest New Drugs. 2010 doi: 10.1007/s10637-010-9455-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar AP, Garcia GE, Slaga TJ. 2-methoxyestradiol blocks cell-cycle progression at G2/M phase and inhibits growth of human prostate cancer cells. Mol Carcinog. 2001;31:111–24. doi: 10.1002/mc.1046. [DOI] [PubMed] [Google Scholar]
- 24.Ghosh R, Ott AM, Seetharam D, Slaga TJ, Kumar AP. Cell cycle block and apoptosis induction in a human melanoma cell line following treatment with 2-methoxyoestradiol: therapeutic implications? Melanoma Res. 2003;13:119–27. doi: 10.1097/00008390-200304000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Perez-Stable C. 2-Methoxyestradiol and paclitaxel have similar effects on the cell cycle and induction of apoptosis in prostate cancer cells. Cancer Lett. 2006;231:49–64. doi: 10.1016/j.canlet.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 26.D’Amato RJ, Lin CM, Flynn E, Folkman J, Hamel E. 2-Methoxyestradiol, an endogenous mammalian metabolite, inhibits tubulin polymerization by interacting at the colchicine site. Proc Natl Acad Sci USA. 1994;91:3964–3968. doi: 10.1073/pnas.91.9.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Attalla H, Makela TP, Adlercreutz H, Andersson LC. 2-Methoxyestradiol arrests cells in mitosis without depolymerizing tubulin. Biochem Biophys Res Commun. 1996;228:467–473. doi: 10.1006/bbrc.1996.1683. [DOI] [PubMed] [Google Scholar]
- 28.Jordan MA, Wilson L. Microtubules and actin filaments: Dynamic targets for cancer chemotherapy. Curr Opinion Cell Biol. 1998;10:123–130. doi: 10.1016/s0955-0674(98)80095-1. [DOI] [PubMed] [Google Scholar]
- 29.Mollinedo F, Gajate C. Microtubules, microtubule-interfering agents and apoptosis. Apoptosis. 2003;8:413–450. doi: 10.1023/a:1025513106330. [DOI] [PubMed] [Google Scholar]
- 30.Wendell KL, Wilson L, Jordan MA. Mitotic block in HeLa cells by vinblastine: ultrastructural changes in kinetochore-microtubule attachment and in centrosomes. J Cell Sci. 1993;104(Pt 2):261–274. doi: 10.1242/jcs.104.2.261. [DOI] [PubMed] [Google Scholar]
- 31.Vasquez RJ, Howell B, Yvon AM, Wadsworth P, Cassimeris L. Nanomolar concentrations of nocodazole alter microtubule dynamic instability in vivo and in vitro. Mol Biol Cell. 1997;8:973–985. doi: 10.1091/mbc.8.6.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang TH, Popp DM, Wang HS, Saitoh M, Mural JG, Henley DC, Ichijo H, Wimalasena J. Microtubule dysfunction induced by paclitaxel initiates apoptosis through both c-Jun N-terminal kinase (JNK)-dependent and -independent pathways in ovarian cancer cells. J Biol Chem. 1999;274:8208–8216. doi: 10.1074/jbc.274.12.8208. [DOI] [PubMed] [Google Scholar]
- 33.Morgan DO. Principles of CDK regulation. Nature. 1995;374:131–134. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- 34.Zoubine MN, Weston AP, Johnson DC, Campbell DR, Banerjee SK. 2-methoxyestradiol-induced growth suppression and lethality in estrogen-responsive MCF-7 cells may be mediated by down regulation of p34cdc2 and cyclin B1 expression. Int J Oncol. 1999;15:639–646. doi: 10.3892/ijo.15.4.639. [DOI] [PubMed] [Google Scholar]
- 35.Du B, Zhao Z, Sun H, Ma S, Jin J, Zhang Z. Effects of 2-methoxyestradiol on proliferation, apoptosis and gene expression of cyclin B1 and c-Myc in esophageal carcinoma EC9706 cells. Cell Biochem Funct. 2012;30:158–165. doi: 10.1002/cbf.1830. [DOI] [PubMed] [Google Scholar]
- 36.Joubert A, Marais S. In vitro effects of 2-methoxyestradiol on cell morphology and Cdc2 kinase activity in SNO oesophageal carcinoma cells. Cell Biochem Funct. 2007;25:357–362. doi: 10.1002/cbf.1409. [DOI] [PubMed] [Google Scholar]
- 37.Agarwal N, Tochigi Y, Adhikari AS, Cui S, Cui Y, Iwakuma T. MTBP plays a crucial role in mitotic progression and chromosome segregation. Cell Death Differ. 2011;18:1208–1219. doi: 10.1038/cdd.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elledge SJ, Harper JW. Cdk inhibitors: on the threshold of checkpoints and development. Curr Opin Cell Biol. 1994;6:847–852. doi: 10.1016/0955-0674(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 39.Xiong Y, Zhang H, Beach D. Subunit rearrangement of the cyclin-dependent kinases is associated with cellular transformation. Genes Dev. 1993;7:1572–1583. doi: 10.1101/gad.7.8.1572. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Benezra R. Identification of a human mitotic checkpoint gene: hsMAD2. Science. 274:246–248. doi: 10.1126/science.274.5285.246. [DOI] [PubMed] [Google Scholar]
- 41.Maurer M, Komina O, Wesierska-Gadek J. Roscovitine differentially affects asynchronously growing and synchronized human MCF-7 breast cancer cells. Ann N Y Acad Sci. 2009;1171:250–256. doi: 10.1111/j.1749-6632.2009.04717.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.