

Abstract
The tetracyclic indolo[3,2-b]quinoline ring system constitutes an important structural moiety in natural products exhibiting numerous biological activities. In particular, indolo [3, 2-b]quinoline, commonly known as linear quindo-line is of particular interest, because of its rigid structure and scope of derivatization. Although the core linear quindoline skeleton shows little or no activity in several biological systems, introduction of a methyl group on the N-5 atom leading to cryptolepine induces remarkable activity against a broad spectrum of biological targets. A number of analogs of quindoline and cryptolepine have been synthesized, incorporating various functional groups on the core quindoline skeleton leading to improved biological activities. In this review, we describe various synthetic methodologies leading to the quindoline scaffold, the biological activities and the structure activity relationships (SAR) of quindoline derivatives toward different disease states to give a better picture of the importance of this moiety in medicinal chemistry.
Keywords: Cryptolepine, indoloquinolines, synthesis, structure-activity relationship (SAR) studies
INTRODUCTION
Cryptolepine or 5-methyl-5H-indolo[3, 2-b] quinoline (1) is an example of a natural product whose synthesis was reported prior to its isolation from plants. The first synthesis was reported in 1906 by Fichter and coworkers [1] before its isolation from Cryptolepis triangularis N. E. Br by Clinquart in 1929 [2] and again by Delvaux [3] soon thereafter. Cryptolepine has also been isolated as the major alkaloid from Sida sp. (Malvaceae) medicinal plants used by indigenous practitioners of Sri Lanka [4].
However, cryptolepine was first isolated from the shrub Cryptolepis sanguinolenta by Gellert from root samples obtained in Nigeria [5] and again by Dwuma-Badu and co-workers from root samples obtained from Ghana [6]. In traditional folk medicine of West and Central Africa, a decoction of the root of the plant C. sanguinolenta is used to treat fevers (including fever caused by malaria), upper respiratory infections and venereal diseases. Apart from cryptolepine, neocryptolepine {cryptotackieine, 5-methyl-5H-indolo[2, 3-b]quinoline} (2), isocryptolepine {cryptosanguinolentine, 5-methyl-5H-indolo[3, 2-c]quinoline} (3), quindoline (4) and several other isomeric indoloquinolines complete the 13 characterized alkaloids isolated from this plant Fig. (1).
Fig. 1.
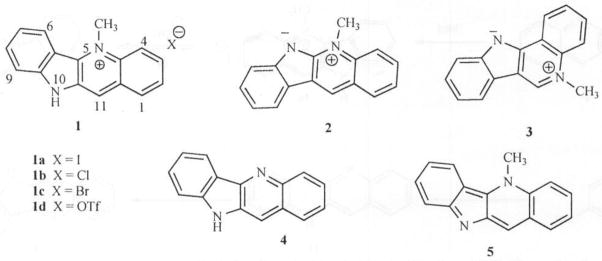
Indoloquinolines.
Under acidic conditions, these tetracyclic compounds exist in their salt forms possessing a broad spectrum of biological activities. However, under basic conditions, the N-5 atom in these alkaloids assumes sp3 hybridized form and the alkaloids exist as free bases as represented (5). Thus, a quaternary nitrogen is able to undergo a reversible acid – base transformation from yellow to pink respectively Fig. (2).
Fig. 2.
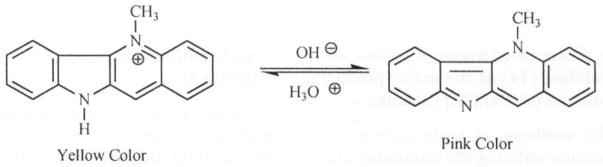
Reversible transformation of Cryptolepine under acidic and basic conditions.
Biological testing of Cryptolepine has led to its identification as having a broad spectrum of activities including antibacterial, [7] antimalarial, [8] antifungal, [9] antithrom-botic, [10] vasodilation, [11] antihyperglycemic, [12] anticancer and cytotoxic, [13] anti-inflammatory, [14] hypotensive and antipyretic, [15] presynaptic α-adrenoceptor blocking action, [16] and anti-muscarinic [17] properties. The mechanism by which cryptolepine produces these biological activities is not completely known. However, several lines of evidence suggest that DNA intercalation and stabilization of topoisomerase II-DNA complex may play some role [18]. As a result of these interesting biological activities, many groups world-wide have designed new methodologies to synthesize derivatives of quindoline (4) and have attempted to study the structure activity relationship of these indoloquinolines towards various disease states. Thus, the purpose of this review was to assemble the various synthetic strategies leading to the construction of the indoloquinoline structure as well as the biological activities and structure-activity relationships of the indoloquinoline alkaloids to date.
METHODS OF CHEMICAL SYNTHESES OF INDOLO[3,2-B]QUINOLINE (QUINDOLINE)
Several routes are now available for the synthesis of cryptolepine analogues. The most versatile method reported by Holt and Petrow [19] was based on the synthesis of linear quindoline (4) in which isatin (6) is condensed with 3-acetylindoxyl (7) under basic and oxygen-free conditions and the resulting quindoline-11-carboxylic acid (8) is decarboxylated by heating in diphenylether or mineral oil to yield 4. (Scheme 1).
Scheme 1.
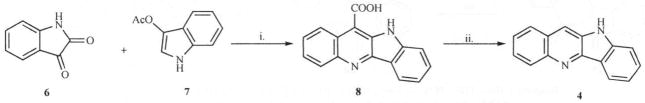
Reagents. i. KOH, N2. 10 days; ii. Mineral Oil/Ph2O, 250–300 °C, 2 hrs.
Better yields are obtained by using indoxyl-1, 3-diacetate (9) Fig. (3) as starting material instead of 7. However, strongly electron withdrawing substituents on isatin such as a nitro group inhibit the first step in the synthesis.
Fig. 3.
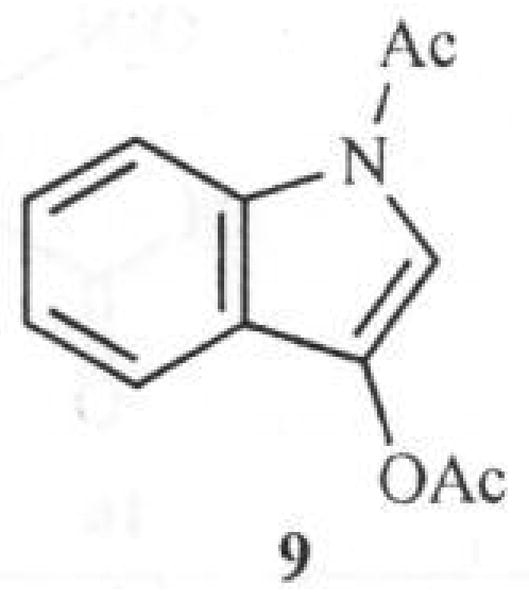
Structure of Indoxyl-1, 3-diacetate.
Quindoline can be methylated at the N-5 position by methyl iodide using the method described by Fichter [1] to yield cryptolepine as the hydroiodide salt (Scheme 2). An other method, developed by Bierer and coworkers [20] for large scale reactions involves the use of methyl triflate as the alkylating agent to obtain hydrotriflate salts of cryptolepine. Conversion from the hydrotriflate or hydroiodide salts to other salt forms can be achieved by reaction with a base and treatment of the resulting free base with ethereal HCl solution for example, to provide cryptolepine hydrochloride salt. Hence, the quaternary N-5 atom does not behave in the normal way as a permanent positively charged atom. These acid-base changes are also associated with color changes. While cryptolepine free base (5) is deep purple, the salt form (1a) is yellow or orange depending on the solvent and pH of the solution (Scheme 2).
Scheme 2.
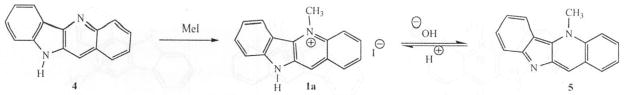
A major disadvantage of the method developed by Holt and others is the long reaction times (≥ 10 days) during the first step of the reaction, i.e., preparation of the quindoline-11-carboxylic acid (8) (Scheme 1). To reduce the reaction times and generate quindoline rapidly, we developed a new and more efficient synthesis of quindoline [21] which requires only two steps and was completed in one day with an overall yield of 22 %. The synthesis involved N-arylation of 3-aminoquinoline (10) with triphenyl-bismuth diacetate in the presence of metallic copper to yield 3-anilinoquinoline (11) in excellent yield. A similar procedure using triphenyl-bismuth in the presence of copper diacetate also produced 11 but with low yields. Cyclization of 11 was effected by palladium (II) acetate in refluxing trifluoroacetic acid to afford 4 (Scheme 3). The cyclization reaction was however low yielding because of the formation of angular quindoline in addition to linear quindoline. The ratio of the regioisomers however, depends on the substituents on the rings (Personal observations). Separation of the regioisomers is easily achieved by flash chromatography.
Scheme 3.
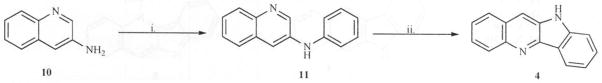
Reagents: i. Ph3Bi(OAc)2, Cu, CH2Cl2, 10 hrs. ii. Pd(OAc)2/CF3COOH, 90 °C, 40 min.
More recently, a similar method was reported albeit in low yields, wherein aniline was condensed with 3-bromo-quinoline (12) resulting in intermediate (13) which on oxidative photocyclization in the presence of traces of iodine, afforded angularly-fused quindoline (14) as the major product (51 %) and 4 as the minor product (16 %) [22] (Scheme 4).
Scheme 4.
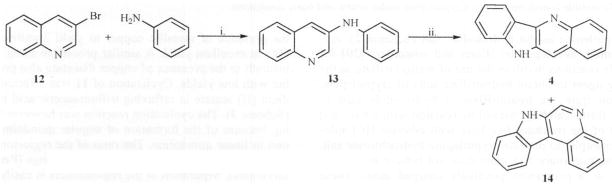
Reagents: i. 200 °C. ii. hv, C6H6: CH3OH:H2SO4 (60:30:1, v/v/v), l2
Cooper [23] reported the synthesis of hydroxycryptole-pine, cryptolepine and quindoline utilizing the intramolecular β-nucleophilic substitution of 1-phenylsulfonyl-2-acylindole (Scheme 5).
Scheme 5.
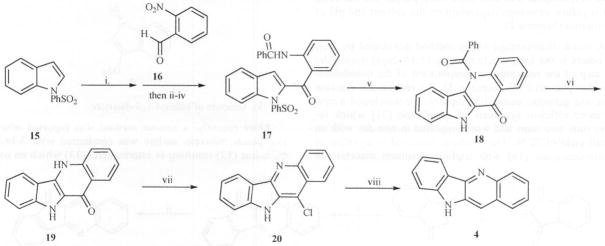
Reagents: i. BuLi, THF, −78 °C; ii. MnO2, CH2Cl2, RT; iii. H2, Pd-C; iv. PhCO.Cl, PhNMe2, RT; v. NaH, THF, Reflux; vi. NaOH, MeOH, heat; vii. POCl3, Reflux; viii. H2, Pd-C, EtOH.
In this process, 1-phenylsulfonylindole (15) was lithiated at the 2 position, reacted with 2-nitrobenzaldehyde (16) and the resulting alcohol was oxidized using MnO2. Subsequent catalytic reduction of the nitro group and N-benzoylation gave amido-ketone (17). N-Deprotonation using NaH allowed ring closure to 18 and hydrolysis produced 19. Heating 19 in POCl3 produced 20 and catalytic hydrogenolysis gave quindoline (Scheme 5).
Recently, Ho [24] reported a synthetic procedure leading to the indolo[3,2-b]quinoline system and the isomeric indolo[2,3-b]quinoline scaffold (21) Fig. (4), based on consideration of their accessibility from a common symmetrical intermediate, 1, 3-bis(1-nitrophenyl)propan-2-one (22) which was readily obtained from (2-nitrophenyl)acetic acid (23).
Fig. 4.
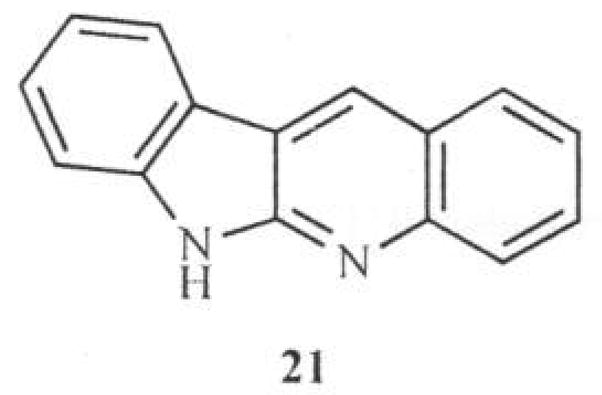
Structure of Indolo[2. 3-b]quinoline.
Reduction of compound 22 with sodium dithionite in aqueous methanol led to the diamino ketone which underwent spontaneous cyclization to produce the first cyclized product 24. Transformation of 24 to 4 was achieved with iodosobenzene diacetate as the oxidizing agent (Scheme 6).
Scheme 6.
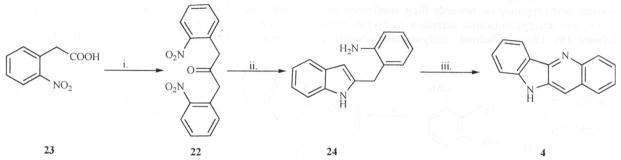
Reagents: i. DCC, THF, DMAP, Δ; ii. sodium dithionate, MeOH D; iii. Phl(OAc)2, THF, RT.
In another synthetic procedure reported by Radl and co-workers [25], 2-nitro phenacyl bromide (25) was reacted with ethyl-2-cyanophenylcarbamate (26) under basic conditions to yield compound 27. Using the nitro group as a leaving group, cyclization of 27 under basic conditions yielded quindolinone (19) which can be converted to quindoline, as shown in Scheme 7.
Scheme 7.
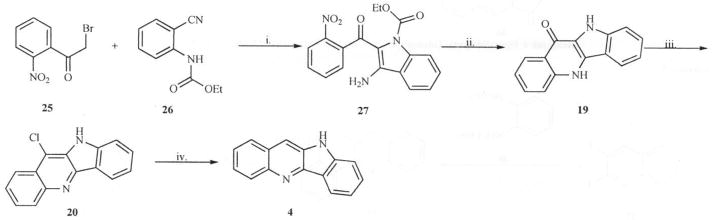
Reagents: i. K2CO3/NaH, DMF. RT; ii. NaH, THF, RT; iii. PCI5, Δ; iv. EtOH.HCl, 10 % Pd-C, MeOH
More recently, Dutta and coworkers [26] reported a simple method for the synthesis of various 2-substituted quindolines starting from 2-nitroacetophenone (28) as shown in Scheme 8. The important steps involved in the synthesis are regioselective thermal cyclization of enaminoimine hydrochloride and generation of a nitrene intermediate followed by its insertion into a sp2 C-H bond. The first step in this process involves treatment of 2-nitroacetophenone (28) with POCl3 in DMF and a Vilsmeier-Haack reaction to produce β-chlorocinnamaldehyde, (29) with the nitro group remaining intact. At 0 °C, chloroaldehyde (29) reacts with aniline in ethanolic HCl to produce the corresponding enaminoimine hydrochloride (30) in a very good yield. Thermal cyclization of 30 at 200–250 °C, produced 2-(2-nitrophenyl)quinoline (31) as the major isolable product. The desired quindoline was synthesized by heating 31 with triethyl phosphate at 160 °C. This step involves intramolecular annulation through a nitrene intermediate.
Scheme 8.
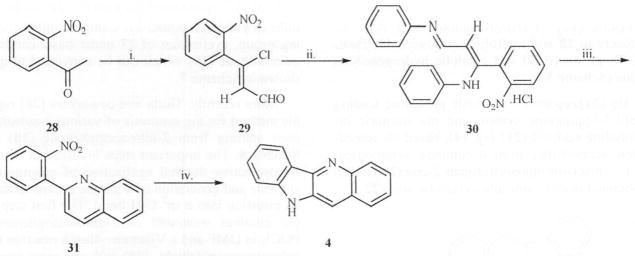
Reagents: i. POCl3, DMF, 0 °C, 1 h then 80 °C, 4h; ii. 2 N ethanolic HCl, arylamine, 0 °C; iii. heat, 200–250 °C, 5 min; iv. P(OEt)3, Δ. 4 h;
Bierer [27] synthesized a methoxy analog of the indoloquinoline by condensation of 1-acetyl-3-oxoindole (32) with 2-nitro-3-methoxybenzaldehyde(33)using a catalytic amount of piperidine to give compound 34 as a mixture of E/Z isomers. Hydrogenation of 34 gave quindoline, 35 in 78–85% yields after chromatography. Hydrolytic cleavage of the acetate group in 35 using KOH, yielded the substituted quindoline (36) in good yield (Scheme 9). Unsubstituted quindoline can also be prepared in principle using the same procedure.
Scheme 9.
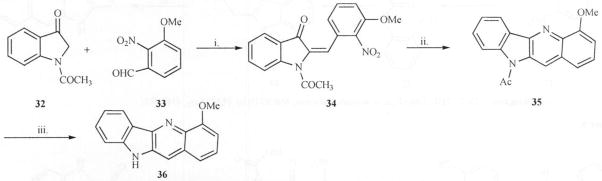
Reagents: i. Piperidine (cat), Toluene/CHCl3, 4 A°, MS. ii. H2, Pd/C, MeOH, iii. KOH
Arzel and co-workers [28] reported a short synthetic route leading to quindoline involving the first reported halogen-dance reaction in the indoloquinoline series. Later, they extended that methodology to synthesize various derivatives of quindoline and cryptolepine towards their studies on antiplasmodial and antitrypanosomal activities of cryptolepine [29] (Scheme 10). Thus, palladium-catalysed cross-coupling reaction (Suzuki coupling) between iodoquinoline (37) and boronic acid (38) afforded the biaryl compound (39) in high yields. Cyclization to quindoline was achieved by heating 39 in boiling pyridinium chloride and hydrolyzing the resulting N-protected quindoline.
Scheme 10.
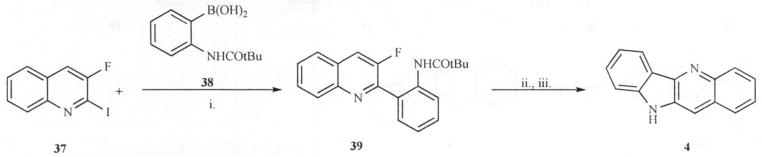
Reagents: i. Pd(PPh3)4/EtOH/toluene, ii. Pyridinium chloride, iii. NH4OH
A widely used method utilizing anthranilic acid (40) as starting material was reported by Bierer in 1998 [27]. The approach involved treating anthranilic acid with bromoacetyl bromide to afford the condensation product (41) which was then reacted with aniline to give compound 42. Acid promoted cyclization of 42 with polyphosphoric acid led to compound 19 which on treatment with phosphorous oxychloride gave 11-chloro quindoline (20). Hydrogenation of the same on Pd/C led to quindoline in good yields (Scheme 11).
Scheme 11.
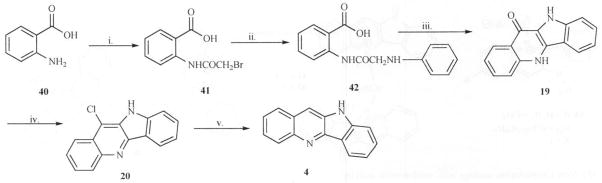
Reagents: i. Bromoacetyl bromide, DMF/dioxane, ii. Anilline, iii. PPA, iv. POCl3, v. H2, Pd/C, MeOH
Using this approach to synthesize halogenated derivatives however, can be problematic. For example, when the 11-chloro quindoline (20) has additional halogen on any other position in the ring, hydrogenation can lead to the stripping of all the halogens on the ring system [27, 30–33]. Thus, although this procedure is widespread and useful for synthesizing substituted quindolines, it is not as useful as some of the other methods when used for halogenated analogs.
BIOLOGICAL ACTIVITIES OF INDOLOQUINOLINES
As noted in the introduction, cryptolepine is by far the most popular and most researched indoloquinoline. In the following sections, several of the biological activities associated with the indoloquinoline scaffold are reviewed.
A. Antibacterial Activity
Bacterial infections such as those associated with Mycobacterium tuberculosis and methicillin- and vancomycin-resistant Staphylococcus aureus (MRSA & VRSA) are of major concern especially for immuno-compromised patients. Given the vast numbers of bacteria, their short generation times and typical gene mutation frequencies of 1 in 107 bacteria, resistance is inevitable. The mechanism of resistance development in these bacteria may include any one or combination of a) destruction of antibiotic by bacterial enzymes, b) bacterial reprogramming of the antibiotic to lowered susceptibility and c) pumping out antibiotics via transmembrane efflux pumps and keeping antibiotic concentration within the bacterial cell below toxic threshold concentrations [34]. To solve the problem of resistance development associated with the treatment of bacterial infections there are three broad strategies. The first and foremost is to develop new antimicrobial agents with a broad spectrum of activity including the production of a super drug that has more than one target in the offending pathogen. Secondly, the search for novel screening approaches and therapeutic agents that have different mechanisms of action from the antibacterials currently available. Thirdly, containment of resistance by avoiding its emergence and preventing its transmission [35]. Certainly, there is a continuing need for a pipeline of new agents to combat multi-drug resistant (MDR) and new emerging opportunistic bacteria.
Bioactive plant extracts or constituents are rich sources for development of chemotherapeutic agents in the management and treatment of opportunistic and resistant antibacterial infections. Many plant-based antimicrobials have produced remarkable antibacterial activity [36] and plant sources of indoloquinoline alkaloids are notable in this regard. In 1979, Boakye-Yiadom and others [37] reported antibacterial activity of cryptolepine. It was noted that this activity was both bacteriostatic and bactericidal against Staphylococcus aureus (S. aureus). They further speculated that cryptolepine may act in this manner, by causing lysis to S. aureus. It was also reported that cryptolepine was more active against Gram-positive bacteria (MIC < 100 μg/mL) than Gram-negative ones [38] and its killing effect on S. aureus was subsequently studied [39]. When cryptolepine was exposed to the bacteria, lysis was observed and the surface morphological appearance of the cells was also altered. A similar effect on morphology and survival of other bacteria and fungi viz., Escherichia coli, Candida albicans and Saccharomyces Cerevisiae was observed [40, 41]. Both the hydrochloride salt of cryptolepine and its free base form were assessed against M. fortuitum (an alternative screening model to M. tuberculosis) and the salt form was found to be ten-fold less active than the free base [38b]. Recent screening of cryptolepine hydrochloride salt against a panel of six species of fast growing mycobacteria showed a significant activity against M. aurum and M. Phlei (MIC = 2 and 4 μg/mL) as compared to the standard antibiotics Ethambutol (1 μg/mL) and Isoniazid (1 μg/mL). Activity against M. fortuitum was shown to be moderate (MIC = 16 μg/mL) as compared to Ethambutol (8 μg/mL) and Isoniazid (0.5 μg/mL) [42]. Another recent report indicates that novel analogs of cryptolepine (43-46, Fig. (5)) displayed rather broad antimicrobial spectra [43]. Compounds 43, 45 and 46 showed more expansive antimicrobial/antiparasitic spectra than cryptolepine. In particular, activity against methicillin-resistant Staphylococcus aureus (MRSA) was significant. The IC50 values were reported to be 2.0, 3.0 and 6.0 μg/mL respectively for compounds 43, 45 and 46 against the standard IC50 value of 0.15 μg/mL for ciprofloxacin. All four compounds showed significant potency against Mycobacterium intracellulare. However, only 45 showed significant potency against Pseudomonas aeruginosa (IC50 = 1.5 μg/mL) as compared to an IC50 value of 0.06 μg/mL for ciprofloxacin [43].
Fig. 5.
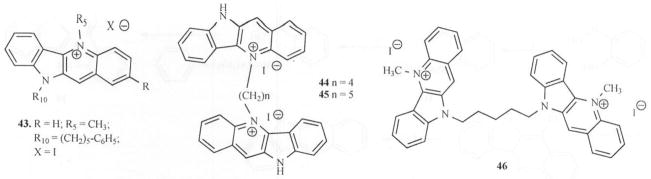
New Cryptolepine analogs with antibacterial activity.
B. Antiprotozoal Activity
Malaria continues to be a major cause of morbidity and mortality in developing nations. A primary factor for this is related to the increasing prevalence of P. falciparum resistant to chloroquine and other antimalarial agents. In one study, it was found that an 11-fold increase in mortality in children under the age of 4 years occurred within 6 years of the emergence of chloroquine-resistance [44]. Another insect borne disease, African sleeping sickness, also known as trypanosomiasis, is an infection endemic to Sub-Saharan Africa. Although drugs like Pentamidine and Suramin are used in initial stages of treatment and Melarsoprol was used in the second phase of the disease, no agent has been found to treat the later phases of the disease. These problems have spurred a considerable interest in identifying antiprotozoal agents from plant sources which can be delivered orally. Thus, many plants, especially those reputed as traditional medicines have been evaluated [45].
Wright [8a] has recently published a review article that discussed the potential of cryptolepine analogs as antimalarial drugs. Cryptolepine and its hydrochloride salt (1b) were found to have potent in vitro activity against various P. falciparum strains [8c, 8e, 8f, 46, 47]. Cimanga and coworkers [8f] studied the antiplasmodial activity of cryptolepine, its hydrochloride, 11-hydroxycryptolepine (47) and quindoline, and found that both cryptolepine and its hydrochloride salt showed strong in vitro antiplasmodial activity against P. falciparum chloroquine-resistant strains, K-1 and W-2. However, quindoline was much less active. In vivo tests in infected mice showed cryptolepine hydrochloride as having significant activity against P. berghei yoelii and P. berghei berghei. 11-Hydroxycryptolepine and quindoline were devoid of activity in these assays.
The influence of the structural modifications at N-5 and C-11 positions of the indoloquinoline moiety against T. cruzi Tulahuen strain and chloroquine-resistant K-1 strain of P. falciparum were studied [29]. A methyl substituent at the N-5 position in the ring seems to be essential for activity since quindoline was about 100 times less active against both parasites than cryptolepine. The quindoline dimer 48 showed better activity than quindoline but its index of selectivity (IS) remained in a similar range. Introduction of alkyl groups at the C-11 position alone on the quindoline ring had no significant influence on activity, except that 11-ethyl quindoline (49) was about 10 times more active than quindoline against T. cruzi trypomastigote forms. With the 5-methyl substituent intact, alkyl groups at the C-11 position reduced activity against P. falciparum, and intracellular multiplication of T. cruzi amastigote forms. However, 11-methyl cryptolepine (50) was about 5 times more active against P. falciparum and slightly more active against T. cruzi amastigote forms than the triflate salt of cryptolepine Fig. (6).
Fig. 6.
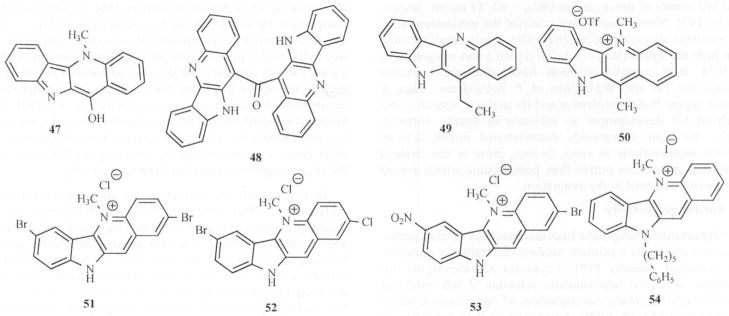
Modified Indolo[3, 2-b]quinolines as Anti-Malarial and Anti-Trypanosomal agents.
Another study involving the syntheses of analogs of cryptolepine and their evaluation showed improved antiplasmodial activity as compared to the parent molecule [46]. In vitro studies of 2- and 7-bromocryptolepine proved to be 2-fold more potent than cryptolepine. The most potent compound revealed was 2, 7-dibromocryptolepine (51) with 10-fold more activity than cryptolepine (as sulfate) (IC50 = 0.049 and 0.44 μM respectively against K-1 and 0.026 and 0.27 μM against HB3). It was also shown in in vivo studies that 51 suppressed parasitemia (P. berghei) by 89 % as compared to untreated infected controls at a dose of 12.5 mg kg−1.
A more elaborate structure-bioactivity study was done on 16 cryptolepine analogs in which six of the analogs demonstrated potent in vitro activities against P. falciparum with IC50 values <0.1 μM, and cytotoxicities 2–4 times greater than that of cryptolepine (Table 1) [48]. Among the compounds evaluated, two novel compounds (52 and 53) were shown to have encouraging antimalarial activities in vivo with suppression of parasitemias (P. berghei) >90 % at doses of 25 mg kg−1 and increased survival of the mice without any apparent toxicity. According to these authors, the antimalarial mode of action in these compounds appears to be similar to that of chloroquine and may involve inhibition of hemozoin formation. It is also possible that the effect of these agents on DNA synthesis, cleavage and inhibition of topoisomerase II may contribute to their action [13d]. A recent study using fluorescence microscopy, suggests that cryptolepine accumulates in parasite structures that may correspond to the parasite nucleus [29].
Table 1.
In Vitro Antiplasmodial Activities (P. falciparum) [48]
| Compound | IC50 μM | Cytotoxic/Antiplasmodial Ratio |
|---|---|---|
| Cryptolepine | 0.440 | 8.98 |
| 1, 2-Dichlorocryptolepine | 0.0880 | 21.7 |
| 2-Chlorocryptolepine | 0.166 | 13.5 |
| 2, 3-Dichlorocryptolepinc | 0.356 | 9.33 |
| 3-Chlorocryptolepine | 0.448 | 3.90 |
| 4-Chlorocryptolepine | 4.69 | 0.76 |
| 8-Chlorocryptolepine | >1 | <8.15 |
| 2-Fluorocryptolepine | 1.21 | 3.96 |
| 2-Methylcryptolepine | 0.419 | 9.81 |
| 2-Methoxycryptolepine | 0.950 | 4.54 |
| 3-Methylcryptolepine | 0.149 | 23.6 |
| 7-Bromo-2-chlorocryptolepine | 0.030 | 57.7 |
| 7-Bromo-2-fluoorocryptolepine | 0.063 | 21.4 |
| 7-Bromo-3-chlorocryptolepine | 0.037 | 30.8 |
| 7-Bromo-3-methylcryptolepine | 0.26 | 20.0 |
| 2, 8-Dichlorocryptolepine | 0.045 | 24.9 |
| 2-Bromo-7-nitrocryptolepine | 0.07 | 14.7 |
| 2, 7-Dibromocryptolepine | 0.049 | 123 |
| Chloroquinc diphosphate | 0.246 | - |
As part of our on-going studies of cryptolepine analogs, we tested several compounds against P. falciparum. One compound (54) showed significant potency against both D6 and W2 clones of the organism (IC50 = 62, 28 ng/mL respectively) [43]. More recently, we reported the evaluation of N-10 isosteres of cryptolepine [9a]. The results demonstrated that both the sulfur (IC50 = 1.6, 2.0 μg/mL) and oxygen (IC50 = 0.28, 0.28 μg/mL) isosteres have moderate activities against the D6 and W2 clones of P. falciparum. Thus, it would appear that cryptolepine and its analogs possess a new scaffold for development as antimalarial agents. Interestingly, the same compounds demonstrated potent activity against leishmaniasis in vitro. In fact, most of the analogs evaluated were more potent than pentamidine which served as a positive control in the evaluation.
C. Antifungal Activity
Opportunistic fungi have become important human pathogens and constitute a primary mode of mortality in immuno-compromised patients [49]. Cryptococcus meningitis for example, is a fatal opportunistic infection if left untreated and is one of the many consequences of opportunistic infections associated with AIDS. Amphotericin B in combination with Flucytosine is the preferred treatment for this disease. However, Amphotericin B lacks oral efficacy and induces several side effects including nephrotoxicity, leukopenia, thrombocytopenia and febrile reactions. Agents from the azole class like ketoconazole and fluconazole were introduced as possible replacements for amphotericin B, but fungal resistance development has limited their broad utility and has inspired the development of new effective drug entities [49].
Cryptolepine re-isolated from C. sanguinolenta, has shown potential antifungal and antimicrobial activities [9]. The reported advantages of cryptolepine over amphotericin B are its relatively low toxicity (LD50 = 146 vs 2.3 mg/Kg weight of mice) and better water solubility (1.2 vs 0.1 mg/mL), making it an attractive lead compound for antifungal drug development. In addition, cryptolepine shows a higher activity against C. neoformans than amphotericin B when tested in agar well diffusion assays [9b]. Several reports have now demonstrated that cryptolepine and several of its analogs have broad spectra of activities against opportunistic fungal and bacterial infections associated with AIDS [37, 38b]. A systematic study was also conducted on the quindoline skeleton to aid the formulation of SAR and the identification of potent agents with a broad spectrum of activity against various pathogens.
Alkylation of the N-5 or/and N-10 atoms in quindoline were contemplated to understand if buckling is a necessary requirement for activity since the flat quindoline without alkylation at N5 is devoid of activity [50]. It was found that compounds 1a and 55 showed good activities (MIC = 12.5 and 6.25 μg/mL) but compound 56 was only marginally active (MIC >62.5 μg/mL) compared to Amphotericin B (0.39 μg/mL) which suggests that the ionized form of cryptolepine may be the active form and that buckling is not a necessary requirement for anticryptococcal activity Fig. (7) [50]. Following the establishment that N-5 methylation of quindoline is a prerequisite for antifungal activity, homologation of the alkyl chain was carried out by Ablordeppey [50] to obtain the chain length necessary for optimum activity.
Fig. 7.
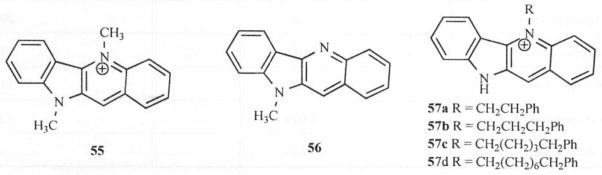
Structures of compounds 55-57.
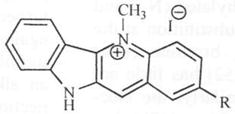
In this regard, the methyl group on compound 1a was replaced with ethyl, propyl, pentyl and octyl groups and the resulting compounds were evaluated against C. neoformans and C. albicans. There was little change in activity against both organisms through this variation. The effect of electron density on the N-5 atom was also explored. This was achieved by substituting one of the hydrogen atoms on the N-5 methyl group with electron donating or withdrawing groups to yield N-5 substituents such as CH2CN, CH2OCH3, CH2Ph and CH2COPh. Again, it was found that these changes resulted in little or no change in activity. A surprising observation in the activity profile of these compounds was the absence of activity associated with the N-benzyl-substituted analog. This led to the hypothesis that lack of activity may be due to a steric requirement for substituents accommodated close to the N-5 atom. An alternative hypothesis was that high electron density in the N-5 region due to the phenyl ring may be responsible for the lack of activity. Replacement of the phenyl ring with cyclohexyl and cy-clopentyl rings restored activity somewhat (MIC = 62.5 μmg/mL) but further reduction of the ring size (cyclobutyl and cyclopropyl) led to only minimal increase in activity. This suggests that while a flat aromatic ring is disfavored around the N-5 region of quindoline, smaller and flexible ring substituents appear to be tolerated. Extending the phenyl ring away from the N-5 atom (57a-d) restored activity and in fact led to significant activity increase above that of cryptolepine Fig. (7). The optimum chain length was found to be that associated with an ω-phenylpentyl moiety. Subsequently, it was found that substituting a cyclohexyl ring for the phenyl ring led to a similar observation. Thus, compound 57c was found to be about eight times more potent than cryptolepine (MIC = 1.9 μg/mL) and constitutes the most active anticryptococcal agent in the series. Extending the chain length beyond five carbons led to a reduction in activity. Compound 57d with the octyl carbon chain (MIC = 10 μg/mL) is as active as cryptolepine, but five times less active than 57c. Modifying the electron density around the N-5 atom through substitution on other positions on the quindoline moiety was hypothesized as a means of modulating electron density without steric interference [51]. Position 2 was selected for evaluation because it is para to the N-5 atom and can influence the electron density around the N-5 atom through resonance effect. To this end, various electron-donating and electron-withdrawing substituents were introduced at position 2 resulting in activities against both C. neoformans and C. albicans, two important opportunistic pathogens associated with AIDS. The active analogs of cryptole-pine with substitutions at position 2 are listed in Table 2.
Table 2.
Effect of Substitutions at Position 2 on the In Vitro Antifungal Activity [51]
| |||
|---|---|---|---|
| Structure R | Cn MIC (μg/ml) | Ca MIC (μg/ml) | Af MIC(μg/ml) |
| H | 12.5–15.6 | 250 | NT |
| F | 7.8 | 15.6 | NT |
| Cl | 2.0 | 5.5 | 0.2 |
| Br | <1.9 | 3.9 | NT |
| I | 7.8 | 3.9 | NT |
| CN | 20 | 20 | NT |
| NO2 | 0.3 | 20 | NT |
| CH3 | 31.2 | 15.6 | NT |
| OCH3 | <1.9 | <1.9 | NT |
| SCH3 | 3.12 | 1.56 | 7.8 |
| OH | 15.6 | 3.9 | NT |
| PhS | 6.25 | 50 | NT |
| Amphotericin B | 0.39 | 0.39 | NT |
MIC = Minimum Inhibitory Concentration. Cn = C. neoformans; Ca = C. albicans
Af = A. fumigatus; NT = Not tested.
However, the activity profile appears to be complex as both types of substituents enhance activity. For example, it is observed that while strong electron withdrawing groups like CN and NO2 enhanced activity, substitution with sulfoxides and sulfones led to reduction in activity. Hydrophobicity did not seem to play a significant role on the SAR of these compounds. These observations suggest the possibility that multiple mechanisms of fungal inhibition may be involved.
Subsequent attempts to combine the ω-cyclohexylpentyl or ω-phenylpentyl group on the N-5 atom with electron-withdrawing/donating groups at position 2 and unsubstituted/methyl groups on the N-10 atom [51] resulted in a mixed effect on activity. The brominated analog (58) retained activity against both C. albicans and C. neoformans (MIC = 1.9 μg/ml for both) compared to Amphotericin B (0.39 μg/ml). Substitution of the –NO2 group at the 7 and 11 positions (59, 60) showed similar high potency (MIC for C. neoformans 0.25 and for Aspergillus fumigatus = 0.39 μg/ml) compared to 0.39 μg/ml for Amphotericin B Fig. (8). A tetracyclic hybrid (61) was synthesized integrating the pharmacophore of another antifungal natural product onychine (62) [52] and cryptolepine. This compound however. showed no activity against C. albicans and C. neoformans. Similarly, its N-5 methylated analog (63) was also inactive against C. neoformans and only marginally active against C albicans (MIC = 62.5 μg/mL). The structure of the tri-cyclic onychine (9) inspired the synthesis of δ-carbolines 64a and 64b. Introduction of an ω-phenyl or ω-cyclohexyl alkyl moieties at the N-1 position of δ-carboline was evaluated. Here too the ω-cyclohexylpentyl substituted analog (64a) exhibited a broad spetrum of activity (MIC in μg/ml were 6.25 against C. albicans, 0.39 against C. neoformans, 12.5 against A. fumigatus, 12.5 against A. flavus and 6.25 against M. intracellulare) Fig. (9).
Fig. 8.
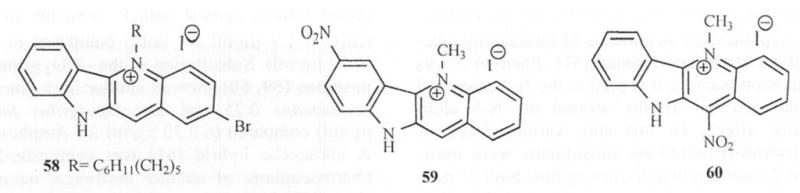
Structures of compounds 58-60.
Fig. 9.
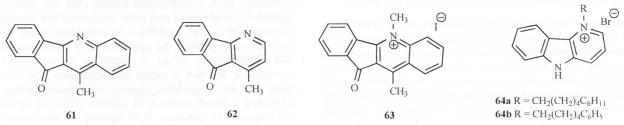
Structures of compounds 61-64.
Thus, the antifungal profile of 64a is somewhat comparable to that of amphotericin B with values at 0.2, 0.2, 0.39 and 0.39 μg/ml against C. albicans, C. neoformans, A. fumigatus and A. flavus respectively. Similar evaluation of ω-phenylpentyl-substituted analog (64b) showed a more subdued activity against C. albicans, C. neoformans and A. fumigatus (MIC = 31.2, 0.98 and 125 μg/ml respectively) [52]. Further evaluation using various combinations of arylalkyl substituents on N-5 and N-10 atoms of the indoloquinoline ring was not very productive [43].
Synthesis and evaluation of doubly methylated (N-5 and N-10) analogs (55, 65a, 65b) showed that substitution at the 2-position with the electron withdrawing bromine (65a) while enhancing anticryptococcal activity [52] has little action on anticandida acitivity Fig. (10). Similarly, the electron-donating methoxy group at the 2-position (65b) has little or no effect on either activity when compared to 55. It was also reported that alkylation of the N-10 atom with ω-cyclohexylpentyl moiety without alkylation on N-5 resulted in no activity against C. neoformans and C. albicans [43]. Since simultaneous alkylation of both N-5 and N-10 appears to be useful, bis-alkylated compounds (66a-c) were synthesized and evaluated for activity. The analog with the ω-phenylpentyl moiety on N-10 and methyl group on N-5 (66a) turned out to be more potent than the reversed substitution against C. neoformans and C. albicans (66a, IC50 = 2.5 & 2.0 μg/ml respectively) [43]. Bis-quindolinium derivatives with an alkyl bridge connecting N5/N5 (67a-b) and those connecting N10/N10 atoms of the quindolines (68) were also synthesized and evaluated Fig. (10). It was observed that a positively charged quaternary nitrogen is also required for activity in bis-quindolinium compounds. Also noted was the fact that a bis-quindolinium compound separated by n-pentyl carbon bridge (67b) was more potent than the corresponding analog separated by four carbon atoms (67a). A similar observation was made about the N-10/N-10 five-carbon connected bis-quindolinium compound (68) [43].
Fig. 10.
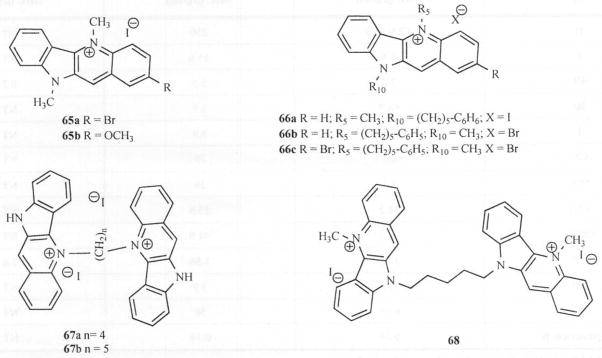
Structures of compounds 65-68.
At the beginning of 2007, we [9a] reported the effect of isosteric replacement of the N-10 in cryptolepine on antifungal activity. It was shown that antifungal activity diminished when the N-10 was replaced by oxygen and carbon but activity was retained when the replacement was sulfur. Interestingly, the benzothienoquinoline (sulfur substituents at position 10) analog of cryptolepine showed a broad spectrum of activity and was significantly less toxic than the parent compound. Thus, the authors suggested that the benzothieno[3,2-b]quinolines might serve as a new scaffold for the development of antifungal drugs.
D. Renal Vasodilation and Inhibition of Platelet Aggregation
The need for new drugs for cardiovascular diseases is underscored by this statement from the World Health Organization: “Cardiovascular disease is the number one cause of death globally and is projected to remain the leading cause of death. An estimated 17.5 million people died from cardiovascular disease in 2005, representing 30 % of all global deaths. Of these deaths, 7.6 million were due to heart attacks and 5.7 million were due to stroke. Around 80% of these deaths occurred in low and middle income countries (LMIC). If appropriate action is not taken, by 2015, an estimated 20 million people will die from cardiovascular disease every year, mainly from heart attacks and strokes.” [53].
Much of the earlier biological investigations on cryptolepine were related to its effect on the cardiovascular system. The effect of cryptolepine on renal vasodilation was reported by Oyekan in 1994 [11]. This effect in rats appears to be endothelium-dependent and involves the release of a non-cyclooxygenase endothelium-dependent relaxing factor (EDRF), possibly nitric oxide (NO). The author further reported that the effector pathway for cryptolepine-induced vasodilation is by cGMP of vascular smooth muscle. In addition, the vasodilatory effect was proposed to be mediated via K+ efflux, notably through activation of tetraethyl ammonium-sensitive but glibenclamide-insenstive K+ channels and Na+/K+-ATPase. Here, unlike diazoxide and acetylcholine, the K+ channels activated by cryptolepine are not coupled to pertussis toxin-sensitive G-proteins. However, many interwoven mechanisms seem to be involved which are directly related to the central mechanism involving the release of EDRF [54]. Cryptolepine inhibits platelet aggregation in vitro and in vivo and stimulates fibrinolysis ex vivo [10a]. The mechanism appears to involve an increase in platelet cAMP levels, possibly through stimulation of platelet adenylate cyclase [10c]. The antiaggregatory effects do not seem to invovlve extracellular or intracellular Ca2+ Cryptolepine also inhibits collagen-induced secondary aggregation through a selective antiphospholipase-like activity. However, there was no effect on platelet cyclooxygenase and lipoxygenase activities of platelets [10d], A limited structure activity relationship was done around the N-5 position of the quindoline ring in inhibiting ADP-induced aggregation and renal vasodilation [55]. Apart from quindoline, 4 analogs were synthesized and tested of which 5-ethyl quindoline was the most potent while quindoline the parent compound, was without effect. In fact, the 5-ethylquindoline was 2.5 times more potent than cryptolepine. The rank order of renal vasodilator and antiplatelet effect following alkyl substitution is: ethyl>methyl>propyl>benzyl. This trend suggests that the N-5 position has limited bulk tolerance and lipophilicity with an ethyl group being the optimum alkyl substituent. Although it was suggested that distortion of the flat quindoline structure due to alkylation at N-5 position may explain the enhanced activity, more recent observations on the antifungal properties may suggest instead the development of the positive charge on N-5 may be a more important factor in the expression of these activities.
E. Anti Hyperglycemic Activity
Hyperglycemia or high level of glucose in blood is one of the classic symptoms of diabetes (I or II) and left untreated can lead to organ damage. People who are older, overweight, have family members with diabetes, or are from certain ethnic groups are at greater risk for developing hyperglycemia [56a]. Hyperglycemia can be caused by insulin resistance, impaired glucose metabolism, medications or the use of some nutritional supplements. Several plants and plant extracts are known to have antihyperglycemic properties and a number of crude plant extracts and purified substances from plants have been under clinical trials. [56b–d]
Bierer and coworkers [20] were the first to report the antihyperglycemic properties of cryptolepine. They have synthesized several salts including the hydroiodide, hydrochloride and hydrotriflate of cryptolepine (1a, 1b and 1d) and tested for their ability to stimulate glucose transport in 3T3-L1 adipocytes. It was observed that cryptolepine showed an increasing ability to stimulate glucose transport beginning at 3 μM. Cryptolepine hydrochloride lowered blood glucose in Non-Insulin-Dependent Diabetes Mellitus (NIDDM) animal models with reduced food intake. Fasting time course studies and a pair-fed study in db/ab mice seem to indicate that this antihyperglycemic effect is separate from the anorexigenic effect. The presence of the methyl substituent at N-5 appears to be critical for activity as the N-10 methyl regioisomer was inactive in vitro and in vivo. Cryptolepine hydrochloride lowered serum glucose concentrations in fructose-fed STZ-treated rat model of NIDDM by 16 %, 34 % and 45 % on days 1, 2 and 3 of dosing respectively. The compound also lowered serum triglyceride concentrations by 30 %, 39 % and 69 % on days 1, 2 and 3 of dosing respectively. There was a significant decline in food consumption (29 %, p < 0.05) however, the body weight gain over the course of the experiment was not altered.
In a preliminary structure-antihyperglycemic activity studies conducted on cryptolepine reported by the same authors [27], a series of substituted cryptolepine analogues were synthesized and their bioactivities were measured in a 3T3-L1 glucose transport assay. Fig. (11). A group of these compounds was further evaluated in genetically altered obesediabetic C57BL/KS-db/ab mice. The results indicated that two derivatives (1b and 69) out of 33 analogs for which SAR studies were conducted, have true antihyperglycemie activity. In vitro studies showed that increasing the lipophilicity of the substituent at N-5 increased the glucose transport activity. A chloro substitution at the C-11 position of the ring reduced the activity of the cryptolepine analog possibly due to a deactivating effect. However, simultaneous substitution of a chloro atom at positions 1 and 11 (70) showed similar activity as the parent compound (1b). Substitution at the 4-position with OMe, (69) or at the 6-position with fluorine (71) improved activity, possibly due to an interaction between the methoxy/fluoro group and the N-5 methyl group, altering the planarity of the molecule. Replacement of the nitrogen at the 10-position with oxygen, sulfur and methylene groups led to a reduction in the glucose transport activity. Later, in vivo studies showed that the methoxy derivative (69) was the most efficacious derivative tested, lowering the plasma glucose level by 58.3 %, at 24 h post dose while showing improved food intake as compared with cryptolepine hydrochloride. The 2-fluoro derivative (72) was also efficacious, lowering the plasma glucose level by 42.5 % at 2 h post dose, with activity still being observed at 24 h. Inter estingly, compound 72 showed no signs of toxicity at higher concentrations (30 mM). However, as with 1b, this effect was accompanied by a significant reduction in both mean body weight and food intake. Substitution at position 11 decreased the ability of the compound to lower plasma glucose. This observation is similar to the SAR observed for antifungal activity and suggests similarity in the mode of action in these biological actions. It was also observed that compound 73, with an 11 -anilino and 2-fluoro groups, showed improvement in food consumption and body weight. Increasing the steric bulk at the N-5 position as in compounds 74 and 75 did not change their ability to lower plasma glucose but produced an improvement in food intake and body weight parameters. The benzyl derivative (74) was observed to have lowered plasma glucose by 15 % at 3 h post dose with no effect on food intake or body weight while the 4-fluoro-benzyl derivative (75) lowered plasma glucose by 36.3 % with improved food intake. This appears to suggest that a fluoro substitution has a beneficial effect on lowering plasma glucose levels in a similar manner irrespective of its position on the scaffold [cf. compound 72, Fig. (11)].
Fig. 11.
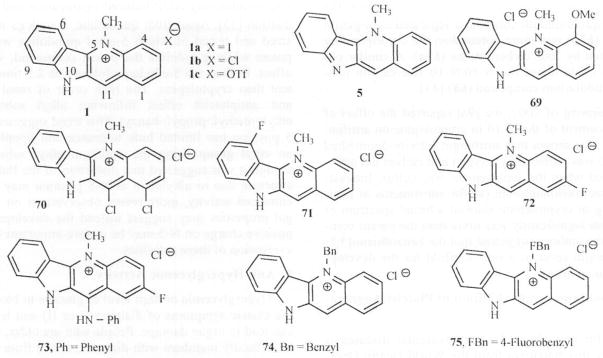
Analogs of Cryptolepine tested for antihyperglycemic Activity.
F. Anticancer Activity and Cytotoxicity
According to the World Health Organization (WHO) reports, more than 11 million people are diagnosed with cancer every year and it is estimated that there will be 16 million new cases every year by 2020. Cancer causes 7 million deaths every year or 12.5% of deaths worldwide. Plant derived drugs such as etoposide, taxol, vincristine and several others have contributed to current treatment modalities. Despite recent advances of cancer chemotherapy, the cytotoxic effects of the present drugs on non-cancerous cells have not been addressed satisfactorily. The need for new drugs is therefore indicated and plant sources continue to be important avenues of new drug discovery.
Cryptolepine has been shown to possess cytotoxic activity (IC50 = 1.3 μM) through the inhibition of DNA synthesis. Its cytotoxic potency is estimated to be 4–5 times more than that of ellipticine towards B16 melanoma cells and therefore a valid candidate for the development of anti-tumor agents [13a]. It easily crosses cell membranes and accumulates selectively in nuclei rather than in the cytoplasm of B16 melanoma cells and blocks the cell cycle in G2/M phase. However, in human lung adenocarcinoma A549 cells, the cell cycle arrest was at either G1 phase or G2/M phase, depending on the concentration [13b]. In addition to its interaction with DNA, cryptolepine is reported to target topoisomerase II in susceptible cells. Cryptolepine binds tightly to DNA and behaves as a typical intercalating agent [18]. The binding affinity is similar to that of other intercalators such as actinomycin D, daunomycin, porphyrin compounds and chromomycin. Specifically, cryptolepine prefers to bind to C-G rich sequences of DNA, with a tendency for non-alternating CC sites. A recent crystal structure of cryptolepine-d(CCTAGG) complex confirms the same [58]. Dassonneville et. al [59] provided direct evidence that cryptolepine induces apoptosis in HL-60 leukemia cells (IC50 = 3.2 μM) and is two times less toxic to HL-60/MX2 cells resistant to the antitumor drug mitoxanthrone. According to these authors, mitochondria and caspases play a central role in the activation of the executioner phase of cryptolepine induced apoptosis.
Cryptolepine also exhibited weak telomerase inhibitory activity (TelIC50 9.3 μM). However, there is moderate PCR inhibition at ~5 μM, suggesting a low degree of selectivity for quadruplex over duplex DNA. It is also suggested that cryptolepine stabilizes topoisomerase II-DNA covalent complex and stimulates the cutting of DNA by topoisorerase II [60]. It is believed that cryptolepine produces cell death signals other than topoisomerase II-mediated DNA breakage and topoisomerase II inhibition may play a minor role in the cytotoxicity of cryptolepine. As with most of the other anti cancer agents, the mechanism of action of cryptolepine is probably pleiotropic.
a) Indoloquinoline Derivatives as DNA Intercalators
Several cryptolepine derivatives based on modifications at C-2, N-5, N-10 and C-11 positions were synthesized and their structure activity relationship towards cytotoxicity studied Fig. (3) [61]. Initially, all the compounds were tested on 1138 yeast strain and selected analogs (1d, 76-81) were tested for cytotoxicity to M109 Madison lung carcinoma cell culture Fig. (12). The most active compounds tested for cytotoxicity against M109 Madison lung carcinoma cell culture were cryptolepine hydrotriflate (1d) and the N-acetyl derivative (78). Compounds 1d and 78 were further subjected to analysis of their effect on cell cycle dependent growth disturbances. The results showed a G1 cell cycle arrest and apoptosis in A2780/p53wt cells at 5 x IC50. More recently, Matsui [13d] and colleagues reported that cryptolepine induces the expression of p21WAF1/CIP1 with growth arrest in p53-mutated human osteocarcinoma MG63 cells. Interestingly, their results showed that cryptolepine inhibited the growth of MG63 cells and caused a G2/M-phase arrest.
Fig. 12.

Indoloquinoline analogs as DNA intercalators.
Higher concentrations showed a dose-dependent increase in apoptosis. Chen and co-workers [62, 63] studied cell growth inhibition for a series of substituted indoloquinoline-6-carboxamides and evaluated their activity in a panel of cell lines including Murine P388 leukemia. Murine Lewis lung carcinoma and Human Jurkat leukemia. Substitution of indolo[1,2-b]quinoline-6-carboxamides (82a and 82b) and 11H-benzothieno[3, 2-b]quinoline-4-carboxamides (83a and 83b) at the pseudo-peri position to the carboxamide side chain provided analogs of significantly increased cytotoxicity; IC50 = 450, 77, 170 and 171 nM respectively on Human Jarkat leukemia JLc as compared to the dual topo I/II inhibitor, N- [2- dimethyl-amino) ethyl]acridine-4-carboxamide (DACA) with an IC50 of 580 nM. The monomeric quindolines (82 and 84) discriminate poorly between cells with varying levels of topoisomerase II. The carboxamide-linkcd bis(quindoline) (85a) was less potent than 84a in P388, but more potent in the human leukemia cell lines (IC50 = 24 vs 230 nM) Fig. (12). The corresponding bis(4-methylquindoline) (85b) had a broadly similar pattern of cytotoxicity. The bis- analogs (86a and 86b) displayed poor activity in P388 and Lewis lung lines, but 86b was unexpectedly more active in the human leukemia cell lines (IC50 = 4.5 nM). In contrast to the monomers, the cytotoxicities of the dimeric quindolines are inversely proportional to the level of topoisomerase II in the cells. Compound 86c was nontoxic (IC50 = 9900 nM) while compound 86d was among the most active analogues (IC50 = 13.9 nM). In all the cases, DACA was used as the standard with an IC50 value of 580 nM. The IC50 ratios of JLA/JLC for the above compounds were less than about 2-fold suggesting a non-topo II mediated mechanism while DACA has the ratio of about 2.0 and behaves as a topo I/II inhibitor. Wright and coworkers [46] identified compound 87 with 4–5 fold more cytotoxicity to MAC15A cells than cryptolepine hydroiodide. Mardenborough [43] synthesized dimeric cryptolepine analogs (45 and 46) Fig. (5), which displayed no cytotoxicity up to 23.8 μg/mL on Vero cells, suggesting the dimeric compounds to be less toxic than their monomeric counterparts.
One concern regarding the use of cryptolepine analogs as anticancer agents is the lack of selectivity between DNA intercalation of cancer and normal cells. A solution to this problem has recently been proposed by Wright and colleagues at the University of Bradford. They proposed the development of bioreducible prodrugs that would be bio-reductively activated by nitroquinone oxido-reductases present in tumor cells at higher concentrations than in normal cells [13e]. With a limited number of nitrocryptolepine derivatives, the authors were able to demonstrate that some of the compounds are cytotoxic and may be substrates for the oxido-reductases (NQO1 & 2). Unfortunately, the compounds did not show significant toxicity towards the cells with high NQO1 (H460 cells) compared to those with low NQO1 (BE cells).
b) Telomerase II Inhibition
Folding of telomeric DNA into a four-stranded structure involving G-quartets has been shown to inhibit enzymes from catalyzing the synthesis of telomeric DNA repeats. Telomerase is expressed in tumor cells, but not in most somatic cells. Synthetic small molecules that induce and stabilize such G-quadruplex structures are potential telomerase inhibitors and could interfere with telomere elongation and replication of cancer cells. Several quindoline derivatives were designed and synthesized by Zhou and coworkers [64] and their interaction with the telomeric G-quadruplex has been studied Fig. (13).
Fig. 13.
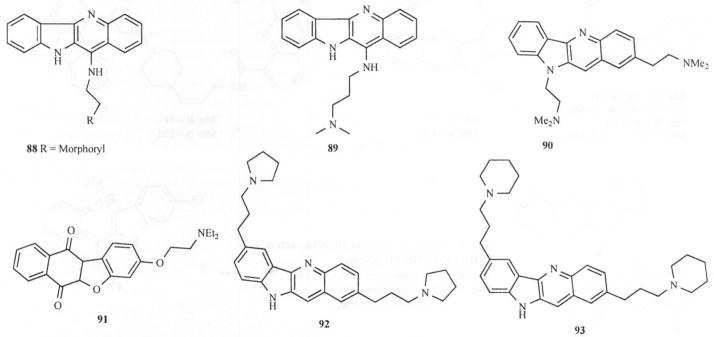
Cryptolepine/lndoloquinoline analogs as Telomerase inhibitors.
Introducing electron-donating groups such as a substituted amino group at position 11 of the quindoline scaffold significantly increased the electron density at the N-5 atom, resulting in easy protonation under physiological pH. EMSA and CD spectral studies showed that these compounds were capable of inducing the formation and stabilization of the G-quadruplex and thus exhibited inhibitory effect on telomerase. Competition dialysis experiments showed that compound 88 interacted preferentially with G-quadruplex and had comparatively weak affinity for duplex DNA. In vitro inhibitory activity studies on these compounds were done using telomerase repeat amplification protocol (TRAP) assay. The results showed that telomerase inhibition properties of the quindoline derivatives under study were significantly improved upon introduction of electron donors into the parent compound. Compounds 88 and 89 (with strong electron donor substituents in the 11 position) showed better TelIC50 values (0.55 μM and 0.44 μM respectively) as compared to quindoline (>138 μM) in that series.
Another report by Capiro [65] observed that 3 has significant activity against human telomerase (TelIC50 16 μM, as compared to 2, 6-dimethylamino-anthraquinone, 4.1 μM), a level comparable to several nucleoside inhibitors of reverse transcriptase activity. Compound 90 is approximately 10-fold less active than the best quadruplex-mediated inhibitors based on tricyclic chromophores and is 2-fold less active than the standard compound (91) (TelIC50 7.0 μM). More recently. Guyen [66] reported 2, 7-dialkylamine substituted quindolines (92 and 93) have improved potency (TelEC50 values of 6.3 μM and 11.8 μM) and did not exhibit any noticeable PCR inhibition at up to 50 μM Fig (4). This is suggestive of selectivity for quadruplex over duplex DNA Fig. (13).
CONCLUSION
Indolo[2,3-b]quinolines constitute a lead scaffold for drug development. Synthetic strategies for obtaining the tetracyclic structure are now available providing flexibility for introducing various functional groups into the ring system. Such functionalized structures are associated with a variety of pharmacological activities including antiinfective, vasodilation, antiplatelet aggregation, antihyperglycemic, anticancer and several others. Structure activity relationship studies are beginning to emerge. However, a consistent mechanism of action by which the tetracyclic ring is involved in the various biological actions is yet to emerge. Clearly, this is one area of focus for the future.
Acknowledgments
The authors gratefully acknowledge the editorial contributions of Mrs. Barbara Bricker. This work would not have been possible without the continuing financial support of RCMI grant number G12 RR 03020 from NCRR and the Title III grant to SYA.
References
- 1.a) Fitchter F, Boehringer R. Chem Ber. 1906;39:3932. [Google Scholar]; b) Fitchter F, Probst H. Chem Ber. 1907;40:3478. [Google Scholar]; c) Fichter F, Rohner F. Chem Ber. 1910;43:3489. [Google Scholar]
- 2.Clinquart E. Bull Acad Med Belg. 1929;9:627. [Google Scholar]
- 3.a) Delvaux E. J Pharm Belg. 1931;13:955. [Google Scholar]; b) Delvaux E. J Pharm Belg. 1931;13:973. [Google Scholar]
- 4.Gunatilaka AAL, Sotheeswaran S, Balasubramaniam S, Chandrasekara AI, Sriyani HTB. Planta Med. 1980;39:66. doi: 10.1055/s-2008-1074904. [DOI] [PubMed] [Google Scholar]
- 5.Gillert E. Helv Chim Acta. 1951;34:642. [Google Scholar]
- 6.a) Dwuma-Badu D, Ayim JSK, Fiagbe FNIY, Knapp JE, Schiff PL, Jr, Slatkin DJ. J Pharm Sci. 1978;67:433. doi: 10.1002/jps.2600670350. [DOI] [PubMed] [Google Scholar]; b) Ablordeppey SY, Hufford CD, Borne RF, Dwuma-Badu D. Planta Med. 1990;56:416. doi: 10.1055/s-2006-960998. [DOI] [PubMed] [Google Scholar]
- 7.a) Cimanga K, Pieters L, Claeys M, Vanden Berghe D, Vlietinck AJ. Planta Med. 1991;57(Suppl 2):A98. doi: 10.1055/s-2006-957657. [DOI] [PubMed] [Google Scholar]; b) Paulo A, Pimentel M, Viegas S, Pires I, Cabrita J, Gomes ET. J Ethnopharmacol. 1994;44:73. doi: 10.1016/0378-8741(94)90071-x. [DOI] [PubMed] [Google Scholar]; c) Paulo A, Duarte A, Gomes ET. J Ethnopharmacol. 1994;44:127. doi: 10.1016/0378-8741(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 8.a) Wright CW. J Pharm Pharmacol. 2007;59:899. doi: 10.1211/jpp.59.6.0017. [DOI] [PubMed] [Google Scholar]; b) Noamesi BK, Paine A, Kirby GC, Warhurst DC, Phillipson JD. Trans R Soc Trop Med Hyg. 1991;85:315. [Google Scholar]; c) Kirby GC, Noamesi BK, Paine A, Warhurst DC, Phillipson JD. Phytother Res. 1995;9:359. [Google Scholar]; d) Cimanga K, De Bruyne T, Lasure A, Vanpoel B, Pieters L, Claeys M, Vanden Berghe D, Kambu K, Tona L, Vlietinck AJ. Planta Med. 1996;62:22. doi: 10.1055/s-2006-957789. [DOI] [PubMed] [Google Scholar]; e) Wright CW, Phillipson JD, Awe SO, Kirby GC, Warhurst DC, Quetin-Leclercq J, Angenot L. Phytother Res. 1996;10:361. [Google Scholar]; f) Cimanga K, De Bruyne T, Pieters L, Vlietinck AJ, Turger CAJ. Nat Prod. 1977;60:688. doi: 10.1021/np9605246. [DOI] [PubMed] [Google Scholar]
- 9.a) Zhu XY, Mardenborough LG, Li S, Khan A, Zhang W, Fan P, Jacob M, Khan S, Walker L, Ablordeppey SY. Bioorg Med Chem. 2007;15:686. doi: 10.1016/j.bmc.2006.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Singh M, Singh MP, Ablordeppey SY. Drug Develop Indust Pharm. 1996;22:377. [Google Scholar]
- 10.a) Oyckan AO, Botting JH, Noamesi BK. Gen Pharmacol. 1988;19:233. doi: 10.1016/0306-3623(88)90067-5. [DOI] [PubMed] [Google Scholar]; b) Oyekan AO, Okafor JP. J Ethnopharmacol. 1989;27:141. doi: 10.1016/0378-8741(89)90086-x. [DOI] [PubMed] [Google Scholar]; c) Oyekan AO, Ablordeppey SYA. Gen Pharmacol. 1993;24:461. doi: 10.1016/0306-3623(93)90333-s. [DOI] [PubMed] [Google Scholar]; d) Oyekan AO, Ablordeppey SYA. Gen Pharmacol. 1993;24:1285. doi: 10.1016/0306-3623(93)90382-8. [DOI] [PubMed] [Google Scholar]
- 11.Oyckan AO. J Cardiovasc Pharmacol. 1994;23:602. doi: 10.1097/00005344-199404000-00012. [DOI] [PubMed] [Google Scholar]
- 12.Luo J, Fort DM, Caarlson TJ, Noamesi BK, Amon-Kotei D, King SR, Tsai J, Quan J, Hobensack C, Lapresca P, Waldeck N, Mendez CD, Jolad SD, Bierer DE, Reaven GM. Diabet Med. 1998;15:367. doi: 10.1002/(SICI)1096-9136(199805)15:5<367::AID-DIA576>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 13.a) Bonjean K, De Pauw-Gillet MC, Defresne MP, Colson P, Houssier C, Dassonneville L, Bailly C, Wright C, Quetin-Leclercq J, Angenot L. Anticancer Res. 1997;17:4068. [Google Scholar]; b) Dassonneville L, Bonjean K, De Pauw-Gillet M-C, Colson P, Houssier C, Quetin-Leclercq J, Angenot L, Bailly C. Biochemistry. 1999;38:7719. doi: 10.1021/bi990094t. [DOI] [PubMed] [Google Scholar]; c) Ansah C, Gooderham NJ. Toxicol Sci. 2002;70:245. doi: 10.1093/toxsci/70.2.245. [DOI] [PubMed] [Google Scholar]; d) Matsui T-A, Sowa Y, Murata H, Takagi K, Nakanishi R, Aoki S, Yoshikawa M, Kobayashi M, Sakabe T, Koichi T, Sakai T. Int J Oncol. 2007;31:915. [PubMed] [Google Scholar]; e) Seville S, Phillips RM, Shnyder SD, Wright CW. Bioorg Med Chem. 2007;75:6353. doi: 10.1016/j.bmc.2007.06.062. [DOI] [PubMed] [Google Scholar]
- 14.a) Bamgbose SO, Noamesi BK. Planta Med. 1981;41:392. doi: 10.1055/s-2007-971733. [DOI] [PubMed] [Google Scholar]; b) Noamesi BK, Bamgbose SO. Planta Med. 1984;44:98. doi: 10.1055/s-2007-969633. [DOI] [PubMed] [Google Scholar]; c) Olajide OA, Heiss EH, Schachner D, Wright CW, Vollmar AM, Dirsch VM. Bioorg Med Chem. 2007;15:43. doi: 10.1016/j.bmc.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 15.a) Hamet RC. R Soc Biol. 1937;126:768. [Google Scholar]; b) Hamet R. Acad Sci Belg. 1938;207:1016. [Google Scholar]; c) Noamesi BK, Bamgbose SOA. Planta Med. 1980;39:51. doi: 10.1055/s-2008-1074902. [DOI] [PubMed] [Google Scholar]
- 16.Noamesi BK, Bamgbose SOA. Planta Med. 1982;44:241. doi: 10.1055/s-2007-971458. [DOI] [PubMed] [Google Scholar]
- 17.Rauwald HW, Kober M, Mutschler E, Lambrecht G. Planta Med. 1992;55:486. doi: 10.1055/s-2006-961531. [DOI] [PubMed] [Google Scholar]
- 18.Bonjean K, De Pauw-Gillet MC, Defresne M-P, Colson P, Houssier C, Dassonneville L, Bailly C, Greimers R, Wright C, Quetin-Leclercq J, Tits M, Angenot L. Biochemistry. 1998;37:5136. doi: 10.1021/bi972927q. [DOI] [PubMed] [Google Scholar]
- 19.a) Holt SJ, Petrow V. Proc Roy Soc B. 1946;148:607. [Google Scholar]; b) Holt SJ, Petrow V. J Chem Soc. 1947:607. [Google Scholar]; c) Holt SJ, Petrow V. J Chem Soc. 1948:919. [Google Scholar]
- 20.Bierer DE, Fort DM, Mendez CD, Luo J, Imbach PA, Dubenko LG, Jolad SD, Gerber RE, Litvak J, LuQing, Zhang P, Reed MJ, Waldeck N, Bruening RC, Noamesi BK, Hector RF, Carlson TJ, King SR. J Med Chem. 1998;41:894. doi: 10.1021/jm9704816. [DOI] [PubMed] [Google Scholar]
- 21.Fan P, Ablordeppey SY. J Heterocycl Chem. 1997;34:1789. [Google Scholar]
- 22.Dhanabal T, Sangeetha R, Mohan PS. Tetrahderon. 2006;62:6258. [Google Scholar]
- 23.Cooper MM, Lovell JM, Joule JA. Tetrahedron Lett. 1996;37:4283. [Google Scholar]
- 24.Ho TL, Jou DG. Helv Chim Acta. 2002;85:3823. [Google Scholar]
- 25.Radl S, Konvicka P, Vachal P. J Heterocycl Chem. 2000;37:855. [Google Scholar]
- 26.Dutta B, Some S, Ray JK. Tetrahderon Lett. 2006;47:377. [Google Scholar]
- 27.Bierer DE, Dubenko LG, Zhang P, Lu Qing, Imbach PA, Garofalo AW, Phuan P-W, Fort DM, Litvak J, Gerber E, Sloan B, Luo J, Cooer R, Reaven GM. J Med Chem. 1998;41:2754. doi: 10.1021/jm970735n. [DOI] [PubMed] [Google Scholar]
- 28.Arzel E, Rocca P, Marsais F, Godard A, Queguiner G. Tetrahedron Lett. 1998;39:6465. [Google Scholar]
- 29.Arzel E, Rocca P, Grellier P, Labaeid M, Frappier F, Gueritte F, Gaspard C, Marsais F, Godard A, Queguiner G. J Med Chem. 2001;44:949. doi: 10.1021/jm0010419. [DOI] [PubMed] [Google Scholar]
- 30.Yamato M, Takeuchi Y, Chang M, Hashigaki K, Tsuruo T, Tashiro T, Tsukagoshi S. Chem Pharm Bull. 1990;38:3048. doi: 10.1248/cpb.38.3048. [DOI] [PubMed] [Google Scholar]
- 31.Yamato M, Takeuchi Y, Chang M, Hashigaki K. Chem Pharm Bull. 1992;40:528. doi: 10.1248/cpb.40.528. [DOI] [PubMed] [Google Scholar]
- 32.Gorlitzer K, Weber J. Arch Pharm (Wemheim) 1981;314:852. [Google Scholar]
- 33.Chang M, Takeuchi Y, Hashigaki K, Yamato M. Heterocycles. 1992;33:147. [Google Scholar]
- 34.Walsh C, Wright G. Chem Rev. 2005;105:391. doi: 10.1021/cr030100y. [DOI] [PubMed] [Google Scholar]
- 35.Brown ED, Wright GD. Chem Rev. 2005;105:759. doi: 10.1021/cr030116o. [DOI] [PubMed] [Google Scholar]
- 36.Gibbons S. Nat Prod Rep. 2004;21:263. doi: 10.1039/b212695h. [DOI] [PubMed] [Google Scholar]
- 37.Boakye-Yiadom K, Heman-Ackah SM. J Pharm Sci. 1979;68:1510. doi: 10.1002/jps.2600681212. [DOI] [PubMed] [Google Scholar]
- 38.a) Sawer IK, Berry MI, Brown MW, Ford JL. J Pharm Pharmacol. 1993;45:1108. [Google Scholar]; b) Cimanga K, De Bruyne t, Lasure A, Vanpoel B, Pieters L, Claeys M, Vanden Berghe D, Kambu K, Tona L, Vlietinck A. Planta Med. 1996;62:22. doi: 10.1055/s-2006-957789. [DOI] [PubMed] [Google Scholar]
- 39.Sawer IK, Berry MI, Ford JL. Lett Appl Microbiol. 2005;40:24. doi: 10.1111/j.1472-765X.2004.01625.x. [DOI] [PubMed] [Google Scholar]
- 40.Sawer IK, Berry MI, Brown MW, Ford JL. J Appl Bacteriol. 1995;79:314. doi: 10.1111/j.1365-2672.1995.tb03143.x. [DOI] [PubMed] [Google Scholar]
- 41.Sawer IK, Berry MI, Ford JL. Lett Appl Microbiol. 1997;25:207. doi: 10.1046/j.1472-765x.1997.00206.x. [DOI] [PubMed] [Google Scholar]
- 42.Gibbons S, Hallah F, Wright CW. Phytother Res. 2003;17:434. doi: 10.1002/ptr.1284. [DOI] [PubMed] [Google Scholar]
- 43.Mardenborough LG, Zhu XY, Fan P, Jacob MR, Khan SI, Walker LA, Ablordeppey SY. Bioorg Med Chem. 2005;13:3955. doi: 10.1016/j.bmc.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 44.Trape J-F, Pison G, Speigel A, Enel C, Rogier C. Trends Parasitol. 2002;18:224. doi: 10.1016/s1471-4922(02)02249-3. [DOI] [PubMed] [Google Scholar]
- 45.Wright CW. Phytochem Rev. 2005;4:55. [Google Scholar]
- 46.Wright CW, Addae-Kyereme J, Breen AG, Brown JE, Cox MF, Croft SLY, Kendrick H, Philips RM, Pollet PL. J Med Chem. 2001;44:3187. doi: 10.1021/jm010929+. [DOI] [PubMed] [Google Scholar]
- 47.Grellier P, Ramiaramanana L, Millerioux V, Deharo E, Schrevel J, Frappier F, Trigalo F, Bodo B, Pousset J-L. Phytother Res. 1996;10:317. [Google Scholar]
- 48.Onyeibor O, Croft SL, Dodson HI, Feiz-Haddad M, Kendrick H, Millington NJ, Parapini S, Phillips RM, Seville S, Snyder SD, Taramelli D, Wright WW. J Med Chem. 2005;48:2701. doi: 10.1021/jm040893w. [DOI] [PubMed] [Google Scholar]
- 49.Ablordeppey SY, Fan P, Ablordeppey JH, Mardenborough L. Curr Med Chem. 1999;6:1151. [PubMed] [Google Scholar]
- 50.Ablordeppey SY, Fan P, Clark AM, Alison N. Biorg Med Chem. 1999;7:343. doi: 10.1016/s0968-0896(98)00244-2. [DOI] [PubMed] [Google Scholar]
- 51.Ablordeppey SY, Fan P, Li S, Clark AM, Hufford CD. Biorg Med Chem. 2002;10:1337. doi: 10.1016/s0968-0896(01)00401-1. [DOI] [PubMed] [Google Scholar]
- 52.Mardenborough LG, Fan PC, Ablordeppey SY. Med Chem Res. 1999;92:118. [PubMed] [Google Scholar]
- 53.http://www.who.int/cardiovascular_diseases/en/
- 54.Oyekan AO. Eur J Pharmacol. 1995;285:1. doi: 10.1016/0014-2999(95)00289-w. [DOI] [PubMed] [Google Scholar]
- 55.Oyekan AO, Ablordeppey SYA. Med Chem Res. 1996:602. [Google Scholar]
- 56.a) Must A, Spadano J, Coakley EH, Field AE, Colditz G, Dietz WH. J Am Med Assoc. 1999;282:1523. doi: 10.1001/jama.282.16.1523. [DOI] [PubMed] [Google Scholar]; b) Anhauser M. Drug Discov Today. 2003;8:868. doi: 10.1016/s1359-6446(03)02856-3. [DOI] [PubMed] [Google Scholar]; c) Atta UrR, Zaman K. J Ethnopharmacol. 1989;26:1. doi: 10.1016/0378-8741(89)90112-8. [DOI] [PubMed] [Google Scholar]; Izzo AA, Ernst E. Drugs. 2001;67:2163. doi: 10.2165/00003495-200161150-00002. [DOI] [PubMed] [Google Scholar]
- 57.Zhu H, Gooderham N. J Tox Sci. 2006;91:132. doi: 10.1093/toxsci/kfj146. [DOI] [PubMed] [Google Scholar]
- 58.a) Lisgarten JN, Coll M, Portugal J, Wright CW, Aymami J. Nat Struct Biol. 2002;9:57. doi: 10.1038/nsb729. [DOI] [PubMed] [Google Scholar]; b) Lisgarten JN, Pous J, Coll M, Wright CW, Aymami J. Acta Crystallogr D: Biol Crystallogr. 2002;58:312–313. doi: 10.1107/s0907444901018960. [DOI] [PubMed] [Google Scholar]
- 59.Dassonneville L, Lansiaux A, Wattelet A, Wattez N, Mahieu C, Miert SV, Pieters L, Bailly C. Eur J Pharmacol. 2000;409:9. doi: 10.1016/s0014-2999(00)00805-0. [DOI] [PubMed] [Google Scholar]
- 60.Guittat L, Alberti P, Rosu F, Miert SV, Thetiot E, Pieters L, Gabelica V, De Pauw E, Ottaviani A, Riou J-F, Mergny J-L. Biochimie. 2003;85:535. doi: 10.1016/s0300-9084(03)00035-x. [DOI] [PubMed] [Google Scholar]
- 61.Yang S, Abdel-kader M, Malone S, Werkhoven MCM, Wisse JH, Bursuker I, Neddermann K, Fairchild C, Raventos-Suarez C, Menendez AT, Lane K, Kingston DGI. J Nat Prod. 1999;62:976. doi: 10.1021/np990035g. [DOI] [PubMed] [Google Scholar]
- 62.Chen J, Deady LW, Desneves J, Kave AJ, Finlav GJ, Baguley BC, Denny WA. Biorg Med Chem. 2000;8:2461. doi: 10.1016/s0968-0896(00)00179-6. [DOI] [PubMed] [Google Scholar]
- 63.Chen J, Deady LW, Kaye AJ, Finlay GJ, Baguley BC, Denny WA. Biorg Med Chem. 2002;10:2381. doi: 10.1016/s0968-0896(02)00067-6. [DOI] [PubMed] [Google Scholar]
- 64.Zhou J-L, Lu Y-J, Ou T-M, Zhou J-M, Huang Z-S, Zhu X-F, Du C-J, Bu X-Z, Ma L, Gu L-Q, Li Y-M, Chan AS-C. J Med Chem. 2005;48:7315. doi: 10.1021/jm050041b. [DOI] [PubMed] [Google Scholar]
- 65.Capiro V, Guyen B, Opoku-Boahen Y, Mann Y, Gowan SM, Kelland LM, Read MA, Neidle S. Bioorg Med Chem Lett. 2000;10:2063. doi: 10.1016/s0960-894x(00)00378-4. [DOI] [PubMed] [Google Scholar]
- 66.Guyen B, Schultes CM, Haze P, Mann J, Neidle S. Org Biomol Chem. 2004;2:981. doi: 10.1039/b316055f. [DOI] [PubMed] [Google Scholar]