

Abstract
Squamous cell carcinoma (SCC) is a significant cause of cancer morbidity and mortality worldwide, with an incidence of up to 166 cases per 100 000 population. It arises in the skin, upper aerodigestive tract, lung, and cervix and affects more than 200 000 Americans each year. We report here that a microarray experiment comparing 41 SCC and 13 normal tissue specimens showed that Id2, a gene that controls the cell cycle, was significantly up-regulated in SCC. Enforced expression of Id2 in vitro stimulated the proliferation of SCC cells and up-regulated the transcription of nuclear factor kappa B (NF-κB) and cyclin D1. Enhancement of the NF-κB activity with p65 significantly increased the cell proliferation and the transcription of cyclin D1, whereas inhibition of the NF-κB activity with I kappa B alpha mutant (IκBα M) and pyrroline dithiocarbamate (PDTC) abrogated cell proliferation and transcription of cyclin D1. Furthermore, a mutated NF-κB binding site in the cyclin D1 promoter fully abrogated the Id2-induced transcription of cyclin D1. Taken together, these data indicate that Id2 induces SCC tumor growth and proliferation through the NF-κB/cyclin D1 pathway.
Keywords: Id2, head and neck squamous cell carcinoma, NF-κB, cyclin D1, human
The Id gene family encodes 4 related proteins (Id1-4) implicated in cell cycle progression[1] and immortalization[2],[3]. These proteins antagonize the binding of basic helix-loop-helix (bHLH) transcription factors to specific genomic DNA sites that lead to the transcription of genes for lineage commitment, cell differentiation, growth control, and apoptosis[4]–[6]. Id proteins interact with the bHLH family of proteins and act as dominant-negative bHLH transcription factors. Thus, Id proteins have been implicated in neurogenesis[7], hematopoesis[8], and tumorigenesis[9],[10]. Id genes in tumor cells display a general pattern of dysregulation[11]–[13]. Id2 is capable of abrogating the growth-suppressive functions of the tumor suppressors p16, p21, and Rb through a direct interaction with the Rb protein[14],[15].
Overexpression and dysregulation of Id2 protein are observed in many cancers including astrocytic tumors[16], neuroblastoma[17], Ewing family tumors[18], colorectal adenocarcinoma[19], and colon carcinoma[20]. Forced expression of Id1 protein in the mouse small intestinal epithelium triggers the development of adenoma[21] and SCC in the head and neck[22]. In loss-of-function studies, Id2 is key to carcinogenesis of neuroblastoma[17],[23], whereas Id1 and Id3 are indispensable for growth of tumor xenografts and angiogenesis[24].
Id proteins respond to growth factor stimulation[25], promoting cell growth and impairing cell differentiation in vitro. Expression of Id proteins is highest in proliferating cell lines and lowest in mature cells. Overexpression of Id proteins provides an opportunity for cells to escape growth control and potentiates cellular hyperplasia, dysplasia, and tumor development[9]. These referenced studies indicate that Id proteins are involved in tumorigenesis. However, how Id2 is involved in the tumorigenesis of SCC is unclear.
To test this hypothesis, we analyzed Id2 expression in SCC tissues and then modulated Id2 expression in vitro to assess changes in cell proliferation. We also measured the activity of the NF-κB/cyclin D1 pathway, which is critical in SCC, to determine its role in Id2-mediated effects in SCC. We found that Id2 is extensively expressed in SCC tissues and that Id2 overexpression significantly increased the proliferation of SCC cells. Furthermore, the effects of Id2 were dependent upon the NF-κB/cyclin D1 pathway, as NF-κB inhibitors and activators respectively blocked and increased the activity of Id2. In addition, NF-κB binding was required for cyclin D1 transcription. Taken together, these data indicate that the Id2/NF-κB/cyclin D1 pathway is dysregulated in SCC.
Materials and Methods
Tissue specimens, cell lines, and materials
A total of 41 head and neck SCC and 13 normal tissue specimens were collected from patients who underwent head and neck cancer surgery at the Department of Otolaryngology, University of Minnesota Hospital and Clinics. Control specimens were biopsies of normal tissues near the cancer site. Total RNA from all 54 tissue specimens was isolated using Trizol kit (GIBCO BRL Life Technologies). An additional 50 head and neck SCC specimens (embedded in paraffin) were obtained from the Department of Surgical Pathology for histopathologic evaluation in this study.
The CA9-22, SCC9, HOK16B, and Rhek-1A cell lines were used in this study. CA9-22 and SCC9 were established from oral SCC tissues[22],[26]. Rhek-1A was established from dekeratinized squamous cells with transduction of the SV40 oncogene[27] and was a kind gift of Dr. Jhong S. Rhim (Frederick Cancer Research and Development Center, National Cancer Institute, Frederick, MD). HOK16B was established from normal human oral keratinocytes with transduction of type 16 human papillomavirus[22]. CA9-22 and SCC9 cells were maintained in Gibco® RPMI-1640 (Invitrogen, Carlsbad, CA) containing 10% heat-inactivated fetal bovine serum (FBS), 1% penicillin/streptomycin, 1.2% L-glutamine, and 2.5% HEPES buffer. Rhek-1A cells were maintained in Eagle's minimal essential medium (MEM, Invitrogen) supplemented with 10% FBS, 50 µg/mL penicillin/streptomycin, and 1.2% L-glutamine (hereafter referred to as complete medium). HOK-16B cells (from human oral mucosal keratinocytes) were maintained in keratinocyte basal medium (Lon2a). For transient transfection of cells, serum-free Opti-MEM medium with 6 µg/mL Polybrene® (Invitrogen, Carlsbad, CA) (hereafter referred to as transfection medium) was used.
Human Id2 cDNA from head and neck SCC tissue specimens was cloned into a plasmid with enhanced green fluorescent protein (pEGFP, Clontech) using standard protocols. The full-length Id2 cDNA (GenBank accession #213931) was obtained from the NCBI website and the open reading frame (ORF) of the Id2 gene was identified with the Genetic Computer Group program (GCG, Wisconsin). Specific primers for Id2 spanning the ORF were designed with the Oligo 4.0® program, and the specificity of the primers was examined using Amplify 1.2® followed by BLAST search. The primer sequences are as follows: sense 5′-GGC-TTCCTCGCGGTCAGCATG-3′ and antisense 5′-ACTG-TGTGGCTGAATAAGCG-3′. Reverse transcription-poly-merase chain reaction (RT-PCR) was performed on the head and neck SCC tissue RNA to obtain the ORF of Id2 cDNA. PCR products were sequenced using an autosequencer, cloned into pT-Adv vector (Clontech) and subcloned into the pEGFP expression vector to express the Id2 protein. The p65 cDNA expression plasmid (pMT2T-p65) and empty vector (pMT2T), which were previously described[28],[29], were a kind gift of Dr. Keith Brown (National Institute of Allergy and Infectious Disease, NIH, Bethesda, MD)[30],[31].
IκBα M is a mutant form of the IκBα inhibitor in which the serines at positions 32 and 36 have been changed to alanines, preventing phosphorylation and subsequent proteasomal degradation following an NF-κB-activating stimulus[32]. This mutant protein was used as a specific inhibitor of NF-κB activity in this study. PDTC, a proteasome inhibitor purchased from Calbiochem (La Jolla, CA), was also used as an inhibitor of the NF-κB activity[33]. Cyclin D1 luciferase reporter plasmids, generous gifts of Dr. Richard Pestell (Department of Developmental and Molecular Biology, Albert Einstein College of Medicine, Bronx, NY), contain the cyclin D1 promoter upstream from the luciferase gene as previously described[34]. The 1745 cyclin D1 luciferase reporter plasmid contains the full sequence cyclin D1 promoter upstream from the luciferase gene, and the 66NF-κBmut luciferase reporter plasmid contains the cyclin D1 promoter with a site-specific mutation at the NF-κB-binding site. The NF-κB reporter gene contains two immunoglobulin Gk chain NF-κB-binding sites and has been previously described[35]. pCMV-β-galactocidase (β-gal) reporter plasmid was purchased from Stratagene. The β-gal luciferase activity was used as an internal control for NF-κB and cyclin D1 luciferase activities in this study.
Immunohistochemistry
Head and neck SCC tissue sections from the Department of Surgical Pathology were routinely cut to a thickness of 4 microns, deparaffinized, incubated with primary antibodies (Id2, NF-κB, and Cyclin D1) and secondary antibodies (rabbit anti-mouse IgG or goat anti-rabbit IgG), followed by DAB substrate, as previously described[36].
Microarray analysis
Experiments using Affymetrix microarrays were performed as described previously[37]. Briefly, cDNA was prepared from 10 µg total RNA using the double-stranded DNA synthesis kit (Life Technologies, Rockville, MD). cRNA was synthesized from cDNA and labeled with biotin-streptavidin using the BioArray High Yield RNA Transcript Labeling Kit (Enzo Diagnostics, Farmingdale, NY). Approximately 15 µg of cRNA was fragmented and hybridized to human genome set U133A (representing approximately 18 000 genes) arrays. Hybridization signals on the arrays were detected with anti-streptavidin antibody and measured using the Agilent GeneArray Laser Scanner. The expression of the Id2 gene in SCC tissue specimens was identified and measured as a relative light unit. The expression of Id2 in control and SCC cell lines and tissues was confirmed by RT-PCR, real-time PCR, and western blotting.
Transient cell transfection, Trypan Blue exclusion, 3H-thymidine incorporation, and real-time PCR
Approximately 25 000 SCC cells (CA9-22 or Rhek-1A) were plated into each well of a 24-well tissue culture plate and cultured overnight. Cells were transfected with Id2 cDNA plasmids (2.0 µg/mL) in transfection medium for 6 h and incubated in complete medium for 12 h to allow recovery from transfection. A similar protocol was used for co-transfection with Id2 cDNA (1.4 µg/mL) and IκBα M (1.4 µg/mL) to inhibit the NF-κB activity for evaluation of its involvement in the Id2-triggered response. Cells transfected with empty vector, IκBα M or incubated with medium alone served as controls.
For Trypan Blue exclusion, cells were harvested at 8, 16, 24, and 48 h following the recovery incubation. Cells were washed in phosphate-buffered saline (PBS) and incubated in 0.3 mL of 0.05% trypsin/EDTA solution for 10 min. Five microliters of the trypsin solution was mixed with 5 µL of Trypan Blue and transferred to a hemacytometer for counting.
For 3H-thymidine incorporation, cells were harvested at 8, 16, 24, and 48 h following the recovery incubation. Cells were washed in PBS twice and digested in a solution of 0.2 mol/L sodium hydroxide and 2.5% sodium dodecyl sulfate (SDS) at 37°C for 1 h. Afterwards, they were transferred to 1 mL of scintillation fluid (Ecoscent A, National Diagnostics), mixed thoroughly, and digested for 2 h prior to counting in the scintillation counter (Beckman). Cell counting was also performed at the same time points for calculation of radioactivity (counts per minute, CPM) per cell. Results are presented as CPM per 10 000 cells.
For proliferating cell nuclear antigen (PCNA) immunohistochemistry, cells were harvested at 24 h following Id2 transfection. Cells were fixed with 70% ethanol, washed with PBS, and incubated with biotinylated mouse anti-PCNA for 60 min, streptavidin-peroxidase for 10 min, and DAB mixture for 5 min at room temperature, followed by counterstaining with hematoxylin for 1 min. Sections were observed under a light microscope (Nikon E400) and photographed.
For verification of transfection, real-time PCR was performed on CA9-22 with and without Id2 cDNA transfection for 24 h, using Rhek-1A as a normal squamous cell control. RNA was extracted using the Mini-prep RNA extraction kit (Stratagene). Real-time PCR was performed in triplicate using the Taqman® one-step RT-PCR Master Mix Reagent Kit (PE Biosystems) according to the manufacturer's instruction. The specific primers used were as follows: upstream, 5′-TGGACTCGCATCCCACTATTG-3′; downstream, 5′-CCTGGACGCCTGGTTCTG-3′; and probe, 5′-cagcctgcatcaccagagacccg-3′.
Transient co-transfection and luciferase assays
After being cultured in a 12-well plate and reaching 60% confluency, cells were co-transfected with the Id2 cDNA (1.4 µg/mL) and the NF-κB or cyclin D1 luciferase reporters (1.4 µg/mL) for 16 h and incubated in complete medium for 24 h for recovery. To determine whether cyclin D1 transcription was dependent on NF-κB, cells were co-transfected with Id2 (1.4 µg/mL) and IκBα M (1.4 µg/mLI) or p65 (1.4 µg/mL) or incubated with PDTC (50 µmol/L) for 16 h and then incubated in complete medium for 24 h. Cells were then harvested for luciferase assays. Luciferase activity was measured using the dual-light luciferase assay kit (Tropix) on a microplate luminometer (Model TR717, Tropix). β-galactocidase reporter activity was used as an internal control. The activities of luciferase reporters are presented by a ratio of luciferase-to-β-galactocidase reporter activity.
Western blotting
Normal and SCC tissues were harvested for protein isolation. Approximately 30 µg of protein were electrophoresed and transferred to a nitrocellulose membrane. Id2 and GAPDH antigens on the membrane were detected by anti-Id2 (sc-489, Santa Cruz) and anti-GAPDH (ab9485, Novus Biological) antibodies, respectively, and visualized using ECL kit (Amersham Biosciences) according to the manufacturer's instructions.
Statistical analysis
Student's t test was used to evaluate the differences between control and experimental samples in vitro, whereas Fisher's exact test was used to evaluate the differences between control and experimental samples if sample sizes are small. P values less than 0.05 were considered significant.
Results
Expression of Id2 mRNA and protein is significantly higher in SCC tissue specimens than in normal controls
Id2 mRNA transcript levels were significantly higher in 41 head and neck SCC tissue specimens [(1196.94 ± 641.89) relative light units (RLU)] than in control specimens [(655.19 ± 240.95) RLU, P < 0.05], as measured by Affymetrix microarrays (Figure 1A). Similarly, RT-PCR showed that Id2 mRNA transcripts in SCC cell lines were abundant compared to those in control cell lines (Figure 1B). To confirm Id2 expression, we performed western blotting using SCC and control tissue specimens and found that Id2 protein was expressed in SCC but not in control tissues (Figure 1C). We obtained similar results using quantitative PCR (qPCR) to compare SCC and control tissues (Figure 1D). The microarray heatmap demonstrated that Id2, NF-κB, and cyclin D1 were up-regulated in the 41 SCC tissues compared to control tissues (Figure 1E).
Figure 1. Id2 mRNA and protein are up-regulated in head and neck squamous cell carcinoma (SCC) tissue and cell lines.

A, the level of the Id2 mRNA transcripts in SCC tissue specimens was significantly higher than that in control tissues (P < 0.05) as measured with Affymetrix microarrays. B, the expression of Id2 mRNA transcripts was high in SCC cell lines (CA9-22 and SCC9) but low in non-cancerous cell lines (Rhek and HOK) by reverse transcription-polymerase chain reaction (RT-PCR). The fold increase in Id2 mRNA transcripts in the SCC cell lines vs. noncancerous cell lines is presented in the lower panel after normalization with β-actin. C, Id2 protein expression was barely detected in normal tissues but abundant in SCC specimens by Western blotting. The fold increase of protein expression is shown in the lower panel. D, quantitative polymerase chain reaction (qPCR) indicated that Id2 mRNA transcript levels in SCC specimens were significantly higher than those in the control tissues. E, the heatmap of the microarray data indicated that Id2, nuclear factor-kappa B (NF-κB), and cyclin D1 transcript levels were higher in the 41 SCC specimens than in the 13 normal tissues.
Id2-induced cell proliferation of both SCC and squamous cells is dependent upon the activity of NF-κB
Since the expression of Id2 was high in SCC specimens, we hypothesized that Id2 may act as an oncogenic protein in SCC. Therefore, we performed cell proliferation studies in CA9-22 by measuring cell numbers (Trypan Blue exclusion), DNA synthesis (3H-thymidine incorporation), and PCNA expression (immunohistochemistry). The results demonstrated that Id2 increased cell numbers, DNA synthesis, and PCNA expression compared to controls (Figure 2A, B, and C). To determine the pathways that mediate Id2-induced cell proliferation in SCC, we focused on the NF-κB pathway because of its role in cellular proliferation. CA9-22 cells were co-transfected with the Id2 cDNA and IκBα M, incubated with 3H-thymidine for 24 h, and harvested for radioactivity determination. Cells transfected with empty vector, IκBα M, or medium alone served as controls. The results demonstrated that IκBα M significantly blocked the cell proliferation induced by Id2 (Figure 2D). Likewise, Id2 induced cell proliferation in Rhek-1A cells in a similar way (data not shown). Real-time PCR demonstrated that transfection of Id2 cDNA increased the level of the Id2 mRNA transcripts in CA9-22 cells, whereas Id2 mRNA transcript levels were low in Rhek-1A immortalized squamous cells (Figure 2E).
Figure 2. Id2 increases the proliferation of CA9-22 cells via NF-κB.

A, 3H-thymidine incorporation increased in a time-dependent manner and was significantly different in Id2- and empty vector-transfected cells (*, P < 0.05). B, cell number increased in a time-dependent manner and was significantly different in Id2- and empty vector-transfected cells (*, P < 0.05). C, proliferating cell nuclear antigen (PCNA) expression was increased in Id2-transfected cells (Id2) compared with enpty vector-transfected cells (Ctrl). D, inhibition of NF-κB activity with IκBα M abrogated cellular DNA synthesis induced by Id2 (*, P < 0.05,). E, qPCR confirmed that the level of the Id2 mRNA transcripts in Id2 cDNA-transfected cells (Id2) was higher than that in empty vector-transfected cells (vec). Ctrl, control; vec, empty vector; med, medium; IκB, IκBα M.
To test whether Id2 induces the transcription of NF-κB, we performed luciferase assays in Id2-transfected CA9-22 cells. The results indicated that Id2 significantly increased the activity of NF-κB reporter (Figure 3A), suggesting that Id2 may act through the NF-κB pathway to stimulate the proliferation of SCC cells. Cyclin D1 is purportedly induced by diverse mitogenic signaling pathways, including the NF-κB pathway[38]. Thus, we similarly measured the luciferase activity of a cyclin D1 reporter in Id2-transfected cells. The data indicated that the activity of cyclin D1 reporter in Id2-transfected cells was up-regulated compared to empty vector or activation protein 2 (AP2), a luciferase assay control (Figure 3A).
Figure 3. Id2 induces the transcription of cyclin D1 via an NF-κB-dependent mechanism in CA9-22 cells.

A, Id2 significantly increased the activity of both NF-κB and cyclin D1, not AP2, luciferase reporter compared with the empty vector control (Ctrl). B, NF-κB p65 significantly strengthened the activity of the Cyclin D1 luciferase reporter by Id2, whereas both PDTC and IκBα M abrogated the activity of the Cyclin D1 luciferase reporter by Id2. Mutation of the NF-κB-binding site in the Cyclin D1 promoter (NF-κBmt) similarly abrogated Cyclin D1 luciferase reporter activity by Id2. C, transfection of Id2 increased the expression of NF-κB in both cytosol and nuclei of CA9-22 cells (upper panel: Id2-transfected cells; lower panel: empty vector-transfected cells).
Id2 increases the transcription of cyclin D1 via an NF-κB-dependent mechanism in SCC cells
The promoter of cyclin D1 has been found to contain an NF-κB-binding site[39]–[41]. To study whether Id2 induces the transcription of cyclin D1 through NF-κB, we modulated NF-κB activity using the NF-κB p65 subunit to enhance activity or using IκBα M or PDTC to inhibit activity, and we also mutated the NF-κB-binding site in the cyclin D1 promoter. We then measured the cyclin D1 luciferase reporter activity in CA9-22 cells. As expected, p65 increased the Id2-induced activity of cyclin D1, whereas both IκBα M and PDTC abrogated the activity of Id2-induced reporter cyclin D1. Likewise, mutations of the NF-κB-binding site in the cyclin D1 promoter abrogated the luciferase activity of cyclin D1 induced by Id2 (Figure 3B). Without transfection of Id2, neither administration of PDTC nor transfection of IκBα M nor mutation of cyclin D1 at the NF-κB-binding site was sufficient to significantly change the luciferase activity of cyclin D1 compared to empty vector in CA9-22 cells (data not shown). Id2 transfection increased NF-κB expression in both cytosol and nuclei (Figure 3C). Consistent with these findings, the expression of Id2, NF-κB, and Cyclin D1 protein was abundant in human head and neck SCC specimens but not in the normal tissues between SCC cell lumps (Figure 4). These results suggest the importance of the Id2/NF-κB/cyclin D1 pathway in the disease mechanism of human SCC.
Figure 4. Id2, NF-κB, and Cyclin D1 are highly expressed in human SCC tissue sections.

A, the expression of Id2 was abundant in SCC cells (brown lumps) but not in non-cancerous cells (spotty in the normal tissue between SCC lumps). B, NF-κB showed a similar expression pattern as Id2 in human head and neck SCC specimens. C, Cyclin D1 expression was observed in the nuclei in a majority of SCC cells but was not detectable in the normal tissue between SCC lumps (between dashed lines). D, a representative immunohistochemistry control for A, B, and C.
Discussion
In this study, we demonstrated that Id2 is highly up-regulated in head and neck SCC tissues compared to normal tissues at both mRNA and protein levels. Enforced expression of Id2 in both normal squamous (Rhek-1A) and cancer (CA9-22) cells increased cell proliferation and oncogenic signals, suggesting that Id2, an Id family member, plays an important role in the carcinogenesis of head and neck SCC.
In terms of the molecular pathway through which Id2 stimulates SCC cell proliferation, we demonstrated that Id2 induced the transcription of NF-κB, a transcription factor that is frequently involved in the oncogenesis of many cells[42],[43] and a target for cancer treatment[44],[45]. NF-κB itself contributes to a variety of cellular responses, including cell proliferation and apoptosis, depending on the stimuli and/or signaling pathways to which it is linked. For example, the epidermal growth factor receptor (EGFR)/NF-κB pathway frequently causes proliferative responses[46], whereas the tumor-necrosis factor receptor (TNFR)/NF-κB pathway results in cell death[47]. We demonstrated that the up-regulation of NF-κB by Id2 was linked to cell proliferation in both immortalized skin squamous cells (Rhek-1A) and SCC cells (CA9-22), as judged by increased DNA synthesis, cell count, and PCNA expression. The link between Id2/NF-κB and cell proliferation is supported by the finding that inhibition of NF-κB activity with IκBα M abrogated cell proliferation. IκBα M has been shown to be a specific inhibitor of NF-κB activity[48],[49]. This study clearly indicated that Id2 stimulates the proliferation of CA9-22 via an NF-κB-dependent mechanism. Examination of NF-κB protein expression in SCC tissue specimens revealed that cells expressing Id2 also express NF-κB, suggesting that Id2 may regulate the expression of NF-κB in vivo. The above data from SCC tissue specimens support the above notion.
Cyclin D1 acts as an oncogene because it is induced by diverse oncogenic and mitogenic signals in many cancers[50],[51]. Furthermore, because it is frequently (>50%) up-regulated in head and neck SCC[52],[53], cyclin D1 has been considered an important biomarker of head and neck SCC[54]. We demonstrated here that Id2 significantly up-regulated the transcription of cyclin D1. Generally, abnormal activity of the cyclin D/retino-blastoma (Rb) pathway represents a hallmark of human tumors[55]. Cyclin D1, together with cyclin- dependent kinases (cdks 4/6), overcomes Rb-mediated control of cell growth, which is a necessary step for tumor formation. Cyclin D1 is a well-established positive regulator of the early cell cycle, controlling the G0/G1, to S phase transition[56] through phosphorylation of Rb. Phosphorylation of Rb induces dissociation of E2F, a checkpoint protein for S phase entry. Thus, increased expression of cyclin D1 contributes to tumor malignancy via the pRb/E2F pathway[57]. The up-regulation of cyclin D1 in CA9-22 by Id2 may represent a molecular model of the carcinogenesis of head and neck SCC. This is consistent with the results of our immunohistochemistry experiments, which show that Id2 and cyclin D1 or NF-κB and cyclin D1 are indeed expressed in SCC cells.
The up-regulation of both NF-κB and cyclin D1 in CA9-22 cells by Id2 led us to examine the relationship between these two molecules. Since the promoter sequence of cyclin D1 contains a putative NF-κB-binding site, we hypothesized that NF-κB is responsible for the transcription of cyclin D1. Indeed, our results show that mutations of the NF-κB-binding site in the cyclin D1 promoter fully abrogated the transcription of cyclin D1. An increase or decrease of the NF-κB activity with p65 and PDTC/IκBα M, respectively, accordingly affects the luciferase activity of cyclin D1. Taken together, we conclude that Id2 regulates the growth and proliferation of SCC cells through the NF-κB/cyclin D1 pathway in vitro. Id2 expression, which is highly up-regulated in SCC tissue specimens, may represent an impetus for high cyclin D1 activity in head and neck SCC and a driving force for SCC malignancy. The proposed pathway of Id2-induced cell proliferation in CA9-22 is summarized in Figure 5. Because of the aberrant up-regulation of Id2 in SCC, the link of Id2 to the NF-κB/cyclin D1-cdks/Rb pathway, targeting Id2 protein with immunologic and molecular biologic approaches may provide a new avenue for treatment of SCC. This would be an important advance, as SCC is resistant to the current therapeutic protocols (chemotherapy and radiation).
Figure 5. Schematic representation of the Id2-induced cellular proliferation pathway in SCC.
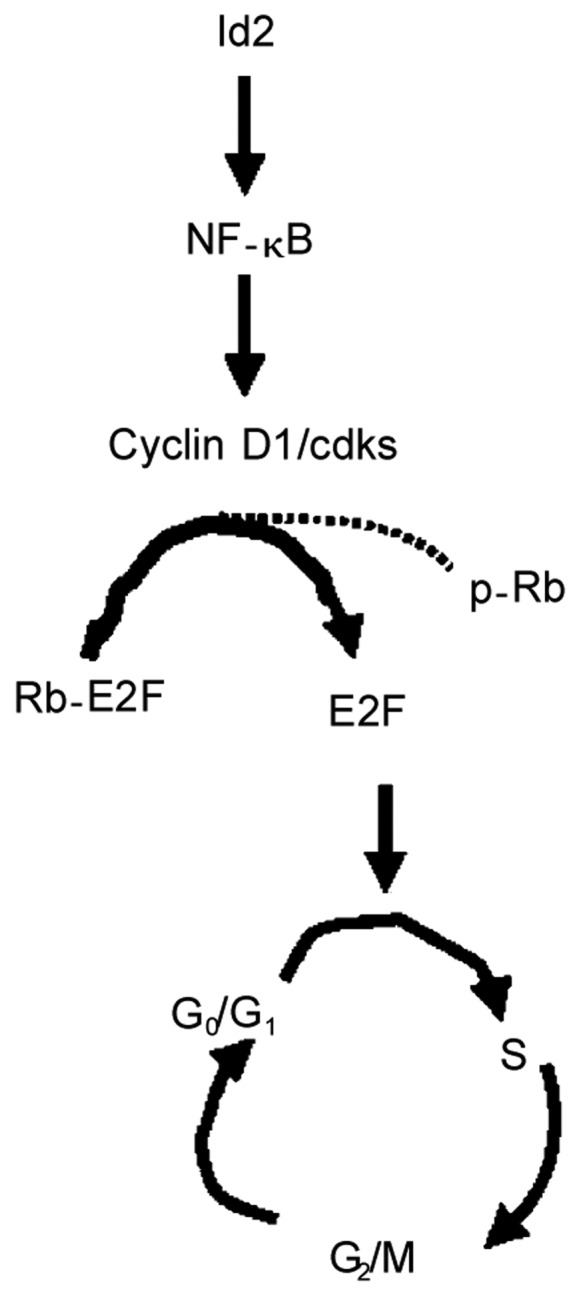
Id2 in head and neck SCC regulates the expression of cyclin D1/cdks via NF-κB. Cyclin D1/cdks increases the progression of head and neck SCC cells by phosphating Rb-E2F complex. This action dissociates Rb-E2f complex and results in the releases of E2F from the Rb-E2F complex. E2F then enters into the nucleus and initiates the progression of cell cycle from G0/G1 to S phase. Cdks, cyclin-dependent kinases (cdks 4/6); Rb, retinoblastoma; p-Rb, phosphorylated Rb; E2F, a transcription factor that drives the transition from G0/G1 to S phase of the cell cycle.
References
- 1.Norton JD, Deed RW, Craggs G, et al. et al. Id helix-loop-helix proteins in cell growth and differentiation. Trends Cell Biol. 1998;8:58–65. [PubMed] [Google Scholar]
- 2.Alani RM, Hasskarl J, Grace M, et al. et al. Immortalization of primary human keratinocytes by the helix-loop-helix protein, Id-1. Proc Natl Acad Sci U S A. 1999;96:9637–9641. doi: 10.1073/pnas.96.17.9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jogi A, Persson P, Grynfeld A, et al. et al. Modulation of basic helix-loop-helix transcription complex formation by Id proteins during neuronal differentiation. J Biol Chem. 2002;277:9118–9126. doi: 10.1074/jbc.M107713200. [DOI] [PubMed] [Google Scholar]
- 4.Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol. 2000;20:429–440. doi: 10.1128/mcb.20.2.429-440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grandori C, Cowley SM, James LP, et al. et al. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Biol. 2000;16:653–699. doi: 10.1146/annurev.cellbio.16.1.653. [DOI] [PubMed] [Google Scholar]
- 6.Baudino TA, Cleveland JL. The Max network gone mad. Mol Cell Biol. 2001;21:691–702. doi: 10.1128/MCB.21.3.691-702.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinsen BJ, Bronner-Fraser M. Neural crest specification regulated by the helix-loop-helix repressor Id2. Science. 1998;281:988–991. doi: 10.1126/science.281.5379.988. [DOI] [PubMed] [Google Scholar]
- 8.Becker-Herman S, Lantner F, Shachar I. Id2 negatively regulates B cell differentiation in the spleen. J Immunol. 2002;168:5507–5513. doi: 10.4049/jimmunol.168.11.5507. [DOI] [PubMed] [Google Scholar]
- 9.Norton JD. ID helix-loop-helix proteins in cell growth, differentiation and tumorigenesis. J Cell Sci. 2000;113:3897–3905. doi: 10.1242/jcs.113.22.3897. [DOI] [PubMed] [Google Scholar]
- 10.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 11.Andres-Barquin PJ, Hernandez MC, Hayes TE, et al. et al. Id genes encoding inhibitors of transcription are expressed during in vitro astrocyte differentiation and in cell lines derived from astrocytic tumors. Cancer Res. 1997;57:215–220. [PubMed] [Google Scholar]
- 12.Deed RW, Bianchi SM, Atherton GT, et al. et al. An immediate early human gene encodes an Id-like helix-loop-helix protein and is regulated by protein kinase C activation in diverse cell types. Oncogene. 1993;8:599–607. [PubMed] [Google Scholar]
- 13.Zhu W, Dahmen J, Bulfone A, et al. et al. Id gene expression during development and molecular cloning of the human Id-1 gene. Brain Res Mol Brain Res. 1995;30:312–326. doi: 10.1016/0169-328x(95)00017-m. [DOI] [PubMed] [Google Scholar]
- 14.Iavarone A, Garg P, Lasorella A, et al. et al. The helix-loop-helix protein Id-2 enhances cell proliferation and binds to the retinoblastoma protein. Genes Dev. 1994;8:1270–1284. doi: 10.1101/gad.8.11.1270. [DOI] [PubMed] [Google Scholar]
- 15.Lasorella A, Iavarone A, Israel MA. Id2 specifically alters regulation of the cell cycle by tumor suppressor proteins. Mol Cell Biol. 1996;16:2570–2578. doi: 10.1128/mcb.16.6.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vandeputte DA, Troost D, Leenstra S, et al. et al. Expression and distribution of id helix-loop-helix proteins in human astrocytic tumors. Glia. 2002;38:329–338. doi: 10.1002/glia.10076. [DOI] [PubMed] [Google Scholar]
- 17.Lasorella A, Boldrini R, Dominici C, et al. et al. Id2 is critical for cellular proliferation and is the oncogenic effector of N-myc in human neuroblastoma. Cancer Res. 2002;62:301–306. [PubMed] [Google Scholar]
- 18.Nishimori H, Sasaki Y, Yoshida K, et al. et al. The Id2 gene is a novel target of transcriptional activation by EWS-ETS fusion proteins in Ewing family tumors. Oncogene. 2002;21:8302–8309. doi: 10.1038/sj.onc.1206025. [DOI] [PubMed] [Google Scholar]
- 19.Wilson JW, Deed RW, Inoue T, et al. et al. Expression of Id helix-loop-helix proteins in colorectal adenocarcinoma correlates with p53 expression and mitotic index. Cancer Res. 2001;61:8803–8810. [PubMed] [Google Scholar]
- 20.Rockman SP, Currie SA, Ciavarella M, et al. et al. Id2 is a target of the beta-catenin/T cell factor pathway in colon carcinoma. J Biol Chem. 2001;276:45113–45119. doi: 10.1074/jbc.M107742200. [DOI] [PubMed] [Google Scholar]
- 21.Wice BM, Gordon JI. Forced expression of Id-1 in the adult mouse small intestinal epithelium is associated with development of adenomas. J Biol Chem. 1998;273:25310–25319. doi: 10.1074/jbc.273.39.25310. [DOI] [PubMed] [Google Scholar]
- 22.Lin J, Guan Z, Wang C, et al. et al. Id1 regulates the survival of HNSCC via the NF-kappaB/survivin and PI3K/Akt signaling pathways. Clin Cancer Res. 2010;16:77–87. doi: 10.1158/1078-0432.CCR-08-2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lasorella A, Noseda M, Beyna M, et al. et al. Id2 is a retinoblastoma protein target and mediates signalling by Myc oncoproteins. Nature. 2000;407:592–598. doi: 10.1038/35036504. [DOI] [PubMed] [Google Scholar]
- 24.Lyden D, Young AZ, Zagzag D, et al. et al. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature. 1999;401:670–677. doi: 10.1038/44334. [DOI] [PubMed] [Google Scholar]
- 25.Langlands K, Down GA, Kealey T. Id proteins are dynamically expressed in normal epidermis and dysregulated in squamous cell carcinoma. Cancer Res. 2000;60:5929–5933. [PubMed] [Google Scholar]
- 26.Kato T, Duffey DC, Ondrey FG, et al. et al. Cisplatin and radiation sensitivity in human head and neck squamous carcinomas are independently modulated by glutathione and trancription factor NF-kappa B. Head Neck. 2000;22:748–759. doi: 10.1002/1097-0347(200012)22:8<748::aid-hed2>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 27.Rhim JS, Jay G, Arnstein P, et al. et al. Neoplastic transformation of human epidermal keratinocytes by AD12-SV40 and Kirsten sarcoma viruses. Science. 1985;227:1250–1252. doi: 10.1126/science.2579430. [DOI] [PubMed] [Google Scholar]
- 28.Franzoso G, Bours V, Azarenko V, et al. et al. The oncoprotein Bcl-3 can facilitate NF-kappa B-mediated transactivation by removing inhibiting p50 homodimers from select kappa B sites. EMBO J. 1993;12:3893–3901. doi: 10.1002/j.1460-2075.1993.tb06067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Israel DI, Kaufman RJ. Highly inducible expression from vectors containing multiple GRE's in CHO cells overexpressing the glucocorticoid receptor. Nucleic Acids Res. 1989;17:4589–4604. doi: 10.1093/nar/17.12.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bours V, Franzoso G, Azarenko V, et al. et al. The oncoprotein Bcl-3 directly transactivates through kappa B motifs via association with DNA-binding p50B homodimers. Cell. 1993;72:29–739. doi: 10.1016/0092-8674(93)90401-b. [DOI] [PubMed] [Google Scholar]
- 31.Bressler P, Brown K, Timmer W, et al. et al. Mutational analysis of the p50 subunit of NF-kappa B and inhibition of NF-kappa B activity by trans-dominant p50 mutants. J Virol. 1993;67:288–293. doi: 10.1128/jvi.67.1.288-293.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brockman JA, Scherer DC, McKinsey TA, et al. et al. Coupling of a signal response domain in I kappa B alpha to multiple pathways for NF-kappa B activation. Mol Cell Biol. 1995;15:2809–2818. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ziegler-Heitbrock HW, Sternsdorf T, Liese J, et al. et al. Pyrrolidine dithiocarbamate inhibits NF-kappa B mobilization and TNF production in human monocytes. J Immunol. 1993;151:6986–6993. [PubMed] [Google Scholar]
- 34.Albanese C, Johnson J, Watanabe G, et al. et al. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem. 1995;270:23589–23597. doi: 10.1074/jbc.270.40.23589. [DOI] [PubMed] [Google Scholar]
- 35.Kanno T, Brown K, Siebenlist U. Evidence in support of a role for human T-cell leukemia virus type I Tax in activating NF-kappa B via stimulation of signaling pathways. J Biol Chem. 1995;270:11745–11748. doi: 10.1074/jbc.270.20.11745. [DOI] [PubMed] [Google Scholar]
- 36.Lin J, Tsuprun V, Kawano H, et al. et al. Characterization of mucins in human middle ear and Eustachian tube. Am J Physiol. 2001;280:L1157–L1167. doi: 10.1152/ajplung.2001.280.6.L1157. [DOI] [PubMed] [Google Scholar]
- 37.Ginos MA, Page GP, Michalowicz BS, et al. et al. Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer Res. 2004;64:55–63. doi: 10.1158/0008-5472.can-03-2144. [DOI] [PubMed] [Google Scholar]
- 38.Guttridge DC, Albanese C, Reuther JY, et al. et al. NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol. 1999;19:5785–5799. doi: 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joyce D, Bouzahzah B, Fu M, et al. et al. Integration of Rac-dependent regulation of cyclin D1 transcription through a nuclear factor-kappaB-dependent pathway. J Biol Chem. 1999;274:25245–25249. doi: 10.1074/jbc.274.36.25245. [DOI] [PubMed] [Google Scholar]
- 40.Henry DO, Moskalenko SA, Kaur KJ, et al. et al. Ral GTPases contribute to regulation of cyclin D1 through activation of NF-kappaB. Mol Cell Biol. 2000;20:8084–8092. doi: 10.1128/mcb.20.21.8084-8092.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westerheide SD, Mayo MW, Anest V, et al. et al. The putative oncoprotein Bcl-3 induces cyclin D1 to stimulate G(1) transition. Mol Cell Biol. 2001;21:8428–8436. doi: 10.1128/MCB.21.24.8428-8436.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharma HW, Narayanan R. The NF-kappaB transcription factor in oncogenesis. Anticancer Res. 1996;16:589–596. [PubMed] [Google Scholar]
- 43.Duffey DC, Chen Z, Dong G, et al. et al. Expression of a dominant-negative mutant inhibitor-kappaBalpha of nuclear factor-kappaB in human head and neck squamous cell carcinoma inhibits survival, proinflammatory cytokine expression, and tumor growth in vivo. Cancer Res. 1999;59:3468–3474. [PubMed] [Google Scholar]
- 44.Darnell JEJ. Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 45.Orlowski RZ, Baldwin ASJ. NF-kappaB as a therapeutic target in cancer. Trends Mol Med. 2002;8:385–389. doi: 10.1016/s1471-4914(02)02375-4. [DOI] [PubMed] [Google Scholar]
- 46.Biswas DK, Cruz AP, Gansberger E, et al. et al. Epidermal growth factor-induced nuclear factor kappa B activation: a major pathway of cell-cycle progression in estrogen-receptor negative breast cancer cells. Proc Natl Acad Sci U S A. 2000;97:8542–8547. doi: 10.1073/pnas.97.15.8542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haridas V, Darnay BG, Natarajan K, et al. et al. Overexpression of the p80 TNF receptor leads to TNF-dependent apoptosis, nuclear factor-kappa B activation, and c-Jun kinase activation. J Immunol. 1998;160:3152–3162. [PubMed] [Google Scholar]
- 48.Henkel T, Machleidt T, Alkalay I, et al. et al. Rapid proteolysis of I kappa B-alpha is necessary for activation of transcription factor NF-kappa B. Nature. 1993;365:182–185. doi: 10.1038/365182a0. [DOI] [PubMed] [Google Scholar]
- 49.Brown K, Gerstberger S, Carlson L, et al. et al. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 50.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 51.Pestell RG, Albanese C, Reutens AT, et al. et al. The cyclins and cyclin-dependent kinase inhibitors in hormonal regulation of proliferation and differentiation. Endocr Rev. 1999;20:501–534. doi: 10.1210/edrv.20.4.0373. [DOI] [PubMed] [Google Scholar]
- 52.Jares P, Fernandez PL, Campo E, et al. et al. PRAD-1/cyclin D1 gene amplification correlates with messenger RNA over-expression and tumor progression in human laryngeal carcinomas. Cancer Res. 1994;54:4813–4817. [PubMed] [Google Scholar]
- 53.Quon H, Liu FF, Cummings BJ. Potential molecular prognostic markers in head and neck squamous cell carcinomas. Head Neck. 2001;23:147–159. doi: 10.1002/1097-0347(200102)23:2<147::aid-hed1010>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 54.Namazie A, Alavi S, Olopade OI, et al. et al. Cyclin D1 amplification and p16(MTS1/CDK4I) deletion correlate with poor prognosis in head and neck tumors. Laryngoscope. 2002;112:472–481. doi: 10.1097/00005537-200203000-00013. [DOI] [PubMed] [Google Scholar]
- 55.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 56.Sherr CJ. Mammalian G1 cyclins. Cell. 1993;73:1059–1065. doi: 10.1016/0092-8674(93)90636-5. [DOI] [PubMed] [Google Scholar]
- 57.Tashiro E, Maruki H, Minato Y, et al. et al. Overexpression of cyclin D1 contributes to malignancy by up-regulation of fibroblast growth factor receptor 1 via the pRB/E2F pathway. Cancer Res. 2003;63:424–431. [PubMed] [Google Scholar]