

Abstract
Although gene therapy was regarded as a promising approach for glioma treatment, its therapeutic efficacy was often disappointing because of the lack of efficient drug delivery systems. Mesenchymal stem cells (MSCs) have been reported to have a tropism for brain tumors and thus could be used as delivery vehicles for glioma therapy. Therefore, in this study, we attempted to treat glioma by using MSCs as a vehicle for delivering replication-competent adenovirus. We firstly compared the infectivity of type 3, type 5, and type 35 fiber-modified adenoviruses in MSCs. We also determined suitable adenovirus titer in vitro and then used this titer to analyze the ability of MSCs to deliver replication-competent adenovirus into glioma in vivo. Our results indicated that type 35 fiber-modified adenovirus showed higher infectivity than did naked type 3 or type 5 fiber-modified adenovirus. MSCs carrying replication-competent adenovirus significantly inhibited tumor growth in vivo compared with other control groups. In conclusion, MSCs are an effective vehicle that can successfully transport replication-competent adenovirus into glioma, making it a potential therapeutic strategy for treating malignant glioma.
Keywords: Mesenchymal stem cell, replication-competent adenovirus, gene therapy, glioma
Glioma, the most common brain tumor, is insensitive to the currently available radiotherapy and chemotherapy. In addition, glioma that typically infiltrates the brain parenchyma cannot be excised surgically, leading to short survival and poor prognosis. In search of an effective approach for treating glioma, researchers adopted several different therapeutic methods, including adenoviral gene therapy. Adenovirus, the most frequently used vehicle in gene therapy, not only kills target cells by self-replicating, but acts as a vehicle to carry and express various anticancer genes. Thus, adenovirus is regarded as a promising antitumor therapy. However, for highly invasive gliomas, effective delivery of therapeutic genes is a great challenge. In previous studies, investigators directly injected adenovirus into encephalic tumor tissues. However, the replication-competent adenovirus only infected tumor tissues surrounding the injection site and did not infect distant cancer tissues, leading to undesirable clinical efficacy[1]-[3]. Type 5 adenovirus vehicle infects tumor cells by binding with coxsackie and adenovirus receptor (CAR) expressed on the surface of most cells. Thus, the infection of cancer cells by adenovirus was found to be non-selective[4]. If the adenovirus was rendered by a tropism, the anti-cancer efficacy would be further improved.
Mesenchymal stem cells (MSCs) are pluripotent stem cells recently found to have a tropism for tumor cells; however, the underlying mechanism is still elusive. The tropism is probably associated with the chemotaxis induced by platelet-derived growth factor (PDGF), epidermal growth factor (EGF), and stromal-derived factor-1 in tumor tissues[5]. In 2005, Nakamizo et al.[5] performed gene therapy against brain mesenchymoma using the tropism of MSCs and found that the survival of animals was significantly increased. Subsequently, Loebinger et al.[6] used MSCs expressing tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) to conduct magnetic target therapy against lung metastases and gained desirable clinical efficacy. These studies manifested the prospect of applying MSCs in gene therapy.
In this investigation, we aimed to deliver replication-competent adenovirus into glioma tissues by taking advantage of the tumor tropism of MSCs. We sought to determine if MSCs can act as an effective vehicle and to provide novel evidence for magnetic target gene therapy for malignant tumors.
Materials and Methods
Cell lines and agents
U87MG and U343 human glioma cell lines were purchased from ATCC and cultured in DMEM solution containing 10% fetal bovine serum (GIBCO). BALB/c nude mice were purchased from Shanghai Slack Experimental Animal Co., Ltd [Animal qualified No. SCXK(Shanghai)2003-00031]. Fluorescence quantitative polymerase chain reaction (qPCR) detection kits were purchased from Shanghai Shinegene Molecular Biotechnology Co., Ltd. Fluorescent marker CMTPX (CellTracker™ Red) was purchased from Sigma-Aldrich Co., Ltd.
Tumor chemotaxis assay of MSCs
Fluorescent labeling marker CMTPX (5 mmol/L) was added to MSC culture solution and cultured for 30 min. Cells were then digested with trypsase and centrifuged, and the cell pellet was washed twice with PBS. Fluorescence was observed under a fluorescence microscope. The treated cells were suspended in PBS at a concentration of 5 × 104 cells/µL. Then, 4 µL of cell suspension was extracted using a microsyringe and incubated in the left hemisphere of tumor-bearing mice using stereotaxic apparatus. Two weeks later, the nude mice were euthanized, and frozen sections of brain tissues were analyzed for fluorescence under a fluorescence microscope. Tumor chemotactic capability of MSCs was confirmed by comparison with fluorescence expression of MSC injection site and tumor tissue.
Infection rate of adenovirus vehicles in MSCs
Non-replicating adenoviruses dl-LacZ, dl-3-LacZ, and dl-35-LacZ, as well as replication-competent adenovirus Ad-35 were established according to previous protocols[7],[8]. Non-replicating adenovirus dl-324 was purchased from the Cancer Institute of Yonsei University of South Korea. dl-LacZ was established by inserting LacZ into the adenovirus genome E3 locus. dl-3-LacZ and dl-35-LacZ were established by replacing the dl-LacZ fiber gene with type 3 or type 35 adenovirus fiber genes. The Ad-35 adenovirus was generated by replacing the original type 5 fiber gene with the type 35 adenovirus fiber gene (Figure 1).
Figure 1. Schematic representation of dl-LacZ, dl-3-LacZ, dl-35-LacZ, and Ad-35.

dl-LacZ, dl-3-LacZ, and dl-35-LacZ were E1-deleted replication-incompetent adenoviral vectors. Wild-type fiber in dl-3-LacZ and dl-35-LacZ were replaced by type 3 and 35 adenovirus fiber, respectively. Ad-35 is a replication-competent adenovirus, and the fiber was also replaced by type 35 adenovirus fiber.
dl-LacZ, dl-3-LacZ, and dl-35-LacZ were injected at titers of 1, 2, 5, 10, or 20 MOI into respective wells of MSC cultures in 6-well plates and then incubated for 48 h. The suspension was discarded and the cell pellet was washed twice using PBS, stained with X-gal, fixed in methanol solution, and finally observed for staining status under a light microscope to determine the rate of infection.
Cytotoxicity tests in MSCs
MSCs were cultured in 48-well culture plates. Adenoviruses were supplemented into culture solution at gradient concentrations and then incubated for 10 days. After discarding the suspension, 200 µL of MTT solution was added to culture plates and the cells were cultured for 4 h. After discarding the suspension again, 500 µL of methanol was added to each well and the adenovirus particles were measured by enzyme-linked immunosorbent assay at an absorbance of 540 nm.
Viral particle replication in MSCs
MSCs were cultured in 48-well culture plates. Adenoviruses were supplemented into culture solution at 1 and 5 MOI and then incubated for 10, 12, and 14 days, respectively. The obtained cells or cancer tissues were lysed, and DNA was extracted by using DNA extraction kits (Tianjin Hao Yang Biological Manufacturer Co., Ltd). Equal quantities of extracted DNA (50 ng) were quantitatively analyzed and measured using fluorescence qPCR. The adenovirus E1 gene segment was used as a target to design PCR primers using the real-time fluorescence qPCR system (Applied Biosystems, USA). Experiment was performed according to the methods previously reported[4].
In vivo antitumor effect of replication-competent adenovirus delivered by MSCs
U87MG or U343 cells were lysed with 0.125% trypsin and centrifuged. The supernatant was discarded. After being washed twice with PBS, cells were counted and the suspension solution was adjusted to a concentration of 2 × 104 cells/µL in PBS. A 4-µL aliquot of cell suspension was injected into the right brain hemisphere of nude mice with a syringe. After incubation, the nude mice were fed in a protective system under stable temperature, stable humidity, and sterile conditions. One week later, nude mice were randomly divided into four groups and underwent MSC injection. Ad-35 with a titer of 5 MOI was supplemented into MSC culture solution and cultured for 2 h, after which the cells were digested with trypsin. The mixture was centrifuged and the supernatant was discarded. After being washed twice with PBS, cells were suspended in PBS to a concentration of 5 × 104 cells/µL The cell suspension (4 µL) was injected into the left brain hemisphere of tumor-bearing nude mice by using a syringe. The mice in the control group were injected with PBS, non-viral infected MSCs, or naked Ad-35 (1 × 106 VP) into the left brain hemisphere. After 21 days of breeding, all nude mice were euthanized. Tumor volume was calculated according to the formula V = 1/2 ab2, with a representing the longest diameter and b representing the shortest diameter. The sizes of the brain tumors were measured. Three mice from each group were randomly selected for DNA extraction and fluorescence qPCR detection. The survival time was calculated from tumor cell implantation to death.
Statistical analyses
All data are expressed as mean ± standard error. The data among groups were compared with two-tailed t-test. Survival analysis was performed with the Kaplan-Meier method. The results were compared by log-rank test. SPSS statistical software was used for data analysis (SPSS 13.0 software, SPSS, Chicago, IL). A P value < 0.05 was considered significant.
Results
Phenotype analysis of MSCs
Human MSCs, purchased from the Cell Line Bank of Yonsei University of South Korea, were cultured in DMEM solution containing 10% FBS. The phenotype of MSCs was analyzed using flow cytometry. The results revealed that the MSCs highly expressed MSC-related antigens CD29, CD44, CD73, CD90, and CD105, but showed little to no expression of hematopoietic cell- or endothelial cell-surface antigens CD14, CD34, CD45, CD31, and CD106 (Figure 2).
Figure 2. FACS analysis of mesenchymal stem cells (MSCs) used in this study.

These cells highly express MSC-associated markers CD29, CD44, CD73, CD90, and CD105, but exhibit weak or no expression of non-MSC-associated markers CD14, CD31, CD34, CD45, and CD106.
Migratory capacity of MSCs in glioma orthotopic mouse models
MSCs were labeled with CMTPX. A high fluorescent expression rate was confirmed by fluorescence microscopy and flow cytometry (Figures 3A and 3B). MSCs with fluorescence labeling were implanted in the opposite brain hemisphere of the tumor site in glioma orthotopic mouse models. Three weeks later, the brain tissues were collected for frozen section and observed for fluorescence expression level under a fluorescence microscope. As shown in Figure 3C, a small amount of red fluorescence was expressed at the MSC injection site, whereas a substantial amount of red fluorescence was observed on the opposite side of the brain (non-injection site), suggesting that MSCs had good chemotaxis and migratory capacity towards tumor sites.
Figure 3. Migratory capacity of MSCs in vivo.

A, MSCs co-incubated with the CMTPX probe for 30 min and observed by fluorescence microscopy. Most MSCs are positive for red fluorescence. B, labeling efficiency was confirmed by FACS analysis. Over 95% of MSCs are positive for red fluorescence (CMTPX). C, tumor cells were injected into the right brain hemisphere of nude mice, and CMTPX-labeled MSCs were injected into the left brain hemisphere 1 week after tumor cell injection. Two weeks after MSC injection, brain tissues were harvested, snap-frozen, and sectioned on 10-µm thickness. The CMTPX expression was confirmed by fluorescence microscopy. More red-positive cells are observed in the tumor tissue area than in the MSC-injected site.
Comparison on the infection rates of dl-LacZ, dl-3-LacZ, and dl-35-LacZ in MSCs
MSCs were infected with various titers of dl-LacZ, dl-3-LacZ, or dl-35-LacZ, and the observed infection rate was significantly different between groups. As shown in Figure 4, more X-gal-positive spots were observed in the microscopic field in dl-35-LacZ(20 MOI) group (23.7 ± 2.8) than in dl-LacZ group (1.5 ± 0.6) and dl-3-LacZ group (14.5 ± 2.4), indicating that the infectious efficiency of dl-35-LacZ was significantly higher than those of dl-LacZ and dl-3-LacZ. (dl-35-LacZ vs. dl-3-LacZ, P = 0.012; dl-35-LacZ vs. dl-LacZ, P < 0.001). These results indicated that the infection capacity of adenovirus in MSCs depended upon the fiber subtype of the viral particle surface. Furthermore, the adenovirus with type 35 fiber was superior to those with type 5 fiber.
Figure 4. Comparison of the infectivity of dl-LacZ, dl-3-LacZ, and dl-35-LacZ in MSCs by X-pal staining.
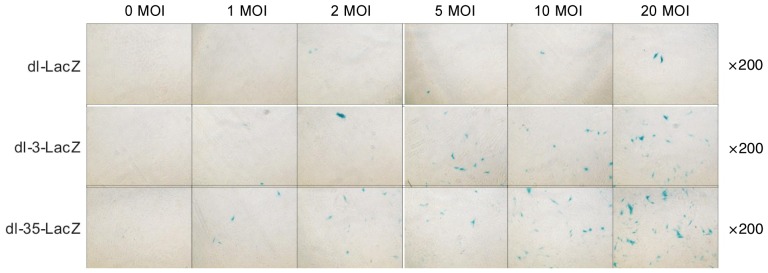
MSCs were infected with different doses of dl-LacZ, dl-3-LacZ, and dl-35-LacZ. After 48 h of infection, X-gal staining was performed. More X-gal-positive spots are observed in the microscopic field in dl-35-LacZ (20 MOI) group (23.7 ± 2.8) than in dl-LacZ group (1.5 ± 0.6) and dl-3-LacZ group (14.5 ± 2.4), indicating that the infectious efficiency of dl-35-LacZ is significantly higher than those of dl-LacZ and dl-3-LacZ (dl-35-LacZ vs. dl-3-LacZ, P = 0.012; dl-35-LacZ vs. dl-LacZ, P < 0.001; dl-3-LacZ vs. dl-LacZ, P < 0.001).
Cytotoxicity and replication of replication-competent adenovirus Ad-35 in MSCs
Previous studies hinted that it took MSCs approximately 10 days to migrate to encephalic tumors from other sites. Thus, we used various titers of Ad-35 to infect MSCs and performed MTT 10 days after infection. The results indicated that almost all (over 95%) MSCs died 10 days after infection with over 10 MOI of adenovirus, whereas 40% survived after infection with 10 MOI and approximately 90% were alive after infection with 1 MOI (Figure 5A). Hence, we selected the titers of 1 MOI and 5 MOI to perform cytotoxicity and replication tests.
Figure 5. Cytotoxicity and replication of replication-competent adenovirus in MSCs in vitro.

A, cytotoxicity of Ad-35 in MSCs was determined by using MTT assay. B, cytotoxicity of 1 MOI and 5 MOI of Ad-35 in MSCs with time-dependent manner was detected by using MTT assay. C, qPCR analysis of replication of Ad-35 in MSCs. Results indicate that more viral particles were produced by MSCs that were infected with 5 MOI of Ad-35 than those by MSCs infected with 1 MOI of Ad-35.
The results revealed that 80% and 70% of MSCs survived 14 days after infection with 1 MOI and 5 MOI of adenovirus, respectively (Figure 5B). More MSCs died over time after infection with 5 MOI of adenovirus compared to 1 MOI. The fluorescence qPCR assay showed a slight increase in the number of viral particles 10 to 14 days after infection with 1 MOI of adenovirus, whereas the amount of viral particles significantly increased after infection with 5 MOI (on the 10th day, 1.6 × 104 VP vs. 4 × 104 VP, P = 0.036; on the 14th day, 5.3 × 104 VP vs. 12.6 × 104 VP, P = 0.049). Especially, on the 12th day after infection, the number of viral particles in the 5 MOI group (20 × 104 VP) was approximately 4 times that of the 1 MOI group (5.2 × 104 VP) (P = 0.001). Moreover, the number of viral particles in the 5 MOI infection group detected on the 14th day was smaller than that observed on the 12th day, and this was probably due to reduced replication capacity as a result of the death of most MSCs. To maximize the clinical efficacy, we chose 5 MOI as the infection viral titer in MSCs.
In vivo antitumor effect of replication-competent adenovirus delivered by MSCs
U87MG and U343 glioma orthotopic models were established in the right brain hemisphere of nude mice (Figure 6). MSCs infected with 5 MOI of Ad-35 were implanted into the left brain hemisphere 1 week later. The brain tissues were collected from nude mice and the tumor size was measured 2 weeks later. As indicated in Figure 7, the injection of PBS, Ad-35, or MSCs failed to suppress the growth of brain tumor, whereas the injection of Ad-35-infected MSCs significantly reduced the tumor size compared to the PBS, Ad-35, or MSCs alone treated groups. The median survival time of the mice implanted with Ad-35-infected MSCs(35 days) was significantly prolonged compared with that of the mice in PBS (30 days), Ad-35 (30 days), or MSC (28 days) alone treated groups (P < 0.05).
Figure 6. An orthotopic human glioma model in nude mice.

Brain tissues were harvested 1 week after tumor cell implantation, embedded in paraffin, and sectioned at a thickness of 4 µm for hematoxylin and eosin (HE) staining. Visible tumor area was observed in the right brain hemisphere of nude mice.
Figure 7. In vivo antitumor effect of an replication-competent adenovirus delivered by MSCs.

A, tumor volume in the U87MG orthotopic models was measured 2 weeks after treatment. Ad-35 delivered by MSCs significantly inhibited U87MG tumor growth compared to other treatments (PBS vs. MSCs + Ad-35, P = 0.001; Ad-35 vs. MSCs + Ad-35, P = 0.005; MSCs vs. MSCs + Ad-35, P < 0.001). B, survival of U87MG tumor-bearing mice after treatment. Results indicate that median survival time was significantly prolonged in mice that underwent MSC-based delivery of Ad-35 compared to mice that underwent other treatments (35 days vs. 30, 30, and 28 days, all P < 0.05). C, survival of U343 tumor-bearing mice after treatment. Results indicate that Ad-35 delivered by MSCs significantly prolonged median survival time compared to PBS, Ad-35, and MSCs alone (29 days vs. 23, 22, and 20 days, all P < 0.05).
To further identify the underlying mechanism of the observed antitumor effect, encephalic brain tumors were collected for DNA extraction and fluorescence qPCR. As shown in Figure 8, a larger amount of adenovirus particles were observed in the nude mice injected with Ad-35-infected MSCs (6400 VP) compared to PBS (94 VP), Ad-35 (128 VP) or MSC (88 VP) alone treated groups, indicating that replication-competent adenovirus had infected tumor cells and eventually inhibited tumor growth.
Figure 8. Quantification of viral particles in tumor tissue from mice treated with PBS, Ad-35, MSC, or MSC + Ad-35.

DNA was extracted from tumor tissue and harvested 3 weeks after treatment. Fluorescence qPCR analysis determined that the number of viral particles was significantly higher in MSCs + Ad-35 group than in PBS, Ad-35, and MSCs groups (all P < 0.001).
Discussion
The tropism of MSCs to tumors has been identified in only a few cancer models. At present, the glioma model is the most frequently used in clinical practice. Recent studies found that MSCs implanted in orthotopic ovarian cancer models via intraperitoneal injection could migrate into tumor tissues[9]. The migratory capacity of MSCs possibly ascribes to the extracellular signal from cancer tissues. MSCs are known to participate in the reconstruction and formation of extracellular stroma. Cancer tissues resemble traumatic or inflammatory tissues, which accumulate a substantial amount of inflammatory transmitters or relevant chemotactic factors, mistakenly sending a “traumatic repair signal” to MSCs. This enables MSCs to migrate into tumor sites and play a role in the construction of the extracellular matrix in tumor tissues[10].
Although cellular delivery of adenovirus is regarded as a viable approach, it is rarely applied in clinical cases due to its multiple limitations. The cell type selected in the clinical studies is the key factor determining the clinical efficacy. Previous studies used mouse fibroblasts as retrovirus delivery vectors to treat glioma, but the clinical efficacy was undesirable because of low viral transmission efficiency[11]. Li et al. [12] chose various types of cells as viral vectors and compared their efficacy on glioma. The results indicated that the viral transmission efficiency of the neural precursor cells was the highest. Cancer cells and T lymph cells have also been applied in gene therapy as delivery vehicles[13],[14]. Nevertheless, the application of most cells are limited due to scarce cell sources or low migratory efficiency. MSCs became a promising vehicle in gene therapy and gained increasing attention because of abundant sources, ease of separation and culture, and high migratory capacity.
Replication-competent adenovirus, which is widely applied in gene therapy, not only serves as a vehicle for antitumor genes in tumor cells but also kills target cells by self-replicating within tumor cells. Hence, the clinical efficacy of replication-competent adenovirus is superior to non-replicating adenovirus. MSCs differ from other vehicles, including retrovirus or plasmid, in the delivery of replication-competent adenovirus; replication-competent adenovirus can lyse MSCs after multiple replication cycles. Thus, this technique is constrained by migration time and initial viral titer. If the titer of adenovirus is excessively high or the migration time is longer than the MSC splitting time after viral replication, MSCs might die out before arriving at tumor tissues[9]. Previous related investigations found that MSCs required two weeks to migrate to encephalic glioma from other sites[5],[15]. According to these outcomes, we used two weeks as the baseline migration time in this study and adjusted initial infection titer of replication-competent adenovirus. Our results indicated that both 1 MOI and 5 MOI were viable titers. However, more adenoviruses were transmitted into tumor tissues at 5 MOI compared to 1 MOI. To enable a larger amount of replication-competent adenoviruses to arrive at tumor tissues, we selected 5 MOI as the applied viral titer in our experiments. In addition, we discovered that type 5 adenovirus vehicle had a low infection rate in MSCs, but the infection rate increased nearly 20-fold with the type 35 adenovirus fiber, which reduced the toxicity of adenoviral vectors to MSCs.
As members of the stem cell family, MSCs have great differentiation capacity. Therefore, in vivo injection of MSCs might provoke certain risks, including forming novel tumors[16] or promoting the growth of original tumors[17]. Alternative gene delivery vehicles, such as retrovirus, parvovirus, plasmid, and others, would not induce MSC death, which might increase such risks. In contrast, replication-competent adenovirus self-replicates within MSCs, which not only produce and release a larger amount of virus into tumor tissues, but lyse MSCs after multiple replication cycles. Hence, compared with other vectors, MSC-based delivery of replication-competent adenovirus in vivo significantly lowers the risks mentioned above.
To sum up, we used the tropism of MSCs towards tumors to transmit replication-competent adenovirus inside tumor tissues. The results validate that MSCs are an efficacious vehicle in gene therapy that successfully delivers replication-competent adenovirus into tumor sites and causes antitumor effects.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (No. 81160275) and the Claison tumor biotherapy research special funds (2011-14).
References
- 1.Lang FF, Bruner JM, Fuller GN, et al. et al. Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: biological and clinical results. J Clin Oncol. 2003;21:2508–2518. doi: 10.1200/JCO.2003.21.13.2508. [DOI] [PubMed] [Google Scholar]
- 2.Lichtenstein DL, Wold WS. Experimental infections of humans with wild-type adenoviruses and with replication-competent adenovirus vectors: replication, safety, and transmission. Cancer Gene Ther. 2004;11:819–829. doi: 10.1038/sj.cgt.7700765. [DOI] [PubMed] [Google Scholar]
- 3.Sandmair AM, Loimas S, Puranen P, et al. et al. Thymidine kinase gene therapy for human malignant glioma, using replication-deficient retroviruses or adenoviruses. Hum Gene Ther. 2000;11:2197–2205. doi: 10.1089/104303400750035726. [DOI] [PubMed] [Google Scholar]
- 4.Yun CO, Yoon AR, Yoo JY, et al. et al. Coxsackie and adenovirus receptor binding ablation reduces adenovirus liver tropism and toxicity. Hum Gene Ther. 2005;16:248–261. doi: 10.1089/hum.2005.16.248. [DOI] [PubMed] [Google Scholar]
- 5.Nakamizo A, Marini F, Amano T, et al. et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65:3307–3318. doi: 10.1158/0008-5472.CAN-04-1874. [DOI] [PubMed] [Google Scholar]
- 6.Loebinger MR, Eddaoudi A, Davies D, et al. et al. Mesenchymal stem cell delivery of trail can eliminate metastatic cancer. Cancer Res. 2009;69:4134–4142. doi: 10.1158/0008-5472.CAN-08-4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He X, Liu J, Yang C, et al. et al. 5/35 fiber-modified conditionally replicative adenovirus armed with p53 shows increased tumor-suppressing capacity to breast cancer cells. Hum Gene Ther. 2011;22:283–292. doi: 10.1089/hum.2010.058. [DOI] [PubMed] [Google Scholar]
- 8.Murakami M, Ugai H, Wang M, et al. et al. An adenoviral vector expressing human adenovirus 5 and 3 fiber proteins for targeting heterogeneous cell populations. Virology. 2010;407:196–205. doi: 10.1016/j.virol.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 9.Komarova S, Kawakami Y, Stoff-Khalili MA, et al. et al. Mesenchymal progenitor cells as cellular vehicles for delivery of oncolytic adenoviruses. Mol Cancer Ther. 2006;5:755–766. doi: 10.1158/1535-7163.MCT-05-0334. [DOI] [PubMed] [Google Scholar]
- 10.Vaananen HK. Mesenchymal stem cells. Ann Med. 2005;37:469–479. doi: 10.1080/07853890500371957. [DOI] [PubMed] [Google Scholar]
- 11.Ram Z, Culver KW, Oshiro EM, et al. et al. Therapy of malignant brain tumors by intratumoral implantation of retroviral vector-producing cells. Nat Med. 1997;3:1354–1361. doi: 10.1038/nm1297-1354. [DOI] [PubMed] [Google Scholar]
- 12.Li S, Tokuyama T, Yamamoto J, et al. et al. Bystander effect-mediated gene therapy of gliomas using genetically engineered neural stem cells. Cancer Gene Ther. 2005;12:600–607. doi: 10.1038/sj.cgt.7700826. [DOI] [PubMed] [Google Scholar]
- 13.Namba H, Tagawa M, Miyagawa T, et al. et al. Treatment of rat experimental brain tumors by herpes simplex virus thymidine kinase gene-transduced allogeneic tumor cells and ganciclovir. Cancer Gene Ther. 2000;7:947–953. doi: 10.1038/sj.cgt.7700172. [DOI] [PubMed] [Google Scholar]
- 14.Yotnda P, Savoldo B, Charlet-Berguerand N, et al. et al. Targeted delivery of adenoviral vectors by cytotoxic T cells. Blood. 2004;104:2272–2280. doi: 10.1182/blood-2003-11-3803. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura K, Ito Y, Kawano Y, et al. et al. Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther. 2004;11:1155–1164. doi: 10.1038/sj.gt.3302276. [DOI] [PubMed] [Google Scholar]
- 16.Hall B, Dembinski J, Sasser AK, et al. et al. Mesenchymal stem cells in cancer: tumor-associated fibroblasts and cell-based delivery vehicles. Int J Hematol. 2007;86:8–16. doi: 10.1532/IJH97.06230. [DOI] [PubMed] [Google Scholar]
- 17.Karnoub AE, Dash AB, Vo AP, et al. et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]