

Abstract
Enormous mechanistic insight has been gained by studying the behavior of single molecules. The same approaches used to study proteins in isolation are now being leveraged to examine the changes in functional behavior that emerge when single molecules have company.
New phenomena can arise when multiple molecules behave collectively, such as the phase transition that occurs when water molecules in the vapor phase condense into liquid water. Physicists refer to this as ‘emergence’ when the underlying physical process can be described with simple, generalizable mathematical rules, even if all of the molecular details are not certain. For many biological systems, thousands of molecules are not required to observe such collective emergent behavior; just a small cohort generates fundamentally new behavior. Recent advances in single molecule approaches are allowing researchers to reveal interesting and physiologically important biological behavior that emerges even with the smallest jump in system complexity, that is, from one molecule to two. This minireview will focus on three examples: (1) protein-DNA complexes that are apparently very stable in isolation can be remodeled rapidly by facilitated dissociation in the presence of molecules of the same or similar type. (2) misfolding of repeat proteins via mispairing of similar domains (3) different functions can be ascribed to biomolecular complexes of different stoichiometry by the concomitant detection of both the function and stoichiometry of single complexes.
Facilitated dissociation of proteins from DNA
Transcription factors bound to genomic DNA sequences need to be quickly removed or replaced by other factors to ensure rapid responses to cellular signaling and environmental cues. However dissociation times measured in vitro can be minutes or longer. There are examples where small molecule binding can accelerate protein dissociation through allostery (as in the case of the Lac repressor), but many transcription factors have no known small molecule modulators of their DNA binding affinity. In recent years, it has become apparent that measuring the dissociation time scale of protein-DNA complexes by sample dilution does not capture the realistic situation in vivo, where many other proteins are present that can compete for binding to the same DNA. To overcome this limitation in characterizing the molecular details of protein-DNA dissociation, researchers have developed new single molecule assays in which the amount of free proteins in solution surrounding an immobilized protein-DNA complex can be varied from zero to high micromolar concentrations. The disappearance of a fluorescently labeled protein from the DNA over time can then be used as an indicator of protein dissociation so that effects of free proteins in solution on the dissociation rate can be precisely determined.
Several recent studies have employed variations of this single molecule approach to characterize the dissociation of protein-DNA complexes. For the bacterial nucleoid-organizing and gene-regulatory protein Fis, Graham et al have shown that GFP-Fis dissociation from a single lambda phage DNA is extremely slow, retaining 90 % of bound proteins after 30 minutes (Graham et al., 2011). In contrast, when 50 nM unlabeled Fis was added to the assay, GFP-Fis dissociated rapidly, with 50 % of bound proteins leaving the DNA in just 3 minutes. Such facilitated dissociation was also observed with other proteins including HU and NHP6A. The degree of facilitated dissociation was dependent on the concentration of the added protein with the effective rate constant approaching 105 M−1 s−1. In another example, Joshi et al examined the bacterial, metal-dependent transcription factor CueR (Joshi et al., 2012). Cu1+-bound and Cu1+-free (apo) forms of CueR compete for the same binding DNA binding sites in order to regulate the transcription of genes required to protect cells from copper-induced stress. The effective dissociation rate (koff′) of apo CueR measured in the presence of ~ 10 nM Cu1+-CueR was a factor of two larger than the intrinsic dissociation rate (koff) measured without free CueR present in the surrounding. The concentration dependence of koff′ showed that the effective rate constant for facilitated dissociation of CueR is as high as 108 M−1 s−1. Remarkably, the rate constant for CueR exchange is 15 times larger than the rate constant for de novo binding of CueR to the recognition sequence on naked DNA, suggesting that DNA-bound CueR facilitates its own displacement by recruiting a second CueR(Joshi et al., 2012). Collectively, these results suggest that koff′ for a protein-DNA complex measured in the presence of competing proteins in solution can be much larger than the intrinsic dissociation rate (koff) measured for that complex in isolation.
Recent single molecule studies have provided important clues to the mechanism of facilitated dissociation (Loparo et al., 2011; McCauley et al., 2013). The facilitated dissociation of Fis and CueR must involve the formation of a ternary complex between Protein 1 (P1), DNA (D) and Protein 2 (P2) that is ultimately resolved into either state P2-D (Fig. 1a). One possibility, proposed based on High-Mobility Group B (HMGB) protein dynamics on single lambda phage DNA (McCauley et al., 2013), is that a protein may undergo micro-dissociation, detaching itself from the DNA with a small but real physical separation from the DNA such that other nearby proteins can invade the space and occupy the same position on the DNA (Fig. 1a). In the absence of additional proteins in solution, the micro-dissociated protein would have a high probability of reassociation with the DNA, such that if one measured dissociation by sample dilution, an extremely stable protein-DNA complex would be detected. Another possibility, proposed in the case of DNA polymerase exchange in a functional replisome (Loparo et al., 2011), requires bivalent or multivalent protein-DNA interactions. If Protein 1 and Protein 2 each bind to DNA with two or more separate binding interfaces, partial dissociation of P1 (e.g. by breaking one of the binding interfaces and keeping the other) could allow a similarly partially dissociated P2 to occupy the vacated DNA binding site (Fig. 1a). The ensuing random walk of the interface is resolved either when P1 returns to full occupancy and ejects P2, or when P2 completely replaces P1 (Fig. 1a). Again, if there is no P2 protein in solution, P1 dissociation, which would require simultaneous breaking of all of the multivalent binding interactions between P1 and DNA, will be much slower. For proteins that bind to specific DNA sequences or structures, the second model is more attractive because enhanced expression of only those proteins that share the sequence- or structure-specificity would influence the dissociation of the incumbent protein. For non-specific binders such as HMGB, a micro-dissociation mechanism is easier to rationalize.
Figure 1.

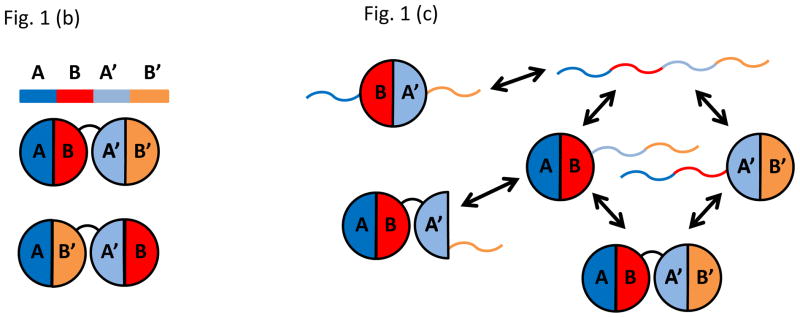

(a) Protein 1 (P1) bound to DNA dissociates slowly unless nearby Protein 2 (P2) facilitates P1 dissociation or exchange. Facilitated dissociation may occur via micro-dissociation or multivalent interactions. (b) A tandem repeat protein made of two domains, one with A and B motifs and the other with A′ and B′ motifs, may fold properly into AB-A′B′ or misfold into AB′-A′B. Interdomain misfolding can be significant if the sequence identify between the two domains is high. (c) Six different conformations of two tandem domains of calmodulin are illustrated along with their connectivity. Folded motifs are denoted as semi-circles and unfolded motifs as curves. (d) A single UvrD protein translocates on ssDNA and stops when it encounters a junction with dsDNA. When a second UvrD joins, the two proteins can unwind the DNA processively. Kymogram shows UvrD position as a function of time and the brightness is a measure of UvrD stoichiometry. The force vs. time curve shows that DNA shortening due to conversion of dsDNA into ssDNA initiates upon the arrival of second UvrD. Reproduced from Lee et al. with permission from Nature Publishing Group.
The recently discovered phenomenon of “allostery through DNA” is another new behavior revealed by single molecule measurements (Kim et al., 2013). In this study, binding of the T7 RNA polymerase to a nearby location on a DNA sequence was found to either increase or decrease the effective dissociation rate (koff′) of the Lac repressor bound to its target sequence in a manner depending on the distance between the two proteins. The modulation of koff′ showed a periodicy of 10 bp of DNA double helix and compelling evidence was presented that this effect is not due to any direct physical contact between the polymerase and the Lac repressor. The detailed microscopic mechanism of allostery through DNA is not yet completely clear, but the mechanism appears to be different from the facilitated dissociation models outlined in Fig. 1a. The fine-tuning of protein dissociation by such “action at a distance” mechanisms could have a profound influence on cellular fitness and evolutionary mechanisms.
What could be the functional importance of these facilitated displacement phenomena? A cell could potentially utilize the process of facilitated dissociation to modulate the effective koff′ of these slowly dissociating transcription factors simply by tuning the amount of other competing proteins. In this scenario, facilitated dissociation of transcription factors would allow the law of mass action to take hold rapidly.
Interdomain misfolding of repeat proteins
Proteins that have two or more repeats of similar domains are ubiquitous in nature. If one domain has two primary motifs, A and B, required for its folding, and an adjacent domain has the highly similar but different motifs, A′ and B′, then there is a possibility that these motifs may mispair during folding to yield the misfolded AB′-A′B conformation instead of the proper AB-A′B′ conformation. The probability of obtaining such misfolded configurations increases as the number of tandem repeat domains increases. Misfolded configurations arising this way are difficult to characterize in bulk solution because each subspecies cannot be clearly isolated or unambiguously identified in many cases. Therefore, appropriately designed single molecule experiments are needed to sort through the potential complexity of these systems and to determine whether any of the potential misfolded configurations actually occur, and if so, with what probability.
In the first demonstration of its kind (Borgia et al., 2011), Borgia et al used single molecule FRET (fluorescence resonance energy transfer) to distinguish between misfolded (AB′-A′B) and folded( AB-A′B′) forms of a tandem repeat of immunoglobulin domains (Fig. 1b). A judicious choice of fluorescent labeling positions brought the donor and acceptor fluorophores into close proximity (high FRET) in the misfolded state and farther away from each other (low FRET) in the folded state. Using a non-natural construct harboring two tandem domains with identical amino acid sequence, a small, stably misfolded population (5 %) was clearly identified as a high FRET species. In contrast, when a native protein sequence containing two tandem domains with only 24% sequence identity was used, no misfolded state was detected. With an intermediate level of sequence identity (42%), the misfolded state could once again be detected as ~ 5% of the total population. These results clearly demonstrated that tandem protein domains of identical sequence have a higher probability of misfolding via mispairing of motifs present in both domains.
In another example (Stigler et al., 2011), Stigler et al used an optical trap to examine the complex folding landscape of calmodulin, which consists of two globular domains, one made of A and B motifs and the other of A′ and B′ motifs, connected by an alpha helical linker. Optical traps can be used to apply pico Newton forces to biomolecules, allowing the measurement of nanometer scale displacements, such as those associated with the unfolding of calmodulin and other typical protein domains. Recent advances in high resolution optical traps have pushed the spatial precision to the angstrom (0.1 nm) level. Because calmodulin can be unfolded under force, multiple refolding and misfolding events could be observed from a single molecule over a long period of time. Remarkably, in this experiment, six different species could be identified (depicted schematically in Fig. 1c with semicircles and lines denoting folded and unfolded motifs, respectively). Note that here A′ as a single motif can fold by itself, and that some of the other possible misfolded conformations such as AB′-A′B and B-A′B′ were not detected. Real time observation of the interconversion between these states also allowed Stigler et al to establish how each of the six states is connected to each other and thus to determine the interconversion pathway and kinetics precisely.
In vivo co-translational folding should help to ensure the vectorial folding of in many cases to minimize interdomain mispairing. However, a protein such as calmodulin, which may unfold and refold multiple times in response to fluctuations in Ca2+ concentration, or a muscle protein titin which may do the same under tension, could potentially sample some of the misfolded states detected in the single molecule studies by Borgia et al and Stigler et al. Support for the existence of such interdomain misfolding in vivo lies in the observation that the amino acid sequence identity between nearest neighboring domains tends to be lower than between non-nearest neighboring domains in multi-domain proteins, suggesting selective pressure to avoid mispairing between adjacent domains (Borgia et al., 2011; Wright et al., 2005). Similarly complex alternative pairing between motifs may also be responsible for the diverse structural features of amyloids. Single molecule methods that have been employed to analyze the folding properties of single intrinsically disordered proteins (Mukhopadhyay et al., 2007; Yu et al., 2012) may soon reveal the rules of amyloid formation.
Function vs. stoichiometry
Although it is well known that many proteins form oligomeric structures that are essential for their function, it is not always easy to ascertain which species (monomer, dimer, trimer, hetero-oligomers, etc.) is required for which function. Single molecule fluorescence imaging can directly reveal the number of proteins in a complex and thus provides a unique opportunity to link the stoichiometry of a complex to its function.
A recent example is the characterization of the activity of the bacterial DNA helicase UvrD. Although there is good agreement that a single UvrD monomer can translocate on single stranded (ss) DNA as a motor protein that hydrolyzes one ATP per nucleotide translocated, there has been a debate as to whether a UvrD monomer can unwind double stranded (ds) DNA processively (Lohman et al., 2008). In a recent study (Lee et al., 2013), UvrD translocation along a long (multi-kilo base), laterally stretched ssDNA was directly visualized by tracking the motion of a fluorophore attached to the protein, revealing that UvrD stops translocating when it encounters a dsDNA duplex (Fig. 1d). When another UvrD joined the stalled UvrD (an event detected by the doubling of the fluorescence intensity), the motion of the complex resumed, albeit at a reduced speed. Because unwinding is known to be slower than translocation by about a factor of three, these observations indicate that the two UvrD proteins function as a helicase to unwind dsDNA processively. The concurrent measurement of force within the optical trap in this experiment confirmed this interpretation: the force on the DNA molecule began to decrease at the precise moment of UvrD dimerization, presumably at the onset of the helicase activity that converts duplex DNA to ssDNA which each have different and well-defined mechanical properties (Fig. 1d). Another recent study used two-color single molecule imaging, one color to count the number of UvrD molecules bound to the DNA and the other to detect complete unwinding of DNA. This study also showed that a UvrD monomer cannot unwind even 18 bp of duplex DNA, and that unwinding is observed only when two or more UvrD molecules are present (Yokota et al., 2013). Ultrahigh resolution optical traps combined with single fluorophore sensitivity (Comstock et al., 2011) may allow a more stringent test of whether a single UvrD can unwind DNA at all, and help explain why a monomer is not a good helicase in vitro.
Outlook
Single molecule tools can help us dissect newly emerging behaviors of molecular cohorts. As discussed here, these include (1) facilitated dissociation (2) inter-domain misfolding in proteins, and (3) new functions for molecular complexes. In all of the examples discussed, recent advances in single molecule measurement technologies, for example multi-color fluorescence imaging, ultrahigh resolution optical trap, and combination of fluorescence imaging and mechanical manipulation, were essential for revealing the emerging behaviors of interest. As the single molecule data become more multidimensional and complex, unbiased computational methods such as hidden Markov modeling become important. Computational tools such as molecular dynamics simulations and modeling are also important in guiding the data interpretation as was the case in the assignment of the interdomain misfolding events described above (Borgia et al., 2011),(Stigler et al., 2011) and may help us to visualize some of the hypothesized, short-lived states such as the micro-dissociation state of a protein loosely bound to DNA (McCauley et al., 2013).
One generalizable lesson that can be learned from the single molecule analysis of facilitated protein dissociation from DNA is the power of multivalent interactions. Multivalency can keep a molecular complex very stable by itself because all of the bonds need to be broken for full dissociation. Under physiological conditions, other competing molecules and interactions can break the multivalent interaction little by little and one at a time, allowing rapid changes in molecular compositions and partnerships. One potential advantage of such interactions is that new equilibria dictated by the changes in protein component concentrations in the cell can be rapidly approached. Another advantage is that breaking and reforming of multivalent interactions in small increments would make cellular machines that are composed of multiple molecules much more dynamic. For example, this principle may allow the thermally driven sliding of RecA filaments on DNA (Ragunathan et al., 2012) and assist the filament in sampling multiple sequences without full dissociation (Forget and Kowalczykowski, 2012; Ragunathan et al., 2012). A similar mechanism may also apply to RNA-guided targeting for microRNA processing or CRISPPR-mediated genome processing and to allowing proteins with multiple DNA binding sites to slide on ssDNA (Zhou et al., 2011). It is fully expected that single molecule measurements will continue to provide a unique level of insight into the emergent biological properties of these and other systems.
Acknowledgments
Research in our laboratory was funded by the National Science Foundation (0822613 and 0646550) and the National Institutes of Health (GM065367, AI083025 and RR025341). I would like to thank A. van Oijen and M. Williams for stimulating discussion, C. Thibodeuax for proofreading the manuscript. I apologize for not being able to cite all relevant literature due to space constraints.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Borgia MB, Borgia A, Best RB, Steward A, Nettels D, Wunderlich B, Schuler B, Clarke J. Nature. 2011;474:662–665. doi: 10.1038/nature10099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comstock MJ, Ha T, Chemla YR. Nature Methods. 2011;8:335–340. doi: 10.1038/nmeth.1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forget AL, Kowalczykowski SC. S Nature. 2012;482:423–427. doi: 10.1038/nature10782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JS, Johnson RC, Marko JF. Nucleic Acids Res. 2011;39:2249–2259. doi: 10.1093/nar/gkq1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi CP, Panda D, Martell DJ, Andoy NM, Chen TY, Gaballa A, Helmann JD, Chen P. Proc Natl Acad Sci U S A. 2012;109:15121–15126. doi: 10.1073/pnas.1208508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Brostromer E, Xing D, Jin J, Chong S, Ge H, Wang S, Gu C, Yang L, Gao YQ, et al. Science. 2013;339:816–819. doi: 10.1126/science.1229223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KS, Balci H, Jia H, Lohman TM, Ha T. Nat Commun. 2013;4:1878. doi: 10.1038/ncomms2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohman TM, Tomko EJ, Wu CG. Nat Rev Mol Cell Biol. 2008;9:391–401. doi: 10.1038/nrm2394. [DOI] [PubMed] [Google Scholar]
- Loparo JJ, Kulczyk AW, Richardson CC, van Oijen AM. Proc Natl Acad Sci U S A. 2011;108:3584–3589. doi: 10.1073/pnas.1018824108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauley MJ, Rueter EM, Rouzina I, Maher LJ, 3rd, Williams MC. Nucleic Acids Res. 2013;41:167–181. doi: 10.1093/nar/gks1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Krishnan R, Lemke EA, Lindquist S, Deniz AA. Proc Natl Acad Sci U S A. 2007;104:2649–2654. doi: 10.1073/pnas.0611503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragunathan K, Liu C, Ha T. Elife. 2012;1:e00067. doi: 10.7554/eLife.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stigler J, Ziegler F, Gieseke A, Gebhardt JC, Rief M. Science. 2011;334:512–516. doi: 10.1126/science.1207598. [DOI] [PubMed] [Google Scholar]
- Wright CF, Teichmann SA, Clarke J, Dobson CM. Nature. 2005;438:878–881. doi: 10.1038/nature04195. [DOI] [PubMed] [Google Scholar]
- Yokota H, Chujo YA, Harada Y. Biophys J. 2013;104:924–933. doi: 10.1016/j.bpj.2013.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Liu X, Neupane K, Gupta AN, Brigley AM, Solanki A, Sosova I, Woodside MT. Proc Natl Acad Sci U S A. 2012;109:5283–5288. doi: 10.1073/pnas.1107736109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Kozlov AG, Roy R, Zhang J, Korolev S, Lohman TM, Ha T. Cell. 2011;146:222–232. doi: 10.1016/j.cell.2011.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]