

Abstract
Scope
Central sensitization is implicated in the pathology of temporomandibular joint disorder (TMD) and other types of orofacial pain. We investigated the effects of dietary cocoa on expression of proteins involved in the development of central sensitization in the spinal trigeminal nucleus (STN) in response to inflammatory stimulation of trigeminal nerves.
Methods and results
Male Sprague Dawley rats were fed either a control diet or an isocaloric diet consisting of 10% cocoa powder 14 days prior to bilateral injection of complete Freund’s adjuvant (CFA) into the temporomandibular joint to promote prolonged activation of trigeminal ganglion neurons and glia. While dietary cocoa stimulated basal expression of GLAST and MKP-1 when compared to animals on a normal diet, cocoa suppressed basal calcitonin gene-related peptide levels in the STN. CFA-stimulated levels of protein kinase A, P2X3, P-p38, GFAP, and OX-42, whose elevated levels in the STN are implicated in central sensitization, were repressed to near control levels in animals on a cocoa enriched diet. Similarly, dietary cocoa repressed CFA-stimulated inflammatory cytokine expression.
Conclusion
Based on our findings, we speculate that cocoa enriched diets could be beneficial as a natural therapeutic option for TMD and other chronic orofacial pain conditions.
Keywords: Cocoa, Cytokines, Inflammation, Sensitization, Trigeminal
1 Introduction
Temporomandibular joint disorder (TMD) is the most common type of chronic orofacial pain referred to dentists [1–3]. TMD involve complex symptoms of pain and dysfunction of the masticatory system including the temporomandibular joint (TMJ), muscles of mastication, and adjacent tissues [4]. TMD is a major cause of chronic non-dental pain in the orofacial region, and hence negatively impacts quality of life. The underlying pathology of TMD involves sensitization and activation of the V3 or mandibular branch of the trigeminal nerve that provides sensory innervation to the muscles and joint, and functions to relay nociceptive signals to the spinal trigeminal nucleus (STN) [5, 6]. Sensitization of trigeminal neurons, characterized by a lowering of the activation threshold to chemical, thermal, or mechanical stimuli, is mediated by increased neuron-glia interactions in trigeminal ganglia and upper spinal cord [7–9]. Importantly, development of central sensitization, which involves second order neurons that transmit pain signals to the thalamus leading to a persistent pain state, is facilitated by hyperexcitable spinal astrocytes and microglia [10]. Under physiological conditions, these glial cells modulate neuronal excitability and maintain homeostasis by regulating the extracellular environment around neurons via the uptake of K+ ions and glutamate by selective transporters [11, 12].
Regulation of the excitability state of neurons involved in pain transmission within the spinal cord is known to involve numerous proteins including the neuropeptide calcitonin gene-related peptide (CGRP) [13], the purinergic receptor P2X3, cytokines, and proteins involved in signal transduction pathways [14]. CGRP stimulates glial cells in the spinal cord to release cytokines and other inflammatory molecules leading to a lower threshold of activation and contributing to persistent pain [15]. Increased levels of P2X3 receptors on trigeminal neurons are indicative of central sensitization and increased pain signalling within the spinal cord [14, 16]. In addition, protein kinase A (PKA) promotes development of central sensitization by increasing the activity of glutamate receptors expressed on second order neurons that facilitate pain transmission [17–19]. Furthermore, cytokines are soluble proteins released from activated glial cells that directly sensitize nociceptors and increase neuronal sensitivity by increasing the expression of receptors and ion channels involved in pain and analgesia [20]. The mitogen-activated protein (MAP) kinases, which are a family of signal transduction enzymes activated in response to inflammatory stimuli, are also implicated in central sensitization [21]. The duration and magnitude of the MAP kinase response is regulated by MAP kinase phosphatases (MKP), which are enzymes involved in restoring elevated MAP kinase levels to normal via dephosphorylation [22, 23]. In particular, MKP-1 is a protein that preferentially dephosphorylates p38 and is reported to play a key role in the resolution of inflammation and pain [24, 25].
We have previously reported that inclusion of dietary cocoa was sufficient to suppress expression of proteins and signalling molecules in trigeminal ganglia associated with peripheral sensitization in response to TMJ inflammation [26]. There is emerging experimental evidence of the anti-inflammatory properties of cocoa that may help to explain the beneficial effects of cocoa in a variety of inflammatory ailments including insulin insensitivity and cardiovascular disease [27, 28]. Cocoa is obtained from the Theobroma cacao tree that is native to the tropical regions of the Americas. Historically, cocoa seeds were used in a variety of rituals by the Mayans and Aztecs who believed the seeds had medicinal benefits [29]. Persistent pathological pain, which is characteristic of TMD, is not effectively treated using standard pharmacological therapies. Therefore, the use of natural products offers a novel method for possibly preventing and treating chronic inflammatory diseases such as TMD [30, 31]. To date, most of the anti-inflammatory and anti-nociceptive effects attributed to compounds isolated from cocoa and other plants include the polyphenols catechins, anthocyanidins, and proanthocyanidins [32]. In addition, other bioactive compounds that are found abundantly in cocoa and reported to have anti-inflammatory properties, are phytosterols such as β-sitosterol [33]. The goal of this study was to extend our previous findings on cocoa to determine whether dietary cocoa could mediate cellular changes in the STN to suppress the development and maintenance of central sensitization in a chronic inflammatory model of TMD.
2 Materials and methods
2.1 Ethics statement
All animal studies were approved by the Institutional Animal Care and Use Committee at Missouri State University (Protocol 10004) and were conducted in compliance with all established guidelines in the Animal Welfare Act and National Institutes of Health. A concerted effort was made to reduce the number of animals used and to minimize any suffering.
2.2 Animals and diet
Adult male Sprague-Dawley rats (200–230 g, Charles River Laboratories, Wilmington, MA) had unrestricted access to food and water and were kept on a 12-hour light/dark cycle. The temperature range was 21–23°C and relative humidity was between 40 and 55%. Changes in food and water consumption as well as weight and grooming behaviours were monitored and recorded daily to assess the overall health of the animals. All natural, non-alkalized davao cocoa powder (fat content of 18–20%) was purchased from Askinosie Chocolate (Springfield, MO) and incorporated into a diet based on a modified AIN-76A rodent diet (Research Diets, New Brunswick, NJ) containing 95.45 g cocoa powder/kg pellet weight [10% (g/g) or 6.7% of total energy intake] (Supporting Information Table S1). The polyphenolic content of the cocoa powder was 22.6 mg/g gallic acid equivalents as determined by the Folin-Ciocalteu assay [34]. All diets were isoenergetic and contained equal concentrations of carbohydrates, protein, and fat. Following a three-day acclimation period on the control diet (AIN-76A), rats were randomly placed into two groups receiving a normal control diet or cocoa enriched diet for 14 days. A total of 16 rats were used for all studies, which were equally split between four groups (control diet only, animals receiving cocoa enriched diets only, animals on control diets receiving an injection of complete Freund’s adjuvant (CFA), and animals on cocoa enriched diets that received an injection of CFA). The same animals were used for immunohistochemistry and array analysis.
2.3 Chronic model of TMJ inflammation
Rats were anesthetized by inhalation of 3.5% isoflurane (VetEquip, Pleasanton, CA). After 14 days on the control diet or cocoa enriched diet, some rats received an injection of 50 μL CFA (1:1 in saline; Sigma-Aldrich, St. Louis, MO) in each TMJ capsule. All animals then continued their assigned diets for 7 more days before being sacrificed. The time of treatment, preparation, and amount of stimulatory agent were based on results from previous studies in our laboratory [26, 35]. As controls, some animals on each diet were not injected with CFA.
2.4 Tissue isolation and preparation
Spinal cord encompassing the spinomedullary junction (Vc/C1) transition zone containing the STN was removed from all rats following CO2 asphyxiation and decapitation, trimmed at the obex and 5 mm posterior to the obex, and cut in half laterally along the dorsal ventral axis. A randomly chosen half of the tissue was quickly frozen in liquid nitrogen and used for cytokine array analysis. The other half of the tissue, which was used for immunohistochemistry, was placed in a solution of 4% paraformaldehyde overnight at 4°C. Following paraformaldehyde fixation, tissues were incubated in 15% sucrose in distilled water at 4°C for 1 hour and then 30% sucrose overnight at 4°C. Spinal cord tissues were positioned with the caudal side in contact with the slide, covered with OCT Compound (Sakura Finetek, Torrance, CA), and quickly frozen. Spinal cord sections (14 μm) containing the STN were sectioned transversely at a distance of 4–5 mm posterior to the obex using a cryostat set at −24°C (Microm HM 525, Thermo Scientific, Waltham, MA). All sections were mounted on Superfrost Plus microscope slides (Fisher Scientific, Pittsburgh, PA). To minimize variability, each slide used for immunohistochemistry contained 4 sections, one section from each experimental condition.
2.5 Immunohistochemistry
Slides containing sections of spinal cord were incubated in a solution of 0.1% Triton X-100 and 5% donkey serum (Jackson Immuno Research Laboratories, West Grove, PA) for 30 minutes. Next, sections were incubated with primary antibodies either for 3 hours at room temperature or overnight at 4°C and then with secondary antibodies for 1 hour at room temperature (Table 1). As a control, some sections were incubated with only secondary antibodies. Sections were mounted using Vectashield medium (H-1200) containing 4′, 6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA) to co-stain cell nuclei. Images (200x) were collected using a Zeiss Axiocam mRm camera mounted on a Zeiss Imager Z1 fluorescent microscope equipped with an ApoTome. Image acquisition was performed using Zeiss AxioVision (Rel 4.8, Thornwood, NY).
Table 1.
Summary of Antibodies Used for Immunohistochemistry
| Antibody | Host | Supplier | Dilution |
|---|---|---|---|
| GLAST | Rabbit | Abcam | 1:200 3 hours |
| CGRP | Rabbit | Sigma | 1:1000 3 hours |
| MKP1 | Goat | Everest | 1:500 3 hours |
| P2X3 | Rabbit | Thermosci | 1:1000 3 hours |
| PKA | Rabbit | Epitomics | 1:500 3 hours |
| P-p38 | Rabbit | Cell Signaling | 1:200 overnight |
| GFAP | Rabbit | Dako | 1:1000 30 minutes |
| OX-42 | Mouse | Abcam | 1:200 overnight |
| NeuN | Mouse | Millipore | 1:1000 3 hours |
| Iba-1 | Rabbit | Wako | 1:400 3 hours |
| Alexa 488 | Rabbit, Mouse | Invitrogen | 1:500 1 hour |
| Alexa 594 | Rabbit, Mouse, Goat | Invitrogen | 1:500 1 hour |
2.6 Measurement of staining intensity
Images of the spinal cord tissue containing the STN were used for analysis. Three randomly chosen 200x images containing a similar number of cells as identified by DAPI, were analyzed for each experimental condition, which were repeated in 4 independent experiments, resulting in 16 images being analyzed for the intensity measurements. The relative staining intensity measurements were based on our previously published protocols [26, 36, 37]. Two researchers blinded to the experimental conditions independently performed the measurements. The staining intensity in spinal cord tissue was determined by measuring the mean gray intensity from four regions, in laminas I–III, of staining in the medullary horn and subtracting the intensity from acellular regions of the medullary horn. The fold-change in staining intensity was defined as the mean change in relative intensity in the experimental condition when compared to the mean of the unstimulated control tissue that was made equal to one.
2.7 Cytokine Array Analysis
The intracellular levels of 29 proteins were determined using the Rat Cytokine Array Panel A Array Kit (R&D Systems, Minneapolis MN) per manufacturer’s instructions. Briefly, spinal cord tissue containing the STN (4–5 mm posterior to the obex) from each animal was sonicated in cell lysis solution (Bio-Rad, Madison, WI). Total protein levels in each sample were determined by the Bradford assay. Samples (200 μg) were diluted in Array Buffer 6 and Detection Antibody Cocktail and incubated on pre-blocked membrane arrays overnight at 4°C with gentle shaking. Membranes were then incubated in streptavadin conjugated peroxidase for 30 minutes and exposed to ECL Plus Detection Reagent (GE Healthcare, Pittsburgh, PA) for 5 minutes prior to developing on Kodak Biomax X-ray film (Sigma-Aldrich). Densitometric analysis was performed using ImageJ (Version 1.45, NIH, Bethesda, MD) using Gilles Carpentier’s Protein Array Analysis plugin for ImageJ (Version 1.0, 2010) with rolling ball background subtraction. Spot data were normalized to the average of six positive control spots on each array. The fold-change in dot intensity was defined as the mean change in relative intensity in the experimental condition when compared to mean level of the unstimulated control tissue that was made equal to one.
2.8 Statistical analysis
Changes in immunostaining intensity and cytokine levels were evaluated using the Mann-Whitney U test. For all studies, results were considered significant when p < 0.05. All statistical tests were performed using SPSS (Version 16, IBM, Chicago, IL). Results are reported as mean ± SEM.
3 Results
3.1 Cocoa regulation of basal expression of GLAST; CGRP; and MKP-1 in the STN
While only minimal staining for the glial cell glutamate uptake transporter, GLAST, was observed in glia within the STN in naïve control animals, GLAST expression was increased in glial cells throughout the STN in animals receiving dietary cocoa (Fig. 1A). When compared to control animals (1.00 ± 0.06), cocoa treated animals exhibited a significant increase in staining intensity throughout the STN (2.12 ± 0.1, p < 0.01) (Fig. 1B).
Figure 1.
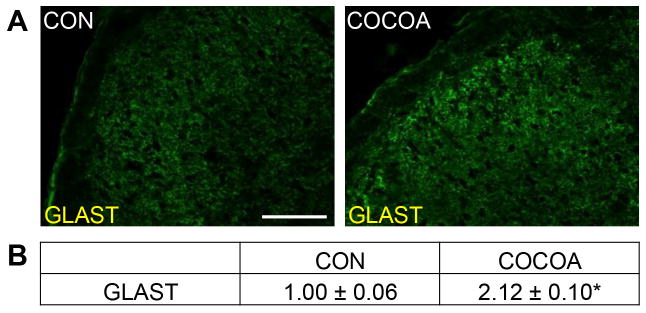
Cocoa enriched diets increase basal GLAST expression in spinal glial cells. Sections of spinal cord containing the STN were obtained from untreated animals (CON) and animals on a 10% cocoa enriched diet for 14 days (COCOA). (A) Representative images of spinal cord containing the STN stained for GLAST are shown. (B) The average fold-change ± SEM of GLAST staining intensity when compared to the mean of the control that was made equal to one is reported (n = 3 independent experiments). * p < 0.01 increase when compared to control. Magnification bar = 50 μm.
In unstimulated control animals, CGRP immunoreactivity was readily detected in neurons located in a discrete band in laminas I and II within the STN (Fig. 2A). However, the expression of CGRP in animals consuming cocoa was decreased when compared to control levels. Inclusion of dietary cocoa caused a significant decrease in CGRP staining intensity (0.64 ± 0.09, p < 0.01) when compared to control animals (1.00 ± 0.12) (Fig. 2B).
Figure 2.
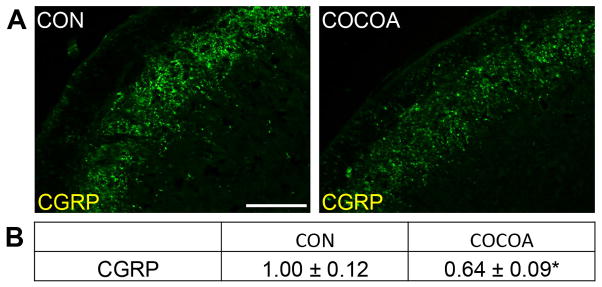
Basal CGRP expression in the STN is suppressed by cocoa- enriched diet. Spinal cord sections containing the STN were obtained from untreated animals (CON) and animals on a 10% cocoa-enriched diet (COCOA). (A) Representative images of spinal cord containing the STN stained for CGRP are shown. (B) The average fold-change ± SEM of CGRP staining intensity when compared to the mean of the control that was made equal to one is reported (n = 3). * p < 0.01 decrease when compared to control. Magnification bar = 50 μm.
A stimulatory effect was seen in the level of immunostaining for the MAP kinase phosphatase MKP-1. Expression of MKP-1 was increased in both neurons and glia in the STN in animals consuming cocoa when compared to levels in naïve control animals (Fig. 3A). While minimal staining was observed in control animals (1.00 ± 0.14), animals consuming cocoa exhibited a significant increase in staining intensity in neurons and glial cells (1.90 ± 0.12, p < 0.01) (Fig. 3B). Importantly, there were no observable differences in weight due to changes in food or water consumption nor were any noticeable differences in grooming or social behaviors in rats on the cocoa enriched diet when compared to rats fed control diets (data not shown).
Figure 3.
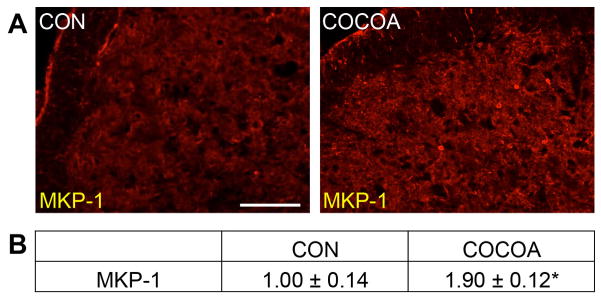
Basal expression of MKP-1 is increased in neurons and glia in the STN of rats on cocoa supplemented diets. (A) Representative images of the STN region in spinal cord obtained from control animals (CON) or animals consuming cocoa stained for MKP-1 are shown. (B) The average fold-change ± SEM of MKP-1 staining intensity when compared to the mean of the control that was made equal to one is reported (n = 3). * p < 0.01 increase when compared to control. Magnification bar = 50 μm.
3.2 CFA mediated elevation in the levels of P2X3; PKA; and P-p38 are repressed by cocoa
Minimal expression of the ATP receptor P2X3 was detectable in neurons within the STN obtained from naïve control animals, and in animals on a cocoa enriched diet (Fig. 4A). In contrast, the level of P2X3 expression was increased in neurons in the STN in response to prolonged TMJ inflammation caused by injection of CFA into the joint capsule. Significantly, staining levels of P2X3 were greatly repressed to near basal levels in animals consuming cocoa for 14 days prior to CFA injection. There was no significant difference in the staining intensity in animals eating cocoa alone (0.96 ± 0.07) when compared to levels in control animals (1.00 ± 0.09) (Fig. 4B). In contrast, CFA treatment caused a significant increase in P2X3 staining intensity (1.82 ± 0.14, p < 0.01) that was repressed in animals consuming cocoa before CFA injection (1.09 ± 0.08, p < 0.01).
Figure 4.

CFA-mediated upregulation of P2X3 expression in STN neurons is repressed in rats receiving cocoa as a dietary supplement. Sections of spinal cord containing STN were obtained from untreated animals (CON), animals on diets supplemented with 10% cocoa for 14 days (COCOA), animals 7 days post CFA injection (CFA), or animals on a cocoa-enriched diet and injected with CFA 7 days prior to harvesting the spinal cord (COCOA + CFA). (A) Images of the STN stained for P2X3 are shown. (B) The average fold change ± SEM of P2X3 staining intensity when compared to the mean of the control that was made equal to one is reported (n = 3). * p < 0.01 increase when compared to control while # p < 0.01 decrease compared to CFA. Magnification bar = 50 μm.
Normal immunostaining levels of the pro-inflammatory signalling marker PKA was observed in neurons and glia in the STN from naïve control animals and those consuming cocoa enriched diets (Fig. 5A). In contrast, PKA expression was increased 7 days after CFA injection in the TMJ. However, the stimulatory effect of CFA on PKA expression was greatly repressed in animals on a cocoa-enriched diet for 14 days prior to CFA injection. There was no significant difference in the staining intensity of rats treated with cocoa (1.07 ± 0.12) when compared to naïve control animals (1.00 ± 0.12) (Fig. 5B). In contrast, CFA caused a significant increase in staining intensity (1.97 ± 0.17, p < 0.01) over levels in control and cocoa animals. The stimulatory effect of CFA on PKA levels was significantly repressed in animals on a cocoa supplemented diet (1.36 ± 0.11, p < 0.01).
Figure 5.

Stimulated PKA expression in STN neurons and glia is repressed by dietary cocoa. (A) Representative images of the STN immunostained for PKA are shown. Tissues were obtained from control animals (CON) or animals on 10% cocoa enriched diets for 14 days (COCOA), animals injected with CFA for 7 days (CFA), or animals on a 10% cocoa diet for 14 days then injected with CFA for 7 days prior to harvesting the spinal cord (COCOA + CFA). (B) The average fold change ± SEM of PKA staining intensity when compared to the mean of the control that was made equal to one is reported (n = 3).* p < 0.01 increase when compared to control while ‡ p < 0.01 decrease from CFA and p < 0.01 increase from control. Magnification bar = 50 μm.
Minimal staining for the active form of the MAP kinase p38 (P-p38) was detected in neurons and glia within the STN obtained from naïve control animals and animals consuming cocoa (Fig. 6A). In contrast, P-p38 expression was increased in neurons and glia in the STN in response to prolonged TMJ inflammation caused by injection of CFA into the joint capsule. However, levels of P-p38 were repressed to near basal levels in animals eating cocoa for 14 days prior to CFA injection. To identify the cell types exhibiting increased P-p38 expression, tissues were costained with antibodies directed against the neuronal protein NeuN, the microglial protein Iba1, or an astrocyte marker glial fibrillary acidic protein (GFAP). Based on our costaining results, elevated levels of P-p38 were only observed in neurons and microglia (data not shown). There was no significant difference in the staining intensity in animals eating cocoa alone (1.15 ± 0.19) when compared to levels in naïve control animals (1.00 ± 0.13) (Fig. 6B). In contrast, CFA treatment caused a significant increase in P-p38 staining intensity (1.74 ± 0.17, p < 0.01) that was repressed in animals consuming cocoa before injection of CFA (1.11 ± 0.15, p < 0.01).
Figure 6.

Increased expression of P-p38 in STN neurons and microglia in response to CFA is repressed by cocoa. (A) Representative images of the STN immunostained for P-p38 are shown. Tissues were obtained from control animals (CON) or animals on 10% cocoa enriched diets for 14 days (COCOA), animals injected with CFA for 7 days (CFA), or animals on a 10% cocoa diet for 14 days then injected with CFA for 7 days prior to harvesting the spinal cord (COCOA + CFA). (B) The average fold change ± SEM of P-p38 staining intensity when compared to the mean of the control that was made equal to one is reported (n = 3). * p < 0.01 increase when compared to control while # p < 0.01 decrease from CFA. Magnification bar = 50 μm.
3.3 Dietary cocoa represses CFA-induced expression of GFAP and OX-42 in the STN
Normal expression of GFAP, a marker of activated astrocytes, was detectable in the STN from naïve control animals and those consuming cocoa enriched diets (Fig. 7A). In contrast, GFAP expression was greatly stimulated in response to CFA injections. However, elevated staining levels of GFAP were greatly repressed in animals receiving cocoa for 14 days prior to CFA. There was no significant difference in the staining intensity of rats treated with cocoa (1.05 ± 0.13) when compared to control animals (1.00 ± 0.10) (Fig. 7B). In contrast, CFA caused a significant increase in staining intensity (1.92 ± 0.19, p < 0.01) over levels in control and cocoa-fed animals. The stimulatory effect of CFA on GFAP levels was significantly repressed in animals on a cocoa supplemented diet (1.20 ± 0.19, p < 0.01).
Figure 7.

Cocoa represses CFA mediated upregualtion of GFAP expression in STN astrocytes. (A) Representative images of the STN immunostained for GFAP are shown. Tissues were obtained from control animals (CON) or animals on 10% cocoa enriched diets for 14 days (COCOA), animals injected with CFA for 7 days (CFA), or animals on a 10% cocoa diet for 14 days then injected with CFA for 7 days prior to harvesting the spinal cord (COCOA + CFA). (B) The average fold change ± SEM of GFAP staining intensity when compared to the mean of the control that was made equal to one is reported (n = 3). * p < 0.01 increase when compared to control while # p < 0.01 decrease from CFA. Magnification bar = 50 μm.
The effect of CFA and cocoa enriched diets on microglia within the STN was investigated using antibodies directed against OX-42, a marker of microglia activation. In tissues from naïve control or animals consuming cocoa, normal expression of OX-42 was observed in microglia (Fig. 8A). However, OX-42 expression was increased in microglia in the STN 7 days after CFA injection into the joint capsule. In contrast, animals pretreated with cocoa for 14 days before CFA injection exhibited much lower levels of OX-42 immunostaining when compared to animals that received CFA injections alone. There was no significant difference in the staining intensity in the STN of animals on a cocoa-enriched diet (1.04 ± 0.14) when compared to levels in naïve control animals (1.00 ± 0.13) (Fig. 8B). In contrast, CFA injection resulted in a significant increase in staining intensity (2.71 ± 0.21, p < 0.01) that was repressed in animals consuming cocoa prior to CFA injections (1.23 ± 0.14, p < 0.01).
Figure 8.

CFA stimulation of OX-42 expression in STN microglia is repressed in rats receiving cocoa supplemented diets. (A) Representative images of the STN immunostained for OX-42 are shown. Tissues were obtained from control animals (CON) or animals on 10% cocoa enriched diets for 14 days (COCOA), animals injected with CFA for 7 days (CFA), or animals on a 10% cocoa diet for 14 days then injected with CFA for 7 days prior to harvesting the spinal cord (COCOA + CFA). (B) The average fold change ± SEM of OX-42 staining intensity when compared to the mean of the control that was made equal to one is reported (n = 3). * p < 0.01 increase when compared to control while # p < 0.01 decrease from CFA. Magnification bar = 50 μm.
3.4 CFA-mediated expression of cytokines is repressed by cocoa
To investigate the effects of cocoa on CFA-mediated increases on cytokine levels, sections of spinal cord containing the STN were used for protein array analysis. CFA stimulation caused a significant (p < 0.05) increase in 14 of the 29 proteins when compared to levels in control tissues obtained from naïve animals (Table 2). While seven proteins (CINC-2α/β, IL-1α, IL-2, IL-4, IL-13, LIX, and RANTES) were increased 2 to 2.4-fold, four proteins (IL-3, IL-17, IP-10, and MIG) were increased 2.5 to 3-fold and an additional three proteins (IL-1ra, TNF-α, and VEGF) were increased greater than 3-fold. However, the elevated levels of eight cytokines were significantly repressed in animals pretreated with cocoa for 14 days before CFA injection, while six cytokines were not appreciably changed by cocoa (Table 2). Animals receiving only cocoa supplemented diets had increases in two cytokines (IL-1ra and MIG) that were both increased 2 to 2.4-fold. A summary of the effect of cocoa on CFA mediated regulation of all 29 cytokines is provided as Supporting Information Table S2. Thus, dietary cocoa differentially regulates the expression of cytokines in the STN in response to prolonged TMJ inflammation.
Table 2.
Relative Fold-Change in Cytokine Levels in the Spinal Trigeminal Nucleus in Response to CFA and Cocoa Enriched Diets. The level of all cytokines were significantly (p < 0.05) elevated above control levels.
| Fold-Change From Control
|
||
|---|---|---|
| CFA | COCOA +CFA | |
|
|
||
| CINC-2α/β | 2.26 ± 0.86 | 1.26 ± 0.69# |
| IL-1α | 2.00 ± 0.56 | 1.58 ± 0.82# |
| IL-1ra | 4.69 ± 2.31 | 3.05 ± 0.80 |
| IL-2 | 2.33 ± 0.60 | 2.33 ± 0.73 |
| IL-3 | 2.58 ± 0.78 | 2.30 ± 0.44 |
| IL-4 | 2.20 ± 0.76 | 1.36 ± 0.44# |
| IL-13 | 2.04 ± 0.46 | 1.25 ± 0.57# |
| IL-17 | 2.83 ± 0.64 | 1.61 ± 0.62# |
| IP-10 | 2.72 ± 0.72 | 3.12 ± 0.83 |
| LIX | 2.41 ± 0.68 | 2.38 ± 0.81 |
| MIG | 2.94 ± 1.35 | 2.30 ± 0.74 |
| RANTES | 2.42 ± 0.72 | 1.83 ± 0.49# |
| TNF-α | 3.23 ± 1.41 | 1.75 ± 0.90# |
| VEGF | 4.27 ±1.71 | 2.57 ± 0.86# |
designates when p < 0.05 when compared to CFA simulation.
4 Discussion
A main finding of our study was that inclusion of cocoa in the diet of Sprague-Dawley rats mediated cellular changes in the STN implicated in central sensitization in response to prolonged inflammation of the TMJ. The amount of cocoa used in our study was equivalent to daily consumption of 33 g of cocoa powder. The injection of CFA into the TMJ capsule, which causes prolonged tissue inflammation and pain mediated via sensitization and activation of trigeminal nerves, is utilized to mimic TMD pathology [38–42]. Excitation of trigeminal neurons results in the release of the neurotransmitter glutamate and neuromodulator CGRP from their terminals within the STN and subsequent activation of second order nociceptive neurons and spinal glia implicated in promoting central sensitization [43]. Importantly, we found that cocoa was able to increase the glial expression of GLAST, a protein that functions to remove the excitatory neurotransmitter glutamate from the external environment around second order neurons [11, 44]. GLAST is member of the excitatory amino acid transporters that function to maintain neuronal homeostasis by controlling the amount of excitatory amino acids localized in the extracellular space around neurons. Another novel finding from our study was that dietary cocoa was sufficient to suppress the expression of the neuropeptide CGRP within the STN. Elevated levels of CGRP within the spinal cord are associated with the development and maintenance of central sensitization [13]. CGRP release within the spinal cord is reported to increase activity of spinal glial cells leading to increased synthesis and secretion of pro-inflammatory molecules including cytokines. The changes in the levels of GLAST and CGRP caused by dietary cocoa would be expected to suppress neuron-glia interactions mediated by glutamate and CGRP in the STN that are implicated in persistent pain states.
In response to trigeminal neuron activation, CGRP release in the spinal cord promotes sensitization of nociceptive neurons via upregulation of the signal transduction protein PKA and P2X3, a receptor associated with nociceptive signalling in neurons, and spinal glia [45–47]. We found that dietary cocoa could suppress the stimulatory effect of CFA-induced inflammation in the TMJ on PKA levels in neurons and glia in the STN. Elevated PKA levels stimulate presynaptic pain neurotransmitter release via phosphorylation of neuronal ion channels, transcription factors, and synaptic transport proteins [48, 49]. Another way that CGRP is implicated in the development of central sensitization is by promoting expression of P2X3 receptors in trigeminal neurons [47, 50]. Elevated levels of ATP in the spinal cord and activation of the ionotropic P2X3 receptors are associated with persistent pain conditions [51, 52]. In support of this notion, P2X3 gene disruption in knockout animals and inhibition of P2X3 receptors with selective antagonists results in reduced pain-related behaviour in inflammatory pain models [49, 51, 53–55]. These findings support an important role of P2X3 in the development and maintenance of chronic pain signalling in sensory neurons. Based on our findings, dietary cocoa represses the prolonged CFA-mediated increases in PKA and P2X3, two proteins known to play an important role in central sensitization.
Another important finding from our study is that dietary cocoa stimulates an increase in the expression of the anti-inflammatory protein MKP-1, while repressing the CFA-induced increase in the level of the active form of p38 in the STN. Increased levels of P-p38 mediate sensitization of primary and second order nociceptive neurons by upregulating ion channel expression and activity and membrane receptor expression [56, 57]. In addition, elevated P-p38 levels stimulate synthesis and secretion of cytokines from spinal glia that promote and maintain a hyperexcitable state of neurons [21, 56, 58, 59]. The cocoa-mediated repression of P-p38 observed in our study was likely due to the increased expression of MKP-1 in the STN since MKP-1 is reported to preferentially suppress active, phosphorylated p38 levels in response to inflammatory stimuli or nerve injury [24]. The activity of MAP kinases is regulated by the family of proteins known as MAP kinase phosphatases that are induced in response to inflammatory stimuli, and thus, function in a compensatory manner to restore normal levels of active MAP kinases [25]. Importantly, data from recent studies provide evidence that mice lacking the MKP-1 gene have elevated MAP kinase activity [60], increased cytokine-induced inflammation [61], and higher susceptibility to inflammatory injuries [23]. Thus, MKPs perform an important function by modulating nociceptive responses to inflammatory stimuli and restoring cellular homeostasis. Interestingly, the cocoa-induced increase in levels of MKP-1 may be partly responsible for the lower levels of CGRP in the STN since CGRP gene expression is regulated by MAP kinases [62, 63].
In this study, we provide evidence that cocoa can suppress the activation of both astrocytes and microglia in the STN, and repress elevated levels of cytokines caused by prolonged joint inflammation. In response to persistent CFA-induced TMJ inflammation, trigeminal neurons are known to release inflammatory mediators that cause activation of spinal glial cells [10]. In agreement with previous studies, we found that immunoreactive levels of GFAP, which is used as a marker of activated astrocytes, and OX-42, a biomarker of activated microglia, were significantly elevated in response to CFA injection into the TMJ [6, 41]. It is now generally accepted that hyperactivation of spinal glia, in particular astrocytes and microglia, contribute to the development and maintenance of inflammatory pain [41, 44, 64]. These cells are thought to promote and sustain inflammatory pain by the release of cytokines that act directly to sensitize nociceptors and increase neuronal sensitivity to chemical, thermal, and mechanical stimuli by increasing the expression of receptors and ion channels involved in pain and analgesia [20, 65]. Interestingly, we found that cocoa, provided as 10% of the total solid diet, repressed the level of many but not all cytokines to baseline levels. Thus, it appears that cocoa functions to modulate the level of these inflammatory mediators but does not completely suppress the inflammatory response following CFA injection in the TMJ. Based on our data, we propose that dietary cocoa can function to inhibit neuron-glia interactions and cytokine release and therefore prevent key cellular events known to promote and maintain central sensitization in response to TMJ inflammation.
Our findings support an important role of dietary cocoa to suppress pathological inflammatory events associated with neurological diseases, cardiovascular disease, diabetes, arthritis, and cancer [66–68]. Our observed changes in protein expression within the upper spinal cord are likely the result of direct effects of cocoa metabolites since dietary plant flavonoids and sterols can cross the blood brain barrier, accumulate in the brain, and modulate neural function [69, 70]. The inhibitory effects of cocoa on nociceptive neurons and glial cells in the spinal trigeminal nucleus are likely mediated by multiple secondary plant metabolites found in cocoa [32, 66]. In support of this notion, flavonoids have been reported to attenuate microglial activation and suppress cytokine release likely by a mechanisms involving inhibition of pro-inflammatory NF-kB and MAP kinase pathways [71, 72]. Flavonoids are also reported to function by preserving cognitive abilities in aging rats and inhibiting programmed neuronal cell death or apoptosis [73, 74]. In addition, there is evidence from multiple studies of the anti-inflammatory effects of β-sitosterol [33, 75, 76]. In summary, there is emerging evidence that plant metabolites from cocoa are neural protective since dietary cocoa has been shown to repress neuronal sensitization of nociceptive neurons, inhibit neuroinflammation, and suppress neurodegeneration.
The findings of this study are in agreement with results from our previous study that provided evidence that dietary cocoa mediated suppression of neuron and glial activity associated with peripheral sensitization of trigeminal nociceptors in response to chronic inflammation [26]. Taken together results from this study and our previous study provide evidence that inclusion of cocoa in the diet of rats is sufficient to modulate expression of proteins implicated in the development of peripheral sensitization and central sensitization, which are pathophysiologic events implicated in TMD pathology. The development of chronic pain, as reported in patients with TMD, is an increasing burden for our society. TMD affects as many as 15% of the adult population [2, 4], is more prevalent in women than men, and is highest during the reproductive years [77]. Given the significant health impact of this disease, it is imperative that treatments are identified. However, orofacial pain diseases such as TMD that are associated with inflammation and pain are reported to be some of the most debilitating human conditions and most difficult to treat effectively [78–80]. To date, there are no FDA approved pharmacological therapies for TMD. While many orofacial pain patients treat their inflammatory pain with nonsteroidal anti-inflammatory drugs (NSAIDs), COX-2 inhibitors, and opioids, the prolonged use of all of these treatments is limited due to the occurrence of side effects [81, 82]. Based on our findings, we propose that regular consumption of cocoa would protect against the development of persistent peripheral and central trigeminal sensitization that can lead to potential pathological responses caused by prolonged joint inflammation. Furthermore, we speculate that dietary cocoa would provide a nutraceutical means for repressing the development of central sensitization, a key component of inflammatory pain, and thus be useful in the treatment of TMD and possibly other chronic orofacial pain conditions.
Supplementary Material
Acknowledgments
This study was funded by NIH (DE017805).
Abbreviations
- CGRP
calcitonin gene-related peptide
- CFA
complete Freund’s adjuvant
- GFAP
glial fibrillary-associated protein
- GLAST
glutamate-aspartate transporter
- PKA
protein kinase A
- MAP
mitogen-activated kinase
- MKP
MAP kinase phosphatase
- STN
spinal trigeminal nucleus
- TMD
temporomandibular disorder
- TMJ
temporomandibular joint
Footnotes
Conflict of Interest
The authors declare that they have no potential conflict of interest.
References
- 1.Dworkin SF, LeResche L, DeRouen T, Von Korff M. Assessing clinical signs of temporomandibular disorders: reliability of clinical examiners. J Prosthet Dent. 1990;63:574–579. doi: 10.1016/0022-3913(90)90079-r. [DOI] [PubMed] [Google Scholar]
- 2.Al-Jundi MA, John MT, Setz JM, Szentpetery A, Kuss O. Meta-analysis of treatment need for temporomandibular disorders in adult nonpatients. J Orofac Pain. 2008;22:97–107. [PubMed] [Google Scholar]
- 3.Isong U, Gansky SA, Plesh O. Temporomandibular joint and muscle disorder-type pain in U.S. adults: the National Health Interview Survey. J Orofac Pain. 2008;22:317–322. [PMC free article] [PubMed] [Google Scholar]
- 4.Herb K, Cho S, Stiles MA. Temporomandibular joint pain and dysfunction. Curr Pain Headache Rep. 2006;10:408–414. doi: 10.1007/s11916-006-0070-7. [DOI] [PubMed] [Google Scholar]
- 5.Bereiter DA, Okamoto K, Bereiter DF. Effect of persistent monoarthritis of the temporomandibular joint region on acute mustard oil-induced excitation of trigeminal subnucleus caudalis neurons in male and female rats. Pain. 2005;117:58–67. doi: 10.1016/j.pain.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 6.Shimizu K, Guo W, Wang H, Zou S, et al. Differential involvement of trigeminal transition zone and laminated subnucleus caudalis in orofacial deep and cutaneous hyperalgesia: the effects of interleukin-10 and glial inhibitors. Mol Pain. 2009;5:75. doi: 10.1186/1744-8069-5-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xie YF, Zhang S, Chiang CY, Hu JW, et al. Involvement of glia in central sensitization in trigeminal subnucleus caudalis (medullary dorsal horn) Brain Behav Immun. 2007;21:634–641. doi: 10.1016/j.bbi.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 8.Damodaram S, Thalakoti S, Freeman SE, Garrett FG, Durham PL. Tonabersat inhibits trigeminal ganglion neuronal-satellite glial cell signaling. Headache. 2009;49:5–20. doi: 10.1111/j.1526-4610.2008.01262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ren K. Emerging role of astroglia in pain hypersensitivity. Jpn Dent Sci Rev. 2010;46:86. doi: 10.1016/j.jdsr.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo W, Wang H, Watanabe M, Shimizu K, et al. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27:6006–6018. doi: 10.1523/JNEUROSCI.0176-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Li GW, Wang C, Gu Y, Huang LY. Mechanisms underlying enhanced P2X receptor-mediated responses in the neuropathic pain state. Pain. 2005;119:38–48. doi: 10.1016/j.pain.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 12.Xie YF. Glial involvement in trigeminal central sensitization. Acta Pharmacol Sin. 2008;29:641–645. doi: 10.1111/j.1745-7254.2008.00801.x. [DOI] [PubMed] [Google Scholar]
- 13.Seybold VS. The role of peptides in central sensitization. Handb Exp Pharmacol. 2009:451–491. doi: 10.1007/978-3-540-79090-7_13. [DOI] [PubMed] [Google Scholar]
- 14.Burnstock G. Purinergic signalling--an overview. Novartis Found Symp. 2006;276:26–48. discussion 48–57: 275–281. [PubMed] [Google Scholar]
- 15.Wang Z, Ma W, Chabot JG, Quirion R. Cell-type specific activation of p38 and ERK mediates calcitonin gene-related peptide involvement in tolerance to morphine-induced analgesia. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2009;23:2576–2586. doi: 10.1096/fj.08-128348. [DOI] [PubMed] [Google Scholar]
- 16.Burnstock G, Wood JN. Purinergic receptors: their role in nociception and primary afferent neurotransmission. Curr Opin Neurobiol. 1996;6:526–532. doi: 10.1016/s0959-4388(96)80060-2. [DOI] [PubMed] [Google Scholar]
- 17.Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aley KO, Levine JD. Role of protein kinase A in the maintenance of inflammatory pain. J Neurosci. 1999;19:2181–2186. doi: 10.1523/JNEUROSCI.19-06-02181.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hucho T, Levine JD. Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron. 2007;55:365–376. doi: 10.1016/j.neuron.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 20.Uceyler N, Schafers M, Sommer C. Mode of action of cytokines on nociceptive neurons. Exp Brain Res. 2009;196:67–78. doi: 10.1007/s00221-009-1755-z. [DOI] [PubMed] [Google Scholar]
- 21.Ji RR, Gereau RWt, Malcangio M, Strichartz GR. MAP kinase and pain. Brain research reviews. 2009;60:135–148. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shanley TP. Phosphatases: Counterregulatory role in inflammatory cell signaling. Crit Care Med. 2002;30:S80–S88. [PubMed] [Google Scholar]
- 23.Dickinson RJ, Keyse SM. Diverse physiological functions for dual-specificity MAP kinase phosphatases. J Cell Sci. 2006;119:4607–4615. doi: 10.1242/jcs.03266. [DOI] [PubMed] [Google Scholar]
- 24.Lasa M, Abraham SM, Boucheron C, Saklatvala J, Clark AR. Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol Cell Biol. 2002;22:7802–7811. doi: 10.1128/MCB.22.22.7802-7811.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clark AR. MAP kinase phosphatase 1: a novel mediator of biological effects of glucocorticoids? J Endocrinol. 2003;178:5–12. doi: 10.1677/joe.0.1780005. [DOI] [PubMed] [Google Scholar]
- 26.Cady RJ, Durham PL. Cocoa-enriched diets enhance expression of phosphatases and decrease expression of inflammatory molecules in trigeminal ganglion neurons. Brain Res. 2010;1323:18–32. doi: 10.1016/j.brainres.2010.01.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grassi D, Lippi C, Necozione S, Desideri G, Ferri C. Short-term administration of dark chocolate is followed by a significant increase in insulin sensitivity and a decrease in blood pressure in healthy persons. The American journal of clinical nutrition. 2005;81:611–614. doi: 10.1093/ajcn/81.3.611. [DOI] [PubMed] [Google Scholar]
- 28.Selmi C, Mao T, Keen C, Schmitz H, Eric Gershwin M. The anti-inflammatory properties of cocoa flavanols. J Cardiovasc Pharmacol. 2006;47:S163–S176. doi: 10.1097/00005344-200606001-00010. [DOI] [PubMed] [Google Scholar]
- 29.Dillinger TL, Barriga P, Escarcega S, Jimenez M, et al. Food of the gods: cure for humanity? A cultural history of the medicinal and ritual use of chocolate. The Journal of nutrition. 2000;130:2057S–2072S. doi: 10.1093/jn/130.8.2057S. [DOI] [PubMed] [Google Scholar]
- 30.Newman DJ, Cragg GM. Natural products as sources of new drugs over the last 25 years. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 31.Vuorelaa P, Leinonenb M, Saikkuc P, Tammelaa P, et al. Natural products in the process of finding new drug candidates. Curr Med Chem. 2004;11:1375–1389. doi: 10.2174/0929867043365116. [DOI] [PubMed] [Google Scholar]
- 32.Katz DL, Doughty K, Ali A. Cocoa and chocolate in human health and disease. Antioxidants & redox signaling. 2011;15:2779–2811. doi: 10.1089/ars.2010.3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jourdain C, Tenca G, Deguercy A, Troplin P, Poelman D. In-vitro effects of polyphenols from cocoa and beta-sitosterol on the growth of human prostate cancer and normal cells. European journal of cancer prevention : the official journal of the European Cancer Prevention Organisation. 2006;15:353–361. doi: 10.1097/00008469-200608000-00009. [DOI] [PubMed] [Google Scholar]
- 34.Singleton V, Rossi J. Colorimetry of total phenolics with phosphomolybdicphosphotungstic acid reagents. Am J Enol Vitic. 1965;16:144–158. [Google Scholar]
- 35.Garrett FG, Durham PL. Differential expression of connexins in trigeminal ganglion neurons and satellite glial cells in response to chronic or acute joint inflammation. Neuron Glia Biol. 2008;4:295–306. doi: 10.1017/S1740925X09990093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cady RJ, Hirst JJ, Durham PL. Dietary grape seed polyphenols repress neuron and glia activation in trigeminal ganglion and trigeminal nucleus caudalis. Mol Pain. 2010;6:91. doi: 10.1186/1744-8069-6-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cady RJ, Glenn JR, Smith KM, Durham PL. Calcitonin gene-related peptide promotes cellular changes in trigeminal neurons and glia implicated in peripheral and central sensitization. Mol Pain. 2011;7:94. doi: 10.1186/1744-8069-7-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harper RP, Kerins CA, McIntosh JE, Spears R, Bellinger LL. Modulation of the inflammatory response in the rat TMJ with increasing doses of complete Freund’s adjuvant. Osteoarthritis Cartilage. 2001;9:619–624. doi: 10.1053/joca.2001.0461. [DOI] [PubMed] [Google Scholar]
- 39.Hutchins B, Spears R, Hinton RJ, Harper RP. Calcitonin gene-related peptide and substance P immunoreactivity in rat trigeminal ganglia and brainstem following adjuvant-induced inflammation of the temporomandibular joint. Arch Oral Biol. 2000;45:335–345. doi: 10.1016/s0003-9969(99)00129-6. [DOI] [PubMed] [Google Scholar]
- 40.Thut PD, Hermanstyne TO, Flake NM, Gold MS. An operant conditioning model to assess changes in feeding behavior associated with temporomandibular joint inflammation in the rat. J Orofac Pain. 2007;21:7–18. [PubMed] [Google Scholar]
- 41.Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20:467–473. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki I, Harada T, Asano M, Tsuboi Y, et al. Phosphorylation of ERK in trigeminal spinal nucleus neurons following passive jaw movement in rats with chronic temporomandibular joint inflammation. J Orofac Pain. 2007;21:225–231. [PubMed] [Google Scholar]
- 43.Ren K. Neuron, Glia and Reciprocal Relationships in Pain Processing. Open Pain J. 2009;2:7–31. doi: 10.2174/1876386301003010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sessle BJ. Peripheral and central mechanisms of orofacial inflammatory pain. International review of neurobiology. 2011;97:179–206. doi: 10.1016/B978-0-12-385198-7.00007-2. [DOI] [PubMed] [Google Scholar]
- 45.Anderson LE, Seybold VS. Calcitonin gene-related peptide regulates gene transcription in primary afferent neurons. J Neurochem. 2004;91:1417–1429. doi: 10.1111/j.1471-4159.2004.02833.x. [DOI] [PubMed] [Google Scholar]
- 46.Sun RQ, Tu YJ, Lawand NB, Yan JY, et al. Calcitonin gene-related peptide receptor activation produces PKA- and PKC-dependent mechanical hyperalgesia and central sensitization. J Neurophysiol. 2004;92:2859–2866. doi: 10.1152/jn.00339.2004. [DOI] [PubMed] [Google Scholar]
- 47.Xin WJ, Weng HR, Dougherty PM. Plasticity in expression of the glutamate transporters GLT-1 and GLAST in spinal dorsal horn glial cells following partial sciatic nerve ligation. Mol Pain. 2009;5:15. doi: 10.1186/1744-8069-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fitzgerald EM, Okuse K, Wood JN, Dolphin AC, Moss SJ. cAMP-dependent phosphorylation of the tetrodotoxin-resistant voltage-dependent sodium channel SNS. J Physiol. 1999;516(Pt 2):433–446. doi: 10.1111/j.1469-7793.1999.0433v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kawasaki Y, Kohno T, Zhuang ZY, Brenner GJ, et al. Ionotropic and metabotropic receptors, protein kinase A, protein kinase C, and Src contribute to C-fiber-induced ERK activation and cAMP response element-binding protein phosphorylation in dorsal horn neurons, leading to central sensitization. J Neurosci. 2004;24:8310–8321. doi: 10.1523/JNEUROSCI.2396-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simonetti M, Giniatullin R, Fabbretti E. Mechanisms mediating the enhanced gene transcription of P2X3 receptor by calcitonin gene-related peptide in trigeminal sensory neurons. J Biol Chem. 2008;283:18743–18752. doi: 10.1074/jbc.M800296200. [DOI] [PubMed] [Google Scholar]
- 51.Nakatsuka T, Tsuzuki K, Ling JX, Sonobe H, Gu JG. Distinct roles of P2X receptors in modulating glutamate release at different primary sensory synapses in rat spinal cord. J Neurophysiol. 2003;89:3243–3252. doi: 10.1152/jn.01172.2002. [DOI] [PubMed] [Google Scholar]
- 52.Burnstock G. Pathophysiology and therapeutic potential of purinergic signaling. Pharmacol Rev. 2006;58:58–86. doi: 10.1124/pr.58.1.5. [DOI] [PubMed] [Google Scholar]
- 53.Ramos KM, Lewis MT, Morgan KN, Crysdale NY, et al. Spinal upregulation of glutamate transporter GLT-1 by ceftriaxone: therapeutic efficacy in a range of experimental nervous system disorders. Neuroscience. 2010;169:1888–1900. doi: 10.1016/j.neuroscience.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rathee PK, Distler C, Obreja O, Neuhuber W, et al. PKA/AKAP/VR-1 module: A common link of Gs-mediated signaling to thermal hyperalgesia. J Neurosci. 2002;22:4740–4745. doi: 10.1523/JNEUROSCI.22-11-04740.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Giniatullin R, Nistri A, Fabbretti E. Molecular mechanisms of sensitization of pain-transducing P2X3 receptors by the migraine mediators CGRP and NGF. Molecular neurobiology. 2008;37:83–90. doi: 10.1007/s12035-008-8020-5. [DOI] [PubMed] [Google Scholar]
- 56.Ji RR. Peripheral and central mechanisms of inflammatory pain, with emphasis on MAP kinases. Curr Drug Targets Inflamm Allergy. 2004;3:299–303. doi: 10.2174/1568010043343804. [DOI] [PubMed] [Google Scholar]
- 57.Ji RR, Suter MR. p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain. 2007;3:33. doi: 10.1186/1744-8069-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Crown ED, Ye Z, Johnson KM, Xu GY, et al. Increases in the activated forms of ERK 1/2, p38 MAPK, and CREB are correlated with the expression of at-level mechanical allodynia following spinal cord injury. Experimental neurology. 2006;199:397–407. doi: 10.1016/j.expneurol.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 59.Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. 2008;28:5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hammer M, Echtenachter B, Weighardt H, Jozefowski K, et al. Increased inflammation and lethality of Dusp1−/− mice in polymicrobial peritonitis models. Immunology. 2010;131:395–404. doi: 10.1111/j.1365-2567.2010.03313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vandevyver S, Dejager L, Van Bogaert T, Kleyman A, et al. Glucocorticoid receptor dimerization induces MKP1 to protect against TNF-induced inflammation. J Clin Invest. 2012;122:2130–2140. doi: 10.1172/JCI60006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Durham PL, Russo AF. Differential regulation of mitogen-activated protein kinase-responsive genes by the duration of a calcium signal. Mol Endocrinol. 2000;14:1570–1582. doi: 10.1210/mend.14.10.0529. [DOI] [PubMed] [Google Scholar]
- 63.Durham PL, Russo AF. Stimulation of the calcitonin gene-related peptide enhancer by mitogen-activated protein kinases and repression by an antimigraine drug in trigeminal ganglia neurons. J Neurosci. 2003;23:807–815. doi: 10.1523/JNEUROSCI.23-03-00807.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Svensson CI, Marsala M, Westerlund A, Calcutt NA, et al. Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem. 2003;86:1534–1544. doi: 10.1046/j.1471-4159.2003.01969.x. [DOI] [PubMed] [Google Scholar]
- 65.Ren K, Torres R. Role of interleukin-1beta during pain and inflammation. Brain research reviews. 2009;60:57–64. doi: 10.1016/j.brainresrev.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Andujar I, Recio MC, Giner RM, Rios JL. Cocoa polyphenols and their potential benefits for human health. Oxidative medicine and cellular longevity. 2012;2012:906252. doi: 10.1155/2012/906252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Faridi Z, Njike VY, Dutta S, Ali A, Katz DL. Acute dark chocolate and cocoa ingestion and endothelial function: a randomized controlled crossover trial. The American journal of clinical nutrition. 2008;88:58–63. doi: 10.1093/ajcn/88.1.58. [DOI] [PubMed] [Google Scholar]
- 68.Ramos-Romero S, Perez-Cano FJ, Ramiro-Puig E, Franch A, Castell M. Cocoa intake attenuates oxidative stress associated with rat adjuvant arthritis. Pharmacological research : the official journal of the Italian Pharmacological Society. 2012;66:207–212. doi: 10.1016/j.phrs.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 69.Jansen PJ, Lutjohann D, Abildayeva K, Vanmierlo T, et al. Dietary plant sterols accumulate in the brain. Biochimica et biophysica acta. 2006;1761:445–453. doi: 10.1016/j.bbalip.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 70.Vanmierlo T, Weingartner O, van der Pol S, Husche C, et al. Dietary intake of plant sterols stably increases plant sterol levels in the murine brain. J Lipid Res. 2012;53:726–735. doi: 10.1194/jlr.M017244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Spencer JP, Vafeiadou K, Williams RJ, Vauzour D. Neuroinflammation: modulation by flavonoids and mechanisms of action. Molecular aspects of medicine. 2012;33:83–97. doi: 10.1016/j.mam.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 72.Williams RJ, Spencer JP, Rice-Evans C. Flavonoids: antioxidants or signalling molecules? Free Radic Biol Med. 2004;36:838–849. doi: 10.1016/j.freeradbiomed.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 73.Nehlig A. The neuroprotective effects of cocoa flavanol and its influence on cognitive performance. British journal of clinical pharmacology. 2012 doi: 10.1111/j.1365-2125.2012.04378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Spencer JP. Flavonoids and brain health: multiple effects underpinned by common mechanisms. Genes & nutrition. 2009;4:243–250. doi: 10.1007/s12263-009-0136-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mahajan SG, Mehta AA. Suppression of ovalbumin-induced Th2-driven airway inflammation by beta-sitosterol in a guinea pig model of asthma. Eur J Pharmacol. 2011;650:458–464. doi: 10.1016/j.ejphar.2010.09.075. [DOI] [PubMed] [Google Scholar]
- 76.Valerio M, Awad AB. beta-Sitosterol down-regulates some pro-inflammatory signal transduction pathways by increasing the activity of tyrosine phosphatase SHP-1 in J774A. 1 murine macrophages. International immunopharmacology. 2011;11:1012–1017. doi: 10.1016/j.intimp.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 77.Bonjardim LR, Lopes-Filho RJ, Amado G, Albuquerque RL, Jr, Goncalves SR. Association between symptoms of temporomandibular disorders and gender morphological occlusion psychological factors in a group of university students. Indian journal of dental research : official publication of Indian Society for Dental Research. 2009;20:190–194. doi: 10.4103/0970-9290.52901. [DOI] [PubMed] [Google Scholar]
- 78.Sessle B. The neurobiology of facial and dental pain: present knowledge, future directions. Journal of Dental Research. 1987;66:962–981. doi: 10.1177/00220345870660052201. [DOI] [PubMed] [Google Scholar]
- 79.Lipton J, Ship JA, Larach-Robinson D. Estimated prevalence and distribution of reported orofacial pain in the United States. Journal of American Dental Association. 1993;124:115–121. doi: 10.14219/jada.archive.1993.0200. [DOI] [PubMed] [Google Scholar]
- 80.Carlsson G, LeResche L. Temporomandibular disorders and related pain conditions. In: Barry J, Sessle PSB, Dionne Raymond A, editors. Progress in pain research and management. IASP Press; Seattle: 1995. pp. 211–226. [Google Scholar]
- 81.Funk CD, FitzGerald GA. COX-2 inhibitors and cardiovascular risk. Journal of cardiovascular pharmacology. 2007;50:470–479. doi: 10.1097/FJC.0b013e318157f72d. [DOI] [PubMed] [Google Scholar]
- 82.Triantafyllou K, Vlachogiannakos J, Ladas SD. Gastrointestinal and liver side effects of drugs in elderly patients. Best practice & research Clinical gastroenterology. 2010;24:203–215. doi: 10.1016/j.bpg.2010.02.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.