

Abstract
Agnoprotein is one of the key regulatory proteins of polyomaviruses, including JCV, BKV and SV40 and is required for a productive viral life cycle. We have recently reported that agnoprotein forms stable dimer/oligomers mediated by a predicted amphipathic α-helix, spanning amino acids (aa), 17 to 42. Deletion of the α-helix renders a replication incompetent virus. Here, we have further characterized this region by a systematic deletion and substitution mutagenesis and demonstrated that a Leu/Ile/Phe-rich domain, (spanning aa 28–39) within α-helix is indispensable for agnoprotein structure and function. Deletion of aa 30–37 severely affects the dimer/oligomer formation and stable expression of the protein. Mutagenesis data also indicate that the residues, 34–36, may be involved in regulation of the splicing events of JCV transcripts. Collectively, these data suggest that the Leu/Ile/Phe-rich domain plays critical roles in agnoprotein function and thus represents a potential target for developing novel therapeutics against JCV infections.
Keywords: Polyomavirus JCV, SV40, BKV, Merkel cell carcinoma virus, Large T antigen, Agnoprotein, Transcription, DNA replication, Progressive multifocal leukoencephalopathy, Splicing, Stable dimer formation, Stable oligomer formation
Introduction
Proteins fulfill many complex functions through highly specific interactions with their binding partners. These interactions can occur between two identical proteins (homo-dimerization/oligomerization); or between two or more different proteins (heterodimerization/oligomerization). Such interactions provide the participant proteins specificity and diversity to perform their functions successfully in many different biological processes including DNA replication, gene transcription and regulation of different pathways.
Viruses are obligatory intracellular parasites and rely heavily on the mechanisms and functions provided by the host cells for their propagation. Some viruses encode a limited number of regulatory proteins and yet successfully complete their replication cycle, suggesting a multi-functional nature for these proteins. This multi-functionality mostly stems from their ability to form diverse but higher ordered dimeric, heteromeric or oligomeric structures. There are many examples of such structures among the viral proteins that have been reported in the literature, including human immunodeficiency virus 1 (HIV-1) proteins (Rev, Vpr, Vif and Vpu) (Bernacchi et al., 2011; Bourbigot et al., 2005; Daugherty et al., 2010a, 2010b; Lu et al., 2010), Hepatitis C virus nonstructural protein 4B (Gouttenoire et al., 2010), Ebola virus VP40 (Hoenen et al., 2010), and large T-antigen (LT-Ag) of polyomaviruses (Cuesta et al., 2010; Foster and Simmons, 2010). Recently, agnoprotein of polyomaviruses was also shown to operate in this fashion. We have reported the formation of highly stable dimeric/oligomeric structures by the agnoprotein of JC virus (JCV), BK virus (BKV) and simian virus 40 (SV40), and roughly mapped the region responsible for this phenomenon to amino acids (aa) spanning from 17 to 42 for JCV agnoprotein (Saribas et al., 2011).
JCV agnoprotein is a small (71 aa long), multifunctional, regulatory protein encoded by the late coding region of the viral genome and was previously shown to interact with a number of cellular and viral proteins, including p53 (Darbinyan et al., 2002), YB-1 (Safak et al., 2002), FEZ1 and HP1-α (Okada et al., 2005), JCV small t-antigen (Sm t-Ag) (Sariyer et al., 2008) and JCV large T-antigen (LT-Ag) (Safak and Khalili, 2001). It has been implicated in many different aspects of the JCV life cycle, including viral transcription (Safak and Khalili, 2001), replication (Safak and Khalili, 2001), functioning as viroporin (Suzuki et al., 2010), encapsidation (Sariyer et al., 2006) and interfering with the process of exocytosis (Johannessen et al., 2011). In addition, this protein was found to deregulate cell cycle progression, where cells that stably express agnoprotein largely accumulate at the G2/M phase of the cell cycle (Darbinyan et al., 2002). Despite these previous reports and intense research, the regulatory function of agnoprotein in viral replication is not fully understood.
JCV agnoprotein shows a high degree of sequence identity and similarity to that of SV40 and BKV (Safak et al., 2001). For example, there is a 60% identity and a 79% similarity between JCV and SV40 agnoprotein sequences respectively, and 82% and 93% respectively between JCV and BKV agnoprotein (Altschuler, 1999). SV40 and BKV agnoprotein, like JCV agnoprotein, have also been previously implicated in different aspects of viral replication cycle (Alwine, 1982; Haggerty et al., 1989; Hay et al., 1982; Hou-Jong et al., 1987; Johannessen et al., 2008; Margolskee and Nathans, 1983; Moens et al., 2011; Myhre et al., 2010; Ng et al., 1985; Unterstab et al., 2010).
Expression of agnoprotein is required for the successful propagation of Orthopolyomaviruses, including JCV, BKV and SV40, because an agnoprotein null mutant of each virus is unable to sustain the replication cycle although mutant viruses were shown to be successfully released from the infected cells (Myhre et al., 2010; Sariyer et al., 2011). Most importantly, even the constitutive expression of LT-Ag, required for polyomavirus DNA replication, is not sufficient to sustain an efficient viral propagation cycle in the absence of agnoprotein, which further emphasizes the importance of agnoprotein in viral life cycle (Sariyer et al., 2011). Agnoprotein mostly resides in cytoplasm with high concentrations accumulating in the perinuclear region of the infected cells. However, a small amount of the protein is also consistently detected in the nucleus, indicating a possible nuclear function for agnoprotein. In fact, our recent DNA binding studies supported this idea demonstrating that agnoprotein stimulates the DNA binding activity of LT-Ag without directly interacting with DNA (Saribas et al., 2012). In addition, we have also recently reported that agnoprotein forms highly stable, SDS-resistant homodimers and oligomers; and the 17–42 aa region of the protein is responsible for this property (Saribas et al., 2011). While the 3D structure of agnoprotein has yet to be determined, computer modeling studies predict that the 17–42 aa, which is important for stable dimer/oligomer formation, is involved in forming an amphipathic α-helical structure (Saribas et al., 2011; Unterstab et al., 2010). This α-helical region contains a Leu/Ile/Phe-rich domain between aa 28 and 39. All three Phe residues (Phe31, Phe35 and Phe39) of JCV agnoprotein localize to this Leu/Ile/Phe-rich domain along with two negatively charged residues, Glu34 and Asp38. The Phe residues of JCV agnoprotein appear to be involved in viral DNA replication (Saribas et al., 2012). With respect to the function of Phe residues, the protein distribution studies with confocal microscopy showed that Phe39 residue of BKV agnoprotein may play a role in localization of the protein to the lipid droplets in infected cells (Unterstab et al., 2010). In addition to in vitro stable dimer/oligomer formation, we have recently reported to dimer formation in vivo in the infected cells (Saribas et al., 2011). Suzuki et al. (2010) have also demonstrated homodimer, homo-oligomer formation in vivo by JCV agnoprotein, by employing intermolecular fluorescence resonance energy transfer (FRET) and chemical cross-linking techniques.
All these studies strongly suggest that the role of agnoprotein in JCV replication cycle is critical and therefore in progressive multifocal leukoencephalopathy (PML), where JCV lytically infects the oligodendrocytes in the central nervous system (CNS). This ubiquitous human polyomavirus, JCV, infects a majority of the human population worldwide (70–80%). Initial infection by this virus is followed by a latency period. The reactivation of the virus mainly occurs in patients with immunosuppressive conditions (1–5%), including in those with cancer, organ transplant, and HIV-1/AIDS (Berger, 2011; Berger and Concha, 1995; Major, 2010; Major et al., 1992). In addition, a small percentage of patients with autoimmune diseases such as multiple sclerosis (MS) and severe psoriasis (Crohn’s disease) who underwent an immunosuppressive therapy with specific monoclonal antibodies (such as natalizumab, rituximab and efalizumab) developed PML (Kleinschmidt-DeMasters and Tyler, 2005; Langer-Gould et al., 2005; Van Assche et al., 2005), suggesting that JCV is an important risk factor in these patients.
In this study, we further examined the region of JCV agnoprotein responsible for formation of highly stable dimers and oligomers using mutagenesis analysis. Our data demonstrate that aa from 30 to 37 are sufficient to confer the dimer/oligomer formation property of agnoprotein. Consistent with our in vitro mapping studies, functional studies demonstrated that the deletion mutants, partially or completely missing the Leu/Ile/Phe-rich domain, showed a significant decrease in viral replication compared to wild-type. In addition, transcriptional analysis studies with the mutants revealed interesting defects in splicing patterns of the viral late transcripts, suggesting a new novel function for agnoprotein in JCV gene regulation at the post-transcriptional level. Collectively, these findings demonstrate that the Leu/Ile/Phe-rich domain is critically important for the dimer/oligomer formation, stability and function of agnoprotein and thus represents a potential target for developing novel therapeutic agents for progressive multifocal leukoencephalopathy.
Results
Structural features of JC virus agnoprotein
Agnoprotein is one of the few regulatory proteins of JCV. It possesses a highly basic aa composition with dominant Arg (R) and Lys (K) residues, which are located at the amino and carboxy ends of the protein (Fig. 1A). However, the middle portion of the protein, encompassing aa from 28 to 39, is composed of mostly hydrophobic residues, including Ile, Leu and Phe. The two negatively charged residues Glu34 (E34) and Asp38 (D38) are also located within this region. It is also interesting to note that all three Phe residues (Phe31, Phe35 and Phe39) of JCV agnoprotein are evenly dispersed and confined within the Leu/Ile/Phe-rich domain (Fig. 1A). We have recently analyzed the functional importance of these Phe residues by site-directed mutagenesis and the results showed that they have a combinatorial effect on viral replication cycle (Saribas et al., 2012). The only cysteine residue of agnoprotein (Cys40) is found towards the middle portion of the protein right after the Leu/Ile/Phe-rich domain. The three dimensional (3D) structure of agnoprotein is yet to be resolved, however, computer modeling studies shown in Fig. 1B using the I-TASSER progam (Roy et al., 2010) suggest that the Leu/Ile/Phe-rich domain is involved in forming an amphipathic α-helix (Fig. 1B and Fig. 1C). The Leu/Ile/Phe-rich domain is highly conserved between agnoprotein of JCV, SV40 and BKV (Fig. 1D). Three D modeling studies also suggest that carboxyl terminal half of the protein encompassing aa from 40 to 71 as well as a small portion of the amino terminal region of the protein may adopt an intrinsically disordered conformation. That is, both regions lack fixed tertiary structures (Fig. 1B). This type of disordered conformation results from their high net charge and low overall hydrophobic character (Dyson and Wright, 2005; Fink, 2005). It has been suggested that such intrinsically disordered regions may provide considerably high flexibility to a protein to interact with its various partners in a given time in a cellular environment so that these types of proteins diversify and perhaps amplify their functions (Dyson and Wright, 2005; Fink, 2005). For example, Ou et al. (2012) recently demonstrated that adenovirus small protein E4-ORF3 dimerizes/oligomerizes and eventually forms a multivalent functional matrix in the infected cells, which leads to (Sariyer et al., 2011) the inactivation of several tumor suppressor proteins including p53, TIM24, MRE11/Rad50/NBS1 (MRN) and promyelocytic leukemia factor (PML). The majority of aa sequences, except those located within the Leu/Ile/Phe-rich domain of agnoprotein, appears to have such intrinsically unstructured properties, which could play important roles in agnoprotein functions with respect to interaction of the protein with its partners in infected cells and thereby contribute significantly to the overall viral replication cycle.
Fig. 1.
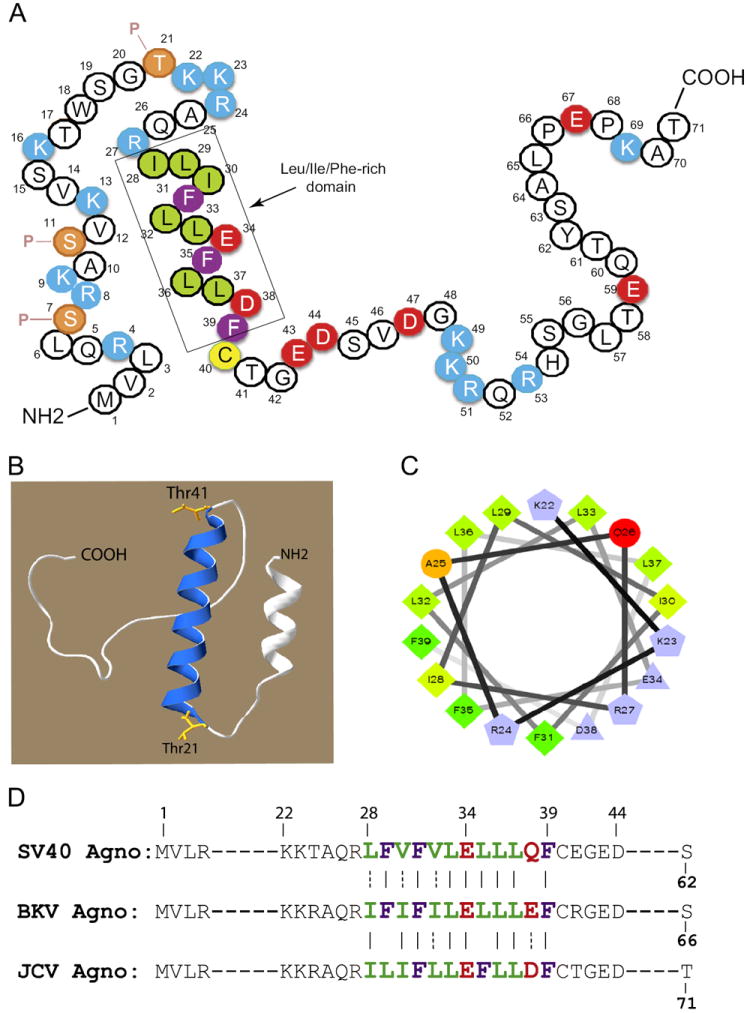
Primary and predicted structural features of JCV agnoprotein. (A) Primary structure of agnoprotein. The Leu/Ile/Phe-rich domain (aa 28 to 39) is indicated. Previously characterized phosphorylation sites (Ser7, Ser11 and Ser21) are also indicated (Sariyer et al., 2001). P stands for phosphorylation. (B) Predicted three dimensional (3D) structure of agnoprotein was generated by I-TASSER program (Roy et al., 2010). This 3D structure indicates that the Leu/Ile/Phe-rich domain of agnoprotein is involved in formation of α-helix (helical wheel). (C) The predicted α-helix has an amphipathic nature. Hydrophobic residues within the α-helix are clustered on one side of the helix and the negatively and positively charged residues are on the opposite site. (D) Amino acid alignments of the Leu/Ile/Phe-rich domain among JCV, BKV and SV40 agnoprotein.
Effect of amino- and carboxy-terminal deletion mutants on stable dimer/oligomer formation by agnoprotein
We have recently reported formation of highly stable dimers/oligomers for agnoprotein of JCV, BKV and SV40 and roughly mapped the region responsible for this phenomenon, for JCV agnoprotein to the aa from 17 to 42. In this study, we sought to further define this region with respect to its ability to form stable dimers and oligomers by employing a systematic mutagenesis analysis of the region. We first started by deleting the region from the amino terminus of agnoprotein. For this purpose, different amino terminal deletion mutants were fused to maltose binding protein (MBP) as described in Materials and methods, expressed in E. coli as MBP-Agno fusion protein and affinity purified. As we recently reported (Saribas et al., 2011), stable dimer and oligomer formation by agnoprotein can be monitored by employing SDS-polyacrylamide gel electrophoresis (SDS-PAGE) under reducing conditions followed by Coomassie blue staining (Fig. 2A). As we previously reported, the full length (FL) agnoprotein readily forms stable dimer and oligomers (Fig. 2A, lane 2) while MBP alone does not (lane 1) (Saribas et al., 2011). Deletion of a 16 aa (17–71 construct, lane 3) or that of a 24 aa region (25–71 construct, lane 4) or a 27 aa region (28–71 construct, lane 5) from the amino terminal did not alter dimer/oligomer formation by agnoprotein. In contrast, a slight increase was observed in the level of stable dimers but not for the oligomers for each construct (17–71, 25–71 and 28–71 constructs). Further deletions of the amino terminus residues including those from the Leu/Ile/Phe-rich domain greatly reduced the ability of agnoprotein to form stable dimers and oligomers (lanes 7–10). For instance, deletion of a 33 aa region from the amino terminus resulted in not only the substantial loss of the level of the dimeric structures (approximately in half) but also completely abrogated the ability of the protein to form oligomers (34–71 construct, lane 6), suggesting that aa 28 to 33 (ILIFLL) are required for stable oligomer formation and in the meantime significantly contribute to dimer formation (34–71 construct, lane 6). Similarly, further elimination of the amino acids belonging to the Leu/Ile/Phe-rich domain from the amino terminus completely abrogated oligomer formation property of agnoprotein and drastically reduced to the levels of dimers to the extent, which are barely detectable (35–71, 36–71 and 37–71 constructs, lanes 7–9, respectively). Elimination of the amino acid residues from the amino terminus including the negatively charged Glu34 severely affected dimer formation. Lastly, as previously reported (Saribas et al., 2011), deletion of the first 42 aa from the amino terminus resulted in a complete abrogation of the formation of dimer and oligomers (lane 10).
Fig. 2.
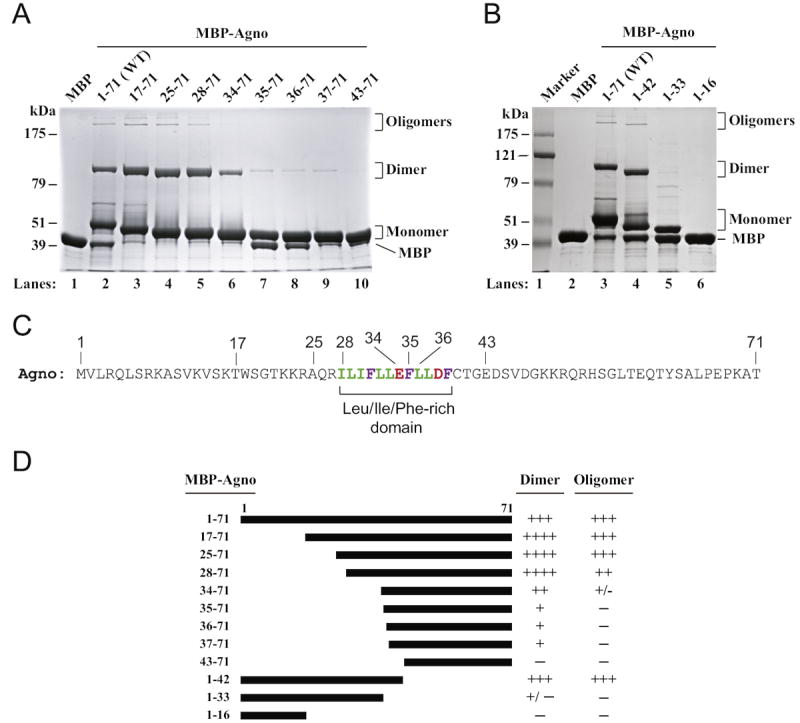
Effect of amino- and carboxy-terminal deletion mutants of agnoprotein on the dimer/oligomer formation. Maltose binding protein (MBP), JCV full length agnoprotein (WT) and amino (A)- and carboxy (B)-terminal deletion mutants of agnoprotein fused to MBP were expressed in bacteria (Escherichia coli) and affinity purified as described in Materials and methods. The ability of each protein to form SDS-resistant dimer and oligomer was tested as described previously (Saribas et al., 2011) by employing an 8%- SDS-PAGE assay followed by Coomassie blue staining. (C) JCV agnoprotein sequence, highlighting the Leu/Ile/Phe-rich domain. (D) Summary of the results with respect to the ability of each protein to form dimer and oligomers relative to those observed for WT. Three plus signs (+++) represent the level of dimer/oligomer formation for the WT agnoprotein. Four plus signs (++++) represent an increased level of dimer formation relative to that of WT. One plus sign (+) represents a substantially reduced level of dimer formation relative to that of WT and a minus sign (−) represents the absence of dimer/oligomer formation.
To further confirm the findings from Fig. 2A, agnoprotein was also subject to a systematic deletion process from the carboxy terminal end and the resulting mutants were analyzed for the stable dimer/oligomer formation as described for Fig. 2A. As reported earlier (Saribas et al., 2011), a mutant, which retains the first 42 aa region, formed a comparable level of dimers and oligomers to wild-type (Fig. 2B, compare lane 4 to 3). Note that this mutant contains the entireLue/Ile/Phe domain and thus supports the idea that a mutant which retains a complete Leu/Ile/Phe-rich domain is capable of forming highly stable dimer and oligomers. In addition, consistent with the results obtained from the amino terminal deletion mutants, deletion of the region, containing 34EFLLDF39 motif, from the carboxyl terminal of the Leu/Ile/Phe-rich domain but retaining the first 33 aa from the amino terminal resulted in a phenotype that almost completely lost the ability to form stable dimer or oligomers (1–33 construct, lane 5) confirming the role of 34EFLLDF39 in dimer/oligomer formation. As previously reported, a mutant retaining the first 16 aa (construct 1–16, lane 6) does not have the ability to form either dimeric or oligomeric structures (Saribas et al., 2011). Taken together, these findings suggest that the Leu/Ile/Phe-rich domain of agnoprotein plays a major role in formation of stable dimers and oligomers. The levels of the dimeric and oligomeric forms of amino terminal deletion mutants relative to those of wild-type are summarized in Fig. 2D.
Deletion of the Leu/Ile/Phe-rich domain resulted in the loss of stable dimer/oligomer formation
We next attempted to further dissect the involvement of the the Leu/Ile/Phe-rich domain in dimer/oligomer formation by creating several internal deletion mutants of this region and testing their ability in oligomerization assays. Deletion of three internal aa (Glu34, Phe35 and Leu36) abrogated the ability of agnoprotein to form stable oligomers and significantly reduced the level of dimer formation (~40% decrease) (Fig. 3A, lane 4). Further widening the deleted region within the Leu/Ile/Phe-rich domain of agnoprotein, [encompassing the aa 30 to 37 (30IFLLEFLL37)] resulted in a phenotype that failed to form both stable dimer and oligomers; and even let the degradation of the monomers (Fig. 3A, lane 5). In addition, as previously reported (Saribas et al., 2011), a wide internal deletion of the region, spanning the amino acids from 17 to 42 also completely abrogated the ability of agnoprotein to form stable dimer, oligomers an monomers (Fig. 3A, lane 6). Taken together, these internal deletion studies further confirm the essential role of the the Leu/Ile/Phe-rich domain in oligomer formation and stability of agnoprotein.
Fig. 3.
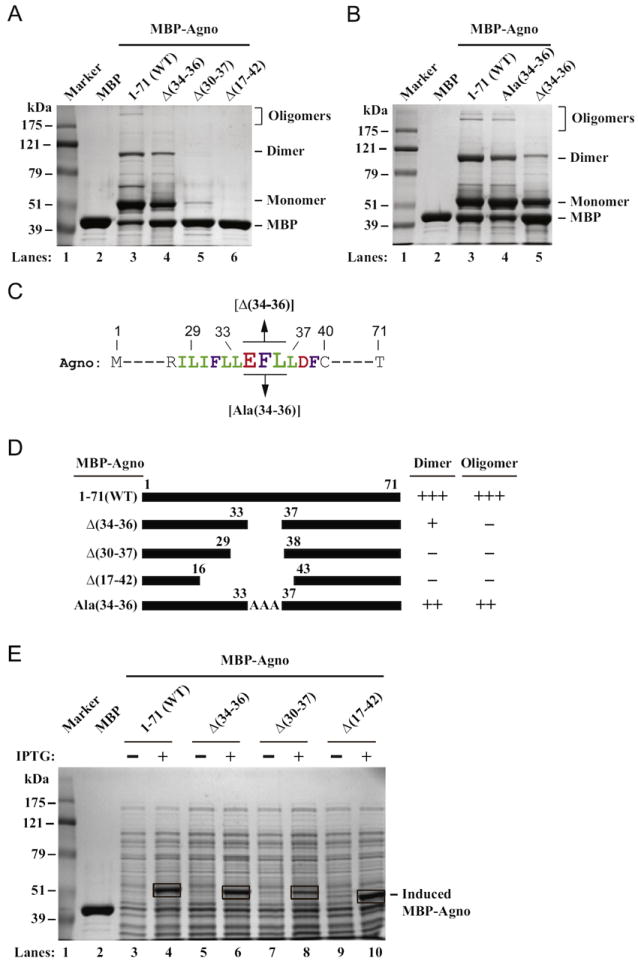
Deletion and substitution analysis of the Leu/Ile/Phe-rich domain of agnoprotein. (A) MBP, MBP-Agno (1–71) and internal deletion mutants of agnoprotein [Δ(34–36), Δ(17–42) and Δ(30–37)] fused to MBP were expressed in E. Coli and affinity purified as described in Materials and methods and stable dimer/oligomer formation by each protein was tested as described in legend for Fig. 2A. (B) Ala substitution analysis of the Glu34, Phe35 and Leu36. Amino acids, Glu34, Phe35 and Leu36, were all substituted with Ala on agnoprotein sequence. Then dimer/oligomer formation property of agnoprotein was tested as described for panel A. (C) Partial agnoprotein primary sequences highlighting the Leu/Ile/Phe-rich domain are shown. (D) Summary of the results with respect to the ability of each protein to form dimer and oligomers relative to those observed for WT as described for Fig. 2D. (E) Direct lysis of the internal deletion mutants of agnoprotein grown in bacteria in sample buffer and analysis of them by 8%-SDSPAGE followed by Coomassie blue staining. The expression plasmids of MBP-Agno WT (1–71) and agnoprotein internal deletion mutants fused to MBP (MBP-Agno Δ(34–36), MBP-Agno Δ(30–37) and MBP-Agno Δ(17–42) were transformed into E. Coli and bacterial cultures (0.25 l each) were grown until OD=0.5 at 37 °C in a culture shaker (225 rpm). One milliliter of bacterial culture (induced with IPTG and uninduced) was pelleted by centrifugation (2 min. at 10,000 × g), resuspended in 50 μL ddH2O, treated with 12.5 μL of SDS loading dye (5 ×) and heated at 98 °C for 5 min. Samples were then spun at 15 krpm for 2 min. Twenty microliters of which were separated on an 8% SDS-PAGE and analyzed by Coomassie blue staining. The bands corresponding to the induced proteins for each construct are indicated by rectangular brackets. In lane 2, purified MBP alone (10 μg) was loaded as a control.
Oligomer formation is lost and dimer formation is attenuated when Glu34, Phe35 and Leu36 are deleted but both properties are restored by Ala substitutions
Internal deletion of, Glu34, Phe35 and Leu36 residues, resulted in the loss of oligomer formation and a substantial reduction in dimer formation (Fig. 3A, lane 4). We wanted to further test whether substitution of these three resides with Ala, which is known to preserve the α-helical nature of a region within a protein, would be able to restore dimer/oligomer formation. In this substitution, our assumption was as follows: A triple Ala substitution will not destroy the α-helical nature of the Leu/Ile/Phe-rich domain and therefore should not significantly affect the dimer/oligomer formation property of agnoprotein. To test this possibility, Glu34, Phe35 and Leu36 all were substituted with Ala residues and the ability of the new mutant to form dimer/oligomers relative to that of WT agnoprotein were investigated by SDS-PAGE/Coomassie blue staining as described in legend to Fig. 3A. Indeed, Ala substitution of three residues (Glu34, Phe35 and Leu36) restored the oligomer formation ability of agnoprotein compared to WT agnoprotein (Fig. 3B compare lanes 3 to 4) although the levels of both were slightly lower than those observed for WT agnoprotein. These results suggest following: (i) The amphipatic nature of the Leu/Ile/Phe-rich domain and (ii) reservation of the α-helical structure of the region are important for the stable dimer/oligomer by agnoprotein. A summary of the results from Fig. 3A and B are shown in Fig. 3D. Finally, the behavior of all the agnoprotein mutants used in this study with respect to their ability to form dimers and oligomers are summarized in Table 1.
Table 1.
The summary of dimer/oligomer formation by JCV Mad-1 WT agnoprotein in MBP fusion form [MBP-Agno (1–71)] and its deletion/substitution mutants.

|
Three plus signs (+++) represent the level of dimer/oligomer formation for the WT agnoprotein. Four plus signs (++++) represent an increased level of dimer formation relative to that of WT. One plus sign (+) represents a substantially reduced level of dimer formation relative to that of WT. Plus/minus sign (+/−) represents a significantly reduced level of dimer formation relative to that of WT and a minus sign (−) represents the absence of dimer/oligomer formation.
Agnoprotein becomes unstable when the Leu/Ile/Phe-rich domain is altered
Elimination of majority of the Leu/Ile/Phe-rich domain [amino acids from 30 to 37 (30IFLLEFLL37)] resulted in not only a complete abrogation of stable dimer and oligomer formation but also greatly reduced the level of monomer production by agnoprotein, suggesting that the Leu/Ile/Phe-rich domain may also play a role in stable expression of agnoprotein. To investigate this possibility, bacterial cultures harboring different MBP-Agno deletion mutants were induced (+) or uninduced (−) by IPTG; and bacterial cell crude extracts were prepared by a rapid boiling and analyzed in a 10%-SDS-PAGE. As shown in Fig. 3E, the induced levels of MBP-Agno WT (Fig. 3E, lane 4) or those of deletion mutants [Agno-Δ (34–36), Agno-Δ(30–37) and Agno-Δ(17–42)] are readily detectable by Coomassie blue staining (Fig. 3E, lanes 6, 8 and 10, respectively). The disappearance of some or all of the monomers, dimers and oligomers on SDS-PAGE on Fig. 3A and B, suggests that these internal deletion mutants [Δ(30–370 and Δ(17–42)] are readily produced upon induction by IPTG, but are rapidly degraded during the protein preparation steps. It should be noted here, that a sufficient amount of protease inhibitors were included in the lysis buffer to inhibit the all the proteases (Fig. 3A). Therefore, the rapid degradation of these two proteins cannot be attributed to a possible case where insufficient amount of protease inhibitors was added to the lysis buffer. As such, one possible explanation for the rapid degradation of the internal deletion mutants of agnoprotein would be that removal of the Leu/Ile/Phe-rich domain renders agnoprotein an intrinsically unstable structure, thereby it leads to degradation, which resembles a previously reported case for heat shock proteins where they undergo a rapid self-degradation process (Mitchell et al., 1985). In addition, there are, of course, other possible ways for rapid degradation of agnoprotein mutants. One of these ways that proteins may also undergo such a degradation process is the selective destruction of the misfolded proteins (Goldberg, 2003). Such a degradation process is also likely for agnoprotein mutants where mutations within agnoprotein Leu/Ile/Phe-rich domain may lead to a misfolding process, which are ultimately detected by bacterial cell degradation systems and eliminated.
Analysis of the Leu/Ile/Phe-rich domain mutants by immunocytochemistry
Next, we analyzed the effect of internal deletion mutants of the Leu/Ile/Phe-rich domain on the expression profile of agnoprotein in the viral background by immunocytochemistry. SVG-A cells were transfected/infected with either Mad-1 WT agnoprotein or a Ala substitution mutant [Mad-1 Agno Ala (34–36)] or two deletion mutants [[Mad-1 Agno Δ(34–36) or [Mad-1 Agno Δ(30–37)], fixed the cell at 5th day posttransfection/infection and probed with primary and secondary antibodies to detect agnoprotein and VP1 as described in the legend to Fig. 4A. Cells were then examined under a fluorescence microscope as described under Materials and methods. As expected, WT agnoprotein displays a heavy cytoplasmic distribution with high concentrations detected around the perinuclear region (Safak et al., 2002) and a small amount of the protein is also localized to the nucleus (Saribas et al., 2012). However, the internal deletion of only three aa of agnoprotein, Δ(34–36) from the viral background drastically reduced the expression profile of the protein to almost undetectable levels (Fig. 4A, Supplement 1). In addition, deletion of aa from 30 to 37 [Δ(30–37)] within the Leu/Ile/Phe-rich domain exhibited a phenotype similar to that observed for a triple deletion mutant [Δ(34–36)], suggesting that this region, aa from 34 to 36 are critical for maintaining the stability of agnoprotein in a stable conformation. In addition to the immunocytochemistry studies shown in Fig. 4A, we also compared the expression profiles of agnoprotein and VP1 at 5th day posttransfection/infection using a lower magnification objective (10 ×) (Supplement 1). Consistent with the results from the high magnification studies (40 ×, Fig. 4A), we also observed an undetectable level of agnoprotein expression for two deletion mutants, [Δ(30–37)] and [Δ(34–36)] at low magnification studies (Supplement 1). Interestingly, the Ala [(34–36)] substitution mutant expressed agnoprotein in a relatively limited number of cells compared to WT (Supplement 1). Observations from these immunocytochemistry assays are consistent with our findings from the in vitro dimerization/oligomerization assays (Fig. 3A). Additionally, our in vivo findings for the [Δ(30–37)] mutant correlates with our in vitro dimer/oligomer formation studies as well. It appears that deletion of majority of the Leu/Ile/Phe-rich domain turned this protein into an intrinsically unstable structure (Fig. 3A, lane 5).
Fig. 4.
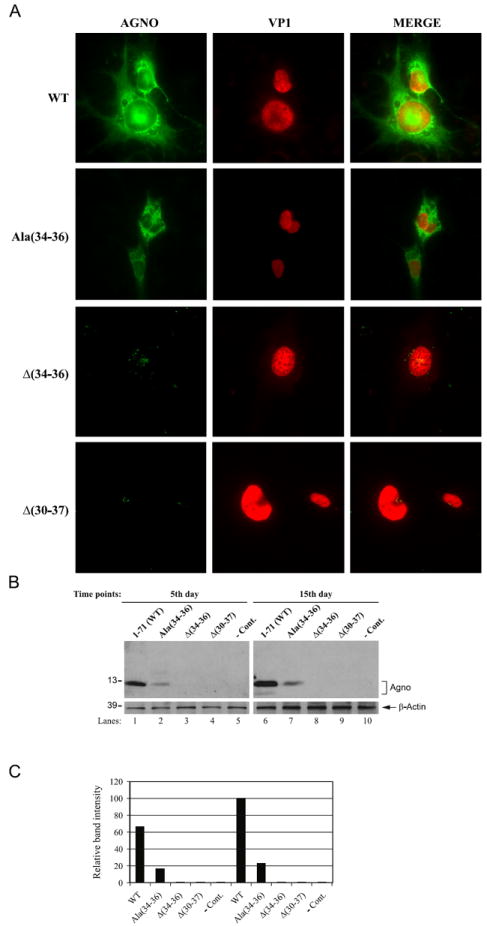
Immunoctyochemistry and Western blot analysis of different mutants of the Leu/Ile/Phe-rich domain of agnoprotein in the viral background. (A) SVGA cells (2 × 106 cells/75 cm2 flask) were transfected with Mad-1 Agno WT, Mad-1 Agno Ala(34–36), Mad-1 Agno Δ(34–36) and Mad-1 Agno Δ(30–37) genomes (8 μg each). The samples were then processed for immunocytochemistry as described under Materials and methods. (B) Detection of agnoprotein by Western blotting. In parallel to the immunocytochemical analysis in (A), whole-cell extracts were prepared from transfected/infected cells at indicated time points and 40 μg of protein extract were separated on a 15% SDS-PAGE and transferred onto a nitrocellulose membrane for 10 min at 250 mA. Blots were then probed with anti-Agno primary polyclonal rabbit antibody (1:1000 dilution) (Del Valle et al., 2002) for 2 h and washed with PBST three times and subsequently incubated with HRP-conjugated anti-rabbit secondary antibody for 45 min. Nonspecific binding of the secondary antibody was eliminated by three times washing of the blots with PBST with 10 min intervals. Blots were finally developed using ECL plus kit (GE HealthCare) according to the Manufacturer’s recommendations for detection of agnoprotein. Blots were also stripped and reprobed for anti-β actin antibody (clone EP1123Y, rabbit monoclonal, Millipore) to monitor the equal loading for each lane. In lane 5 and 10, whole-cell extract prepared from untransfected SVGA cells were loaded as a negative control (− Cont.). (C) Quantitation analysis of the Western blot bands done by a semi-quantitative densitometry method (using NIH Image J program, http://rsb.info.nih.gov/ij) and presentation of the results in arbitrary units. The expression efficiency of each data point was presented relative to that of WT (15th day data point).
In parallel to the immunocytochemical studies, the expression levels of each internal deletion mutant were also analyzed by Western blotting. Whole-cell extracts were separated on a 15% SDS-PAGE, transferred onto a nitrocellulose membrane and probed with an anti-Agno antibody (Del Valle et al., 2002). As shown in Fig. 4B and C, consistent with our results from immunocytochemistry (Fig. 4A), the internal deletions [Δ(34–36) and Δ(30–37)] render agnoprotein an unstable character and therefore it is hard to detect them by Western blotting (Fig. 4B, lanes 3, 4, 8 and 9). Moreover, we also addressed the question of whether substitution of these three residues with Ala will restore a WT-like expression profile for agnoprotein. Ala residues are known to maintain the structure of α-helical integrity if inserted into the region. These Ala substitutions [Ala (34–36)] indeed restored the expression of agnoprotein to an extent which was less than WT. Taken together; these protein expression studies further confirms the importance of the Leu/Ile/Phe-rich domain in stable expression of agnoprotein.
Viral replication was significantly reduced when alterations are made in the Leu/Ile/Phe-rich domain of agnoprotein
We next examined the replication properties of agnoprotein mutants relative to WT by DpnI assays, followed by Southern blotting (Saribas et al., 2011, 2012). JCV Mad-1 WT and mutant viral genomes were individually transfected/infected into SVG-A cells and low molecular weight DNA was isolated at 5th and 15th day posttransfection/infection as described in Materials and methods. In order to obtain a single banded pattern on Southern blots, DNA samples were digested with BamHI enzyme, which cuts JCV genome once and thereby linearize the circular genome. DNA samples were also digested with DpnI enzyme to remove input DNA (bacterially produced and methylated), while keeping newly-replicated viral DNA intact (Hirt, 1967). DNA samples were then analyzed by Southern blotting using a probe prepared from JCV Mad-1 genome as a template. As shown in Fig. 5A and B, the replication efficiency of each mutant is indistinguishable from that of WT (Fig. 5A, lanes 2–5) by the 5th day posttransfection/infection. However, this drastically changed by the 15th day posttransfection/infection. For example, two deletion mutants [Δ (34–36) and Δ(30–37)] substantially lost their replication efficiency (~90% and ~70% respectively), suggesting the importance of Leu/Ile/Phe-rich domain of agnoprotein for JCV replication. Both findings correlate with our results from immunocytochemistry and Western blot analysis studies (Fig. 4A and B), where it was observed that agnoprotein expression was found to be at the undetectable levels for both deletion mutants, [Δ(34–36) and Δ(30–37)]. Additionally, we also tested the ability of Ala(34–36) mutant in restoring the replication efficiency of the virus relative to WT. We did not obtain a full recovery for this mutant compared to WT (Fig. 5A, lane 8) again pointing out the importance of residues within the agnoprotein dimerization domain for the viral DNA replication, particularly to those located at aa 34–36 positions (Glu34, Phe35 and Leu36). In fact, in support of this conclusion, a recent report by our lab demonstrated that substitution of one of these residues, Phe35, with Ala produced a phenotype that replicated less efficiently than WT (Saribas et al., 2012).
Fig. 5.
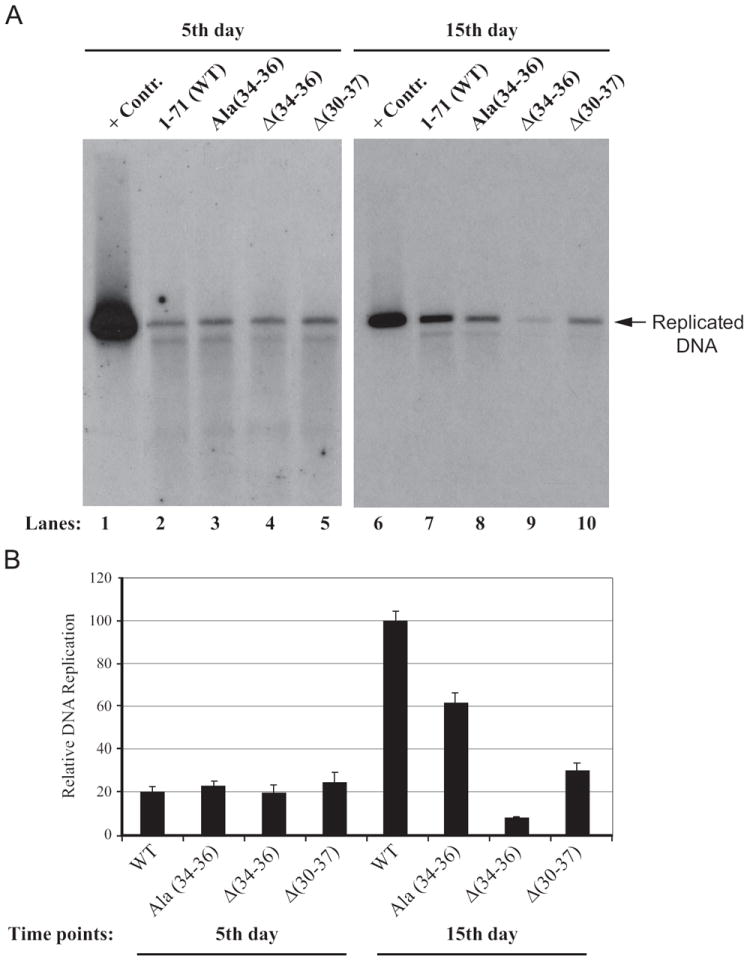
Analysis of the replication efficiency of the Leu/Ile/Phe-rich domain mutants in vivo. In parallel to immunocytochemical analysis of the mutants in Fig. 4A, the low-molecular- weight DNA containing both input and replicated viral DNA was isolated using Qiagen’s spin columns (Ziegler et al., 2004), and digested with BamHI and DpnI restriction enzymes. Digested DNA was separated on a 1% agarose gel, transferred onto a nitrocellulose membrane (Bio-Rad) and probed for detection of the newly replicated DNA using a probe prepared from the JCV Mad-1 WT as a template. In lane 1 and 6, 2 ng of JCV Mad-1 WT linearized by BamHI digestion was loaded as positive control (+Cont.). Replication assays were repeated several times and a representative data is shown here. (B) Quantitation analysis of Southern blots by a semi-quantitative densitometry method (using NIH Image J program) and presentation of the results in arbitrary units. The replication efficiency of each data point was presented relative to that of WT.
Agnoprotein mutants display differential pattern of RNA expression
In vivo protein expression studies clearly demonstrated that two of the deletion mutants, Δ(34–36) and Δ(30–37) did not produce detectable level of agnoprotein (Fig. 3A). We further investigate whether this phenomenon is regulated at transcriptional or posttranscriptional level. To differentiate between these two possibilities, SVG-A cells were separately transfected/infected with either JCV WT or its agnoprotein mutants; and the total RNA was isolated at the indicated time points, and analyzed by Northern blotting.
JC virus late coding region produces a polycistronic message but computer-aided predictions suggest that this message may also undergo an alternative splicing process and leads to the production of two main distinct transcripts, message 1 (M1) and message 2 (M2) as illustrated in Fig. 6A. There are also other possible splicing products generated from the JCV late transcripts (Shishido-Hara et al., 2000). M1 contains the coding regions for all four JCV late proteins, agnoprotein, VP2, VP3 and VP1 and splicing for this message occurs between a splice donor site present right after the 3′-coding region of agnoprotein and a splice acceptor site located right before the 5′-coding region of VP2 (Fig. 6A). M2 message, on the other hand, harbors only the coding regions for agnoprotein and VP1 and splicing for this transcript may occur between a splice donor site present right after the 3′-coding region of agnoprotein and a splice acceptor site located right before the 5′-coding region of VP1 (Fig. 6A). It is noteworthy that agnoprotein coding region is present at the 5′-end of the both alternatively spliced transcripts (Fig. 6A) and thus agnoprotein is expected to be produced from each of these messages.
Fig. 6.
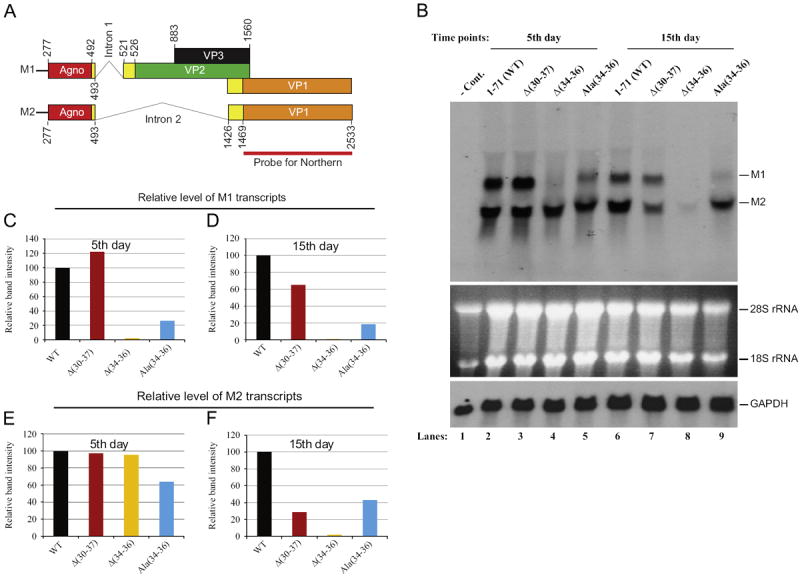
Northern Blot analysis of the viral late transcripts for JCV Mad-1WT and its agnoprotein deletion/substitution mutants. (A) Predicted two main splicing patterns of the JCV late transcripts. The coding regions for agnoprotein, VP2, VP3 and VP1 are indicated by different colors and the regions involved in splicing are colored in yellow. The positions of the intron 1 and intron 2 are indicated. The probe region corresponding to VP1 coding region is designated as a bar. (B) (Top) Total RNA (15 μg of RNA/lane) was prepared from untransfected (lane 1) and transfected/infected cells (SVG-A cells, lanes 2 to 9) as described in Materials and methods and analyzed by Northern blotting for the detection of the viral late transcripts using a radioactive probe corresponding to the JCV VP1 coding region. For the probe preparation, VP1 coding region was PCR-amplified, gel purified, quantitated, and labeled utilizing a ready-prime random labeling kit (Amersham/Pharmacia Biotechnologies, Cat #: RPN1633). Two primary massages detected by the probe are indicated as message M1 and message M2. (Middle) In parallel to the top panel, total RNA for Northern blotting was analyzed on an agarose gel by ethidium bromide staining. Both 18S and 28S rRNA bands indicate the integrity of total RNA analyzed for JCV late messages. (Bottom) Analysis of the same blot with a radiolabeled probe specific to glyceraldehyde-3-phospate dehydrogenase (GAPDH), which is a housekeeping gene and serves as a loading control. (C–F) Quantitation analysis of the Northern blot bands by a semi-quantitative densitometry method (using NIH Image J program) and presentation of the results in arbitrary units. The level of the intensity of WT bands for M1 and M2 message for each figure was set to 100 and the other data points were calculated relative to WT and presented accordingly.
Northern blotting studies showed that both M1 and M2 transcripts are readily detectable for WT when a radioactive probe corresponding to the VP1 coding region is used (Fig. 6A). It was also consistently observed that the level of M1 is less than that of M2 for WT (Fig. 6B, lanes 2 and 6; Supplement 2, lane 3). The levels of M1 and M2 for the mutants, however, deviated significantly from this trend compared to WT and were differentially regulated. For example, the deletion of a group of aa, (30IFLLEFLL37), from the dimerization domain of agnoprotein induced a slight increase in the level of M1 compared to that of WT (lane 3 and 7). More interestingly, the removal of 3 aa [Glu34, Phe35 and Leu36] from the same region of agnoprotein inflicted a more drastic effect on the expression of M1 (lane 4) and we were unable to obtain a detectable level of M1 for both data points (Fig. 6B, lanes 4 and 8, Fig. 6C and D). Similarly, a significant reduction was observed in the level of [Ala(34–36)] mutant (Fig. 6B–D). It is noteworthy to mention here that there is an intriguing discrepancy between Δ(30–37) and Δ(34–36) mutants with respect to their M1 transcript expression levels, because it is expected that the level of expression of M1 for both mutants would be, at least, similar. This unexpected differential regulation of M1 is subject to further investigation.
A differential level of regulation of M2 transcripts was also observed for all three mutants. The levels of M2 for both WT and the mutants were similar to one another at the first data point (5th day) but the deviations from this trend was more prominent among the second data points (15th day) (Fig. 6B, lanes 7–9; Fig. 6F). For example, the level of M2 for Δ(34–36) mutant reduced to barely detectable levels relative to that of WT (Fig. 6B, lane 8; Fig. 6F) and the levels of M2 for Δ(30–37) mutant and A(34–36) mutant were also found to be significantly decreased compared to that of WT (lanes 7 and 9 respectfully). Taken together, all these interesting results suggest that agnoprotein and/or its coding sequences may be involved in regulation of the splicing patterns of the viral late transcripts and the dimerization/oligomerization domain of the protein may play an important role in this process. We plan to further investigate the mechanism of the involvement of agnoprotein in splicing of JCV transcripts using different mutants of this region in the viral background. Furthermore, it also appears that agnoprotein preferentially regulates maintenance of the molar ratios between M1 and M2 transcripts (less production of M1 than M2) and mutations within the dimerization domain greatly affect this regulation. Aforementioned, both M1 and M2 have open reading frames for agnoprotein at their 5′-ends and the expression of agnoprotein is expected to occur upon translation of the each message. In fact, expression of agnoprotein is readily detected for Ala(34–36) mutant, by Western blotting and immunocytochemistry (Fig. 6A and B), although at relatively low protein levels compared to WT. In contrast, we did not detect the expression of agnoprotein for either Δ(34–36) or Δ(30–37) mutant even though the levels of M2 transcripts are detectable on Northern blot at 5th day, suggesting that the regulation of the expression of agnoprotein for the mutants occurs primarily at the level of posttranscription rather than transcription. In other words, agnoprotein is most likely produced but rapidly degraded in cells transfected/infected with these two mutants. It should be also noted here that variations observed in M1 and M2 levels for the mutants cannot be attributed to experimental errors, such as samples are not equally loaded onto the gels or to a possible degradation of total RNA, because results from the control experiments are self-evident and dismiss such possibilities (Fig. 6B).
Dimerization domain mutants show defects in splicing patterns of the viral late transcripts
Results from the Northern blot analysis strongly suggest that agnoprotein may be involved in the splicing events of the viral transcripts. We sought to further examine this possibility by RT-PCR analysis of the viral late transcripts followed by the DNA sequencing of the RT-PCR products.
As mentioned earlier, JCV late genome produces polycistronic transcripts. However, these transcripts may also undergo an alternative splicing process as predicted and produce two primary messages due to removal of the intron 1 and intron 2 (Fig. 6A). We investigated (i) whether these predicted splicing events occur for WT and (ii) whether the mutations made at the dimerization domain of agnoprotein show an effect on these two forms of the splicing patterns of the viral late transcripts.
We first examined the splicing pattern of M1 transcript, where intron 1 is predicted to be spliced out (Fig. 7). The same RNA samples used for Northern blotting were also utilized in RT-PCR as described under legend for Fig. 7, using gene specific primers. RT-PCR products were then analyzed on an agarose gel and DNA bands of interest were excised, gel-purified and subjected to a DNA sequence analysis. Sequencing data revealed that indeed the splicing of the intron 1 occurs for WT as predicted, (Fig. 7B, lanes 3 and 10). A deletion mutant where aa from 30 to 34 are removed [Δ(30–37)] also showed a splicing pattern similar to that observed for WT (lanes 4 and 11). Interestingly, removal of three aa, Glu34, Phe35 and Leu36 from aa 30–37 region (lanes 5 and 12) unexpectedly inhibited splicing of intron 1 from the M1 transcript. This finding suggests that residues, Glu34, Phe35 and Leu36, individually or collectively are involved in regulation of splicing of JCV transcripts. However, we cannot still explain the intriguing behavior of this mutant compared to that of Δ(30–37) mutant, where these three amino acids were also eliminated but obtained a spliced form of the M1 transcript. Similarly, substitution of Glu34, Phe35 and Leu36 with triple Ala residues also resulted in a similar phenotype as observed for Δ(34–36) mutant. In other words, we detected the unspliced forms of the M1 transcript for both mutants (lanes 5 and 6).
Fig. 7.
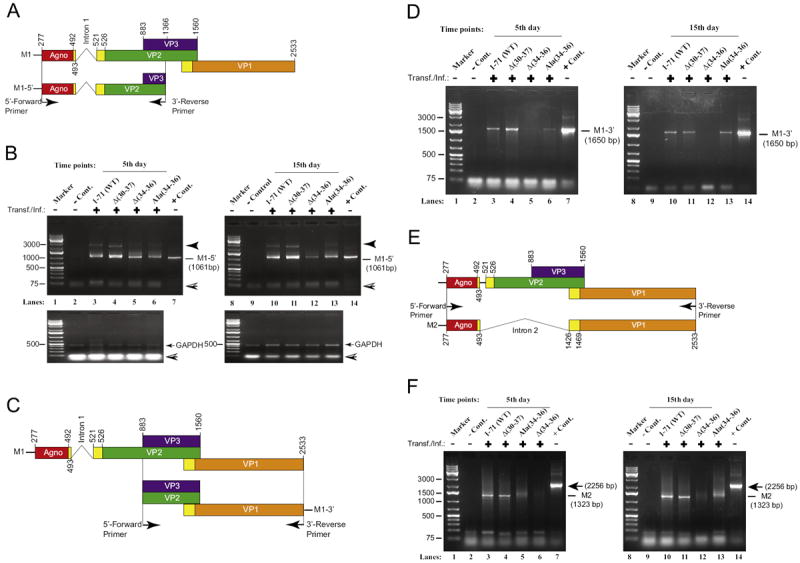
PCR amplification of the reversely transcribed (RT) M1 and M2 transcripts. (A) Graphical presentation of the JCV Mad-1 M1 transcript prior to splicing. Position of the intron 1 between nucleotides 493 and 521 (Mad-1 numbering) is indicated. Location of the primers used in PCR also indicated with arrows. (B) PCR amplification of 5′-end of the RT products for M1 transcript. Two microliter of the RT reaction (described in Materials and methods) for each sample (5th and 15th day data points) were subjected to PCR amplification using Fail Safe™ DNA polymerase mix (Epicentre, PCR-pre mix F, Cat: SF99060) according to the recommendations of the manufacturer. The following primers were used in the PCR reaction: Forward-(JCV Mad-1, 277–303 bp region): 5′-ATGGTTCTTCGCCAGCTGTCACGTAAG-3′. Reverse-(JCV Mad-1, 886–906 bp region): 5′-AGCTTTACAATTATTTAATCCA-3′. PCR conditions were as follows: 94 °C, 1 min; 60 °C, 1 min, 72 °C for 2 min, 35 cycles. Amplified gene products were analyzed by agarose gel electrophoresis (1.5%). Each RT product was also used to PCR-amplify GAPDH cDNA as a control for RT-PCR reactions using the following primers: GAPDH-Forward: 5′- TTCTCCCCATTCCGTCTTCC-3′, GAPDH-Reverse: 5′-GTACATGGTATTCACCACCC-3′. The PCR conditions for GAPDH amplification were the same as the one described for JCV transcripts above. DNA bands of interest for each data point were cut out and purified using QIaquick gel extraction kit (Qiagen, Cat # 28704) and were sequenced by a commercial vendor (Genewiz, http://www.genewiz.com). A closed arrow head points to a nonspecific PCR product. A hatched arrow head points to sample migration front. (C) Graphical presentation of the 3′-end of the M1 transcript. Position of specific 5′- and the 3′-primers used in PCR is indicated. (D). PCR amplification of the 3′-end of the RT products for M1 transcript. Two microliter of the RT reaction for each sample (5th and 15th day data points) were subjected to PCR amplification using the following specific primers: Forward-(JCV Mad-1, 886–909 bp region): 5′-AGCTTTACAATTATTTAATCCA-3′ and Reverse-(JCV Mad-1, 2533–2517 bp region): 5′-TTACAGCATTTTTGTCT-3′. (E) Graphical presentation of the JCV Mad-1 M2 transcript prior and after splicing. Position of the intron 2 between nucleotides 493 and 1426 (Mad-1 numbering) is indicated. Location of the forward and reverse primers used in PCR also indicated. (F) PCR amplification of the RT products for M2 transcript. Two microliter of the RT reaction for each sample (5th and 15th day data points) were subjected to PCR amplification using the following specific primers: Forward-(JCV Mad-1, 277–303 bp region): Forward-(JCV Mad-1, 277–303 bp region): 5′-ATGGTTCTTCGCCAGCTGTCACGTAAG-3′ and Reverse-(JCV Mad-1, 2533–2517 bp region): 5′-TTACAGCATTTTTGTCT- 3′. The PCR amplification, gel analysis and sequencing condition for each sample were the same as described for panel B samples. In lane 1 in panels B, D and F, RT products from SVG-A cells (untransfected cells used as a negative control, - Cont.) was subjected to PCR amplification as described for experimental samples. In lane 7 in panels B, D and F, JCV Mad-1 WT genome (10 ng) was PCR-amplified using specific primers described for the experimental samples and used as a positive control, + Cont., in gel analysis. Transf./Inf.: transfection/infection.
In parallel, 3′-ends of the same late gene transcripts for WT and the mutants were also subjected to analysis by RT-PCR using specific primers as described in legend Fig. 7D. The sequence analysis of the expected RT-PCR fragments (1650 bp) showed no surprising results for WT and the mutants except that the level of the RT-PCR products for Δ(34–36) mutant was observed to be significantly lower than both WT and other two mutants (Fig. 7D, lanes 5 and 12), which is consistent with its M1 levels detected on Northern blot (Fig. 6, lanes 4 and 8).
Next, we also analyzed the splicing patterns of M2 transcripts in parallel to those for M1 by RT-PCR followed by DNA sequencing. Sequencing data showed that the removal of the intron 2 from the primary transcripts indeed occurs at the predicted splice junctions between agnoprotein and VP1 for WT and agnoprotein mutants (Fig. 7E and F). Note that we were unable to obtain a detectable level of RT-PCR product for Δ(34–36) mutant under our RT-PCR conditions, which is most likely due to extremely low levels of specific RNA present at 15th day time point, consistent with the Northern blotting data (Fig. 6, lane 8). Taken together, results from RT-PCR and DNA sequencing are consistent with our findings from the Northern blot analysis of the viral late transcripts for WT and the mutants, demonstrating that agnoprotein is indeed involved in the regulation of the splicing patterns of the viral late transcripts and the dimerization domain of the protein appears to play a critical role in this process. We will further investigate the mechanism of this regulation by creating a combination of different mutants of the dimerization domain in the viral background. In addition, we think that agnoprotein may also be involved in regulation of the viral early transcripts, but it is subject to further investigation.
Discussion
Agnoprotein is a regulatory protein of Orthopolyomaviruses (Johne et al., 2011), including JCV, BKV and SV40 and is essential for the efficient replication cycle of each virus. In the absence of its expression, the rate of the viral replication is significantly reduced for each subsequent replication cycle. As a result, viral propagation halts after 2 or 3 viral life cycles (Myhre et al., 2010; Saribas et al., 2011; Sariyer et al., 2011). In addition, agnoprotein of all three polyomaviruses, JCV, BKV and SV40 forms highly stable SDS-resistant dimeric and oligomeric structures (Saribas et al., 2011), the functions of which are currently unknown. However, it is conceivable that such structures may provide considerable flexibility for agnoprotein to diversify its biological functions in the infected cells.
Our previous mapping studies demonstrated that aa from 17 to 42 are important for dimer/oligomer formation by agnoprotein (Saribas et al., 2011). In the current study, we further characterized the dimer/oligomer formation domain of agnoprotein by deletion/substitution analysis followed by functional studies in the viral background. We demonstrated that Leu/Ile/Phe-rich domain corresponding to aa from 28 to 39 play an essential role in stable dimer/oligomer formation for agnoprotein. Specifically, aa spanning from 30 to 37 are sufficient to confer this phenomenon (Table 1). In vivo data demonstrated that, in the absence of aa spanning from 34 to 36 or from 30 to 37, the mutant JC viruses express agnoprotein poorly (Fig. 4A and B) and replicate at relatively minimal levels compared to WT (Fig. 5A and B), suggesting that Leu/Ile/Phe-rich domain plays an essential role in protein function (Fig. 5A and B) and stability (Table 1). We further investigated whether the undetectable levels of agnoprotein expression is due to the regulation at the transcriptional or posttranscriptional level by Northern blotting (Fig. 6B) and by RT-PCR (Fig. 6) followed by DNA sequencing. Results from these studies let to a novel observation with respect to the agnoprotein function—involvement of the protein in viral gene regulation at the post-transcriptional level. First of all, we detected two primary transcripts namely M1 and M2 regarding the viral late gene expression on Northern blot. RT-PCR studies and subsequent DNA sequencing revealed that M1 transcript is responsible for producing all the JCV late proteins, agnoprotein, VP1, VP2 and VP2 (Fig. 6A). M2 transcripts on the other hand are associated with the production of only agnoprotein and VP1 (Fig. 6A). The novelty of our findings is several fold: (i) Mutations within the dimerization domain of agnoprotein may lead to defects in the splicing patterns of the viral late transcripts. (ii) The levels of M1 and M2 appear to be differentially regulated by agnoprotein and the dimerization/oligomerization domain of the protein appears to play a critical role in this process. (iii) The level of M1 is consistently found to be lower than that of M2 for WT (Fig. 6A, Supplement 2) in the infected cells. These findings may translate into the viral replication cycle as follows: One of the ways that JCV tries to produce a higher number of infectious particles in infected cells is through the regulation of the molar ratios between VP2/VP3 and VP1. It is known that the VP2 and VP3 are located at the inner layers of a virion and VP1 forms the outer layer of an infectious viral particle (Saribas et al., 2010). If one examines the relationship between the size and volume in consideration of a virion, it is apparent that JCV needs less molar amount of VP2/VP3 than VP1 to make up the inner layers of a virion due to the space limitation. Our findings from the Northern blot and RT-PCR studies are consistent with such an analysis. That is, in the absence of a functional agnoprotein, JCV would not be able to maintain the molar ratios between the VP2/VP3 and VP1, which then leads to a defective viral replication cycle. In other words, the efficiency of the virion formation is no longer regulated and the virus may produce mostly empty capsids, as we have previously reported (Sariyer et al., 2011).
Structurally speaking, agnoprotein is not alone among viral proteins with respect to forming dimer and oligomers. Like agnoprotein, there are many examples in the literature of viral proteins that form dimers and oligomers, including human immunodeficiency virus 1 (HIV-1) proteins (Rev, Vpr, Vif and Vpu) (Bernacchi et al., 2011; Bourbigot et al., 2005; Daugherty et al., 2010a, 2010b; Lu et al., 2010), Hepatitis C virus nonstructural protein 4B (Gouttenoire et al., 2010), Ebola virus VP40 (Hoenen et al., 2010), and polyomavirus LT-Ag (Cuesta et al., 2010; Foster and Simmons, 2010). For example, Rev is an RNA binding protein and forms stable dimer/oligomers that regulate the transport of unspliced or partially spliced RNA molecules from nucleus to cytoplasm. Rev initially binds to a highly structured ~350 nucleotide (nt) Rev response element (RRE) RNA, located in the viral introns, in dimeric forms. Recently resolved crystal structure of Rev-RNA complex demonstrates that dimeric forms of Rev cooperatively bind to viral RNA to form an elegant hexameric unit complexed with RNA. This hexameric complex has 500-fold higher affinity to RRE than any monomeric Rev molecule. The Rev-RRE nucleoprotein complex then targets the host export factor Crm1 (XpoI), which shuttles the Rev-RNA complex to cytoplasm. In the cytoplasm, Rev unloads its cargo and free Rev is reimported into the nucleus for further rounds of nuclear export of HIV-1 RNA (Cullen, 2003a, 2003b). As in the case of Rev, we think that dimeric and oligomeric forms of agnoprotein play important roles in its function—perhaps in the transport of JCV transcripts from nucleus to cytoplasm or in the splicing process of the viral transcripts. We will further investigate such possibilities.
In addition to possessing predicted α-helical domains (Fig. 1B), agnoprotein sequences display intrinsically disordered structures. A prime example of such intrinsically disordered structural viral proteins is the HIV-1 Vif protein. This protein plays important roles in the viral pathogenesis by facilitating the degradation of APOBEC3G, an endogenous cellular inhibitor (cytosine deaminase) of HIV-1 replication (Sheehy et al., 2002). These types of proteins, which are known to have extensive regions that lack a fixed tertiary structure, have high net charge and low overall hydrophobicity, which are the main characteristics of such proteins (Dunker et al., 2005; Dyson and Wright, 2005; Fink, 2005; Uversky et al., 2005). It is suggested that these intrinsically disordered regions provide a considerable flexibility to a protein to interact with multiple but structurally diverse partners in the host cells during the viral infection cycle. Predictions from our computer modeling studies indicate that a small portion of N-terminal and C-terminal domains of JCV agnoprotein possess such intrinsic disordered structures (Fig. 1B) (Saribas et al., 2011), implying that these regions may indeed be the executioner domains of agnoprotein to interact with a variety of partners in the infected cells and play significant roles in agnoprotein function.
JCV, like other Orthopolyomaviruses including BKV and SV40, encodes only a limited number of regulatory proteins, one of which is agnoprotein, and yet it successfully goes through its replication cycles. This suggests that regulatory proteins of JCV fulfill more than one function to optimize the viral life cycle. Published reports indicate that JCV agnoprotein is involved in many aspects of viral replication cycle, including viral DNA replication (Safak et al., 2001; Saribas et al., 2011) and transcription (Safak et al., 2001, 2002; Sariyer et al., 2011). If this is the case, then, the question arises “how is such a small protein like agnoprotein able to achieve so many different functions despite its small size?” It appears that different regions of agnoprotein are involved in different functions. For instance, our recent protein–protein interaction studies with agnoprotein and LT-Ag showed that agnoprotein stimulates LT-Ag binding to the viral origin of replication (Ori), through which it contributes to viral replication cycle (Saribas et al., 2012). We also previously reported that alterations in the protein kinase C (PKC) phosphorylation sites of JCV agnoprotein (conversion of Ser7, Ser11 and Thr21 to Ala) resulted in a phenotype that is unable to sustain the viral replication cycle (Sariyer et al., 2006). Moreover, deletion of either the intrinsically disordered C-terminal region (sequences from 51 to 71) (Akan et al., 2006) or that of internal sequences spanning amino acids 17 to 42 (Saribas et al., 2011), which are involved in dimer/oligomer formation by agnoprotein, resulted in phenotypes that are replication incompetent. In this study, beside fine mapping of the multimerization domain of agnoprotein, we have also discovered a novel function for the protein-regulation of the splicing patterns of the viral late transcripts and involvement of the dimerization domain of the protein in this process. All these findings suggest a relationship between agnoprotein structure and its role in splicing of the viral transcripts.
We also investigated the role of the multimerization domain of JCV agnoprotein in viral replication cycle. Amino and carboxy terminal; and internal deletion analysis studies revealed that the Leu/Ile/Phe-rich domain of the protein encompassing amino acids from 28 to 39 plays an essential role not only in stable dimer and oligomer formation (Table 1) but also in stable expression of the protein (Fig. 4A and B). Internal deletions of the Leu/Ile/Phe-rich domain in the viral background significantly hampered the agnoprotein expression (Fig. 4A and B) and viral DNA replication (Fig. 5A and B). For example, deletion of only 3 aa [Δ(34–36)] abrogated oligomer formation and reduced dimer formation in half in vitro (Fig. 3A, lane 4). Further deletion of the region which covers the most of the Leu/Ile/Phe-rich domain resulted in a phenotype that is unable form either stable dimers or oligomers [Δ(30–37)] in vitro (Fig. 3A, lane 5). Consistent with these in vitro observations, in vivo studies with the internal deletion mutants where the Leu/Ile/Phe-rich domain was altered [Δ(34–36) and Δ (30–37)] showed that these internal deletion mutants considerably lost their ability to replicate efficiently due to agnoprotein deficiency (Fig. 5A and B). Amino acid alignment studies showed that the Leu/Ile/Phe-rich domain residues of agnoprotein are substantially conserved among JCV, BKV and SV40 (Fig. 1D). Therefore, it is conceivable that this region serves similar functions in the agnoprotein of BKV and SV40 as well. As such, our results from this study may have broad implications regarding the biology of Orthopolyomaviruses.
Proteins play fundamental regulatory and structural roles in all living organisms, including in viruses. Without their 3D structure, it is difficult to understand their function. We have attempted to obtain the 3D crystal structure of bacterially expressed JCV agnoprotein (MBP-Agno) several times with no success due to the fact that a dynamic process of the conversion of monomers to dimers, then dimers to oligomers (Saribas et al., 2011). We are currently, however, in the process of obtaining its structure by NMR studies using synthetic peptides as similar structural studies were previously reported for HIV-1 Vpr (Bourbigot et al., 2005). The results from these structural studies along with those reported in our current work may provide us new opportunities to develop effective inhibitors against the unique dimerization/oligomerization domain of JCV agnoprotein to hamper the spread of the virus in PML.
Materials and methods
Cell lines
SVG-A cell line was established by immortalization of the primary human fetal glial cells with an origin-defective SV40 mutant (Major et al., 1985). This cell line does not express either SV40 viral capsid proteins (VPs) or agnoprotein, but expresses SV40 LT-Ag and supports JCV propagation. Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and antibiotics [penicillin/streptomycin (100 μg/ml), ciprofloxacin (10 μg/ml]. Cells were maintained at 37 °C in a humidified atmosphere supplemented with 7% CO2.
Plasmid constructs
The cloning of pMAL-c5X-Agno (1–71), pMAL-c5X-Agno (1–16), pMAL-c5X-Agno Δ(17–42), pMAL-c5X-Agno (17–71), pMAL-c5X-Agno (1–42) and pMAL-c5X-Agno (43–71) constructs was previously described (Saribas et al., 2011). Amino terminal [Agno (25–71), Agno (28–71), Agno (34–71), Agno (35–71), Agno (36–71) and Agno (37–71)] and carboxy terminal [Agno (1–33)] deletion mutants of agnoprotein were subcloned into NcoI/Eco RI sites of pMAL-c5X bacterial expression vector to produce MBP-Agno fusion protein by PCR-based cloning. Bluescript KS-JCV-Mad-1 WT plasmid was used as a template for PCR amplifications. PCR products were ethanol-precipitated, digested with NcoI and EcoRI restriction enzymes, gel purified and ligated into pMAL-c5X bacterial expression vector (MBP fusion system, NEB). The resulting plasmids were designated as pMAL-c5X-Agno (25–71), pMAL-c5X-Agno (28–71), pMAL-C5X-Agno (34–71), pMAL-c5X-Agno (35–71), pMAL-c5X-Agno (36–71), pMAL-c5X-Agno (37–71) and pMAL-c5X-Agno (1–33). Amino acids from 34 to 36 and from 30 to 37 were deleted from the virus background by deletion mutagenesis using appropriate primers and these plasmids was designated as Bluescript KS(+)-JCV Mad-1 Agno Δ(34–36) and Bluescript KS(+)-JCV Mad-1 Agno Δ(30–37). Insertion of the wild-type Mad-1 genome into the BamHI site of Bluescript KS (+) vector was previously described (Sariyer et al., 2006). Amino acids from 34 to 36 were substituted with Ala (E34AF35AL36A) in the viral background (JCV Mad-1) using the Quik Change™ site–directed mutagenesis kit (Agilent) using appropriate synthetic oligonucleotides and designated as JCV Mad-1 Agno Ala(34–36). Each mutant agnogene was then subcloned into NcoI/EcoRI sites of pMAL-c5X vector and plasmids were designated as pMAL-c5X-Agno Δ(34–36), pMAL-c5X-Agno Δ(30–37) and pMAL-c5X-Agno Ala (34–36). The integrity of the whole viral genomes in the viral backgrounds and the mutants made in each construct were verified by DNA sequencing.
Expression and purification of recombinant proteins
Bacterial expression plasmids [pMAL-pMAL-c5X alone, pMAL-c5X-Agno (17–71), pMAL-c5X-Agno (25–71), pMAL-c5X-Agno (28–71), pMAL-c5X-Agno (34–71), pMAL-c5X-Agno (35–71), pMAL-c5X-Agno (36–71), pMAL-c5X-Agno (37–71), pMAL-c5X-Agno (43–71), pMALc5X- Agno (1–42) and pMAL-c5X-Agno (1–33). pMAL-c5X-Agno Δ(34– 36), pMAL-c5X-Agno Δ(30–37) and pMAL-c5X-Agno Ala(34–36)] were transformed into Escherichia coli DH5α strain and cultures were grown in 100ml Luria–Bertani broth (LB) supplemented with ampicillin (100 μg/ml) at 37 °C overnight. Next day, cultures were first diluted 1:10 in fresh LB in 0.25 l supplemented with glucose (2 g/l) and ampicillin (100 μg/ml); and grown at 37 °C until at an optical density of 0.5 at 600 nm. Bacterial cultures were then induced with 0.3mM isopropyl-β-thiogalactopyranoside (IPTG) and incubated for an additional 2 h at 28 °C. Bacterial cultures were then harvested by centrifugation and the resulting pellet from the various MBP-Agno fusions were each resuspended in amylose column buffer (20 mM Tris–HCl pH 7.4, 200 mM NaCl and 1mM EDTA). The cell suspension was then incubated on ice for 30min in the presence of lysozyme (0.5mg/ml) and protease inhibitor cocktail (Sigma), after which the cells were lysed by sonication. Clear lysates were obtained after a high speed centrifugation at 15,000 rpm (Thermo Scientific, F21-8 × 50y rotor). The lysates were then incubated with 200 μl of amylose FF (fast flow) resin (1:1, buffer:beads) (New England Biolabs) prepared in amylose column buffer overnight at 4 °C. MBP-Agno fusion proteins bound amylose resins were washed with five column volumes of amylose column buffer and were purified. Purified proteins were either stored on beads at +4 °C or eluted with 10 mM maltose prepared in the same buffer and stored at +4 °C or −30 °C until use. The quality of the proteins was analyzed by SDS-PAGE/Coomassie staining as described previously (Saribas et al., 2011).
Indirect immunofluorescence microscopy
Indirect immunofluorescence microscopy studies were performed as previously described (Sadowska et al., 2003; Sariyer et al., 2011). Briefly, SVG-A cells (2 × 106 cells/75 cm2 flask) were separately transfected/infected with different viral DNA (8 μg each) as indicated in each respective figure legend. At 5th day posttransfection, the cells were seeded at subconfluency on polylysine-coated glass chamber slides. The next day, the cells were washed twice with PBS, fixed in cold acetone and incubated with 5% bovine serum albumin in PBS for 2 h. Chamber slides were then incubated with anti-agnoprotein primary polyclonal antibody (1:200 dilution) (Safak et al., 2002) plus anti-VP1 primary monoclonal (pAB597) antibody (Saribas et al., 2011) (1:200 dilution) overnight. Antibody dilutions and incubations were performed in incubation buffer [PBS-0.01% Tween 20, (PBST)] buffer. Cells were washed three times with PBST buffer for 10 min intervals and subsequently incubated with a Rhodamine-conjugated goat antimouse plus fluorescein isothiocyanate (FITC)-conjugated goat antirabbit secondary antibodies (BD biosciences) for 45 min. Finally, slides were washed three times with PBST buffer, mounted, and examined under a fluorescence microscope (Nikon eclipse TE300; objectives: 10 × 1.25 Ph1 DL and 40 × /1.3 oil, plain flour; eyepiece: 10×; operating software: Slidebook 5.0.) for detection of JCV agnoprotein and VP1.
Replication assays
Replication assays were carried out as previously described (Sariyer et al., 2011). Briefly, the plasmid constructs [Bluescript KS (+)-JCV Mad-1 WT, Bluescript KS(+)-JCV Mad-1 Agno Δ(34–36), Bluescript KS(+)-JCV Mad-1 Agno Δ(30–37) and Bluescript KS (+)-JCV Mad-1 Agno Ala(34–36) were digested with BamHI to liberate the viral genome inserts from the vector [Bluescript KS (+)]. SVG-A cells (2 × 106 cells/75 cm2 flask) were then separately transfected/infected each with the digested DNA (8 mg each) using lipofectamine 2000 according to Manufacturer’s recommendations (Invitrogen). After 5 h incubation with the transfectants, cells were washed with PBS and fed with DMEM supplemented with 10% FBS and 1% ampicillin/streptomycin. Media were replenished every three days posttransfection. At the indicated time points, low-molecular-weight DNA containing both input and replicated viral DNA was isolated using Qiagen spin columns as described previously (Ziegler et al., 2004), digested with BamHI and DpnI enzymes, resolved on 1% agarose gel and analyzed by Southern blotting as described previously (Saribas et al., 2011).
Preparation of whole-cell and nuclear extracts for Western blotting
In parallel to the isolation of DNA samples from the transfected/infected cells as described under replication assay, whole cell extracts were also prepared at the indicated time points (see the respective figure legends) as previously described (Sariyer et al., 2011). Whole cell lysates were analyzed by Western blotting, using an anti-agnoprotein polyclonal antibody as described under each figure legend and described previously (Sadowska et al., 2003).
Northern blotting
Northern blotting was carried out as previously described (Safak et al., 2002). Briefly, SVG-A cells (2 × 106 cell/75 cm2 flask) were separately transfected with different plasmid constructs [Bluescript KS(+)-JCV Mad-1 WT, Bluescript KS(+)-JCV Mad-1 Agno Δ(34–36), Bluescript KS(+)-JCV Mad-1 Agno Δ(30–37) and Bluescript KS(+)-JCV Mad-1 Agno Ala(34–36)] (8 μg/flask), using lipofectamine 2000 according to Manufacturer’s recommendations (Invitrogen). The plasmid constructs were digested with BamHI to liberate the viral genome inserts from the vectors prior to transfection. Total RNA was isolated from untransfected (SVG-A alone), transfected cells (2.2 × 107 cells) at specific time points as indicated in the respective figure legend using the RNeasy Plus Mini Kit (Qiagen, Cat #: 74132). Fifteen micrograms of total RNA was treated with RNase free DNase I to remove the DNA contamination prior to fractionation on a 1% agarose gel and transferred onto a nitrocellulose membrane. The coding region of JCV Mad-1 VP1 was PCR-amplified, [32P]-radiolabeled and incubated with the RNA blots as a probe to detect VP1-associated JCV transcripts.
RT-PCR and DNA sequencing
cDNA synthesis was carried out using SuperScript™ III one step RT-PCR system (Invitrogen, Cat #: 12574-026). Briefly, 300 ng total RNA isolated for Northern blot samples was mixed with 1 ml of oligo dT(12–18) (0.5 μg/μl) and 1 μl of dNTP mix (10 nM each dATP, dCTP, dGTP and dTTP) and the reaction volume was brought up to 13 μl. The samples were then heated at 65 °C for 5 min and cooled on ice for 1 min. After brief centrifugation, 4 μl of 5 × First Strand Buffer, 1 μl DTT (0.1 M) and 1 μl SuperScript™ III reverse transcriptase (200 units/μl, Invitrogen) were added to each reaction and mixed gently by pipetting. The reactions were then incubated at 55 °C for 1 h to synthesize cDNA. Reverse transcriptase was inactivated by heating of the reaction samples at 70 °C for 15 min. Finally RNA complementary to the cDNA was incubated with RNAse H (1 u/μl/reaction) (Invitrogen) at 37 °C for 30 min. Specific PCR reactions were carried out as described under respective figure legends. The purified PCR products were sequenced by a commercial vendor (Genewiz, http://www.genewiz.com).
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We would like to thank past and present members of the Department of Neuroscience and Center for Neurovirology for their insightful discussion and sharing of ideas and reagents. This work was made possible by grants awarded by NIH to MS
Footnotes
Appendix A. Supplementary materials
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.virol.2013.05.003.
References
- Akan I, Sariyer IK, Biffi R, Palermo V, Woolridge S, White MK, Amini S, Khalili K, Safak M. Human polyomavirus JCV late leader peptide region contains important regulatory elements. Virology. 2006;349(1):66–78. doi: 10.1016/j.virol.2006.01.025. [DOI] [PubMed] [Google Scholar]
- Altschuler E. JC virus and chronic lymphocyte leukemia. J Immunother. 1999;22(1):90–91. doi: 10.1097/00002371-199901000-00013. [DOI] [PubMed] [Google Scholar]
- Alwine JC. Evidence for simian virus 40 late transcriptional control: mixed infections of wild-type simian virus 40 and a late leader deletion mutant exhibit trans effects on late viral RNA synthesis. J Virol. 1982;42(3):798–803. doi: 10.1128/jvi.42.3.798-803.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger JR. The basis for modeling progressive multifocal leukoencephalopathy pathogenesis. Curr Opin Neurol. 2011;24(3):262–267. doi: 10.1097/WCO.0b013e328346d2a3. [DOI] [PubMed] [Google Scholar]
- Berger JR, Concha M. Progressive multifocal leukoencephalopathy: the evolution of a disease once considered rare. J Neurovirol. 1995;1(1):5–18. doi: 10.3109/13550289509111006. [DOI] [PubMed] [Google Scholar]
- Bernacchi S, Mercenne G, Tournaire C, Marquet R, Paillart JC. Importance of the proline-rich multimerization domain on the oligomerization and nucleic acid binding properties of HIV-1 Vif. Nucleic Acids Res. 2011;39(6):2404–2415. doi: 10.1093/nar/gkq979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourbigot S, Beltz H, Denis J, Morellet N, Roques BP, Mely Y, Bouaziz S. The C-terminal domain of the HIV-1 regulatory protein Vpr adopts an antiparallel dimeric structure in solution via its leucine-zipper-like domain. Biochem J. 2005;387(Pt 2):333–341. doi: 10.1042/BJ20041759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuesta I, Nunez-Ramirez R, Scheres SH, Gai D, Chen XS, Fanning E, Carazo JM. Conformational rearrangements of SV40 large T antigen during early replication events. J Mol Biol. 2010;397(5):1276–1286. doi: 10.1016/j.jmb.2010.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen BR. Nuclear mRNA export: insights from virology. Trends Biochem Sci. 2003a;28(8):419–424. doi: 10.1016/S0968-0004(03)00142-7. [DOI] [PubMed] [Google Scholar]
- Cullen BR. Nuclear RNA export. J Cell Sci. 2003b;116(Pt 4):587–597. doi: 10.1242/jcs.00268. [DOI] [PubMed] [Google Scholar]
- Darbinyan A, Darbinian N, Safak M, Radhakrishnan S, Giordano A, Khalili K. Evidence for dysregulation of cell cycle by human polyomavirus, JCV, late auxiliary protein. Oncogene. 2002;21(36):5574–5581. doi: 10.1038/sj.onc.1205744. [DOI] [PubMed] [Google Scholar]
- Daugherty MD, Booth DS, Jayaraman B, Cheng Y, Frankel AD. HIV Rev response element (RRE) directs assembly of the Rev homooligomer into discrete asymmetric complexes. Proc Nat Acad Sci U S A. 2010a;107(28):12481–12486. doi: 10.1073/pnas.1007022107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, Liu B, Frankel AD. Structural basis for cooperative RNA binding and export complex assembly by HIV Rev. Nat Struct Mol Biol. 2010b;17(11):1337–1342. doi: 10.1038/nsmb.1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Valle L, Gordon J, Enam S, Delbue S, Croul S, Abraham S, Radhakrishnan S, Assimakopoulou M, Katsetos CD, Khalili K. Expression of human neurotropic polyomavirus JCV late gene product agnoprotein in human medulloblastoma. J Nat Cancer Inst. 2002;94(4):267–273. doi: 10.1093/jnci/94.4.267. [DOI] [PubMed] [Google Scholar]
- Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005;272(20):5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6(3):197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- Fink AL. Natively unfolded proteins. Curr Opin Struct Biol. 2005;15(1):35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Foster EC, Simmons DT. The SV40 large T-antigen origin binding domain directly participates in DNA unwinding. Biochemistry. 2010;49(10):2087–2096. doi: 10.1021/bi901827k. [DOI] [PubMed] [Google Scholar]
- Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426(6968):895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- Gouttenoire J, Penin F, Moradpour D. Hepatitis C virus nonstructural protein 4B: a journey into unexplored territory. Rev Med Virol. 2010;20(2):117–129. doi: 10.1002/rmv.640. [DOI] [PubMed] [Google Scholar]
- Haggerty S, Walker DL, Frisque RJ. JC virus-simian virus 40 genomes containing heterologous regulatory signals and chimeric early regions: identification of regions restricting transformation by JC virus. J Virol. 1989;63(5):2180–2190. doi: 10.1128/jvi.63.5.2180-2190.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N, Skolnik-David H, Aloni Y. Attenuation in the control of SV40 gene expression. Cell. 1982;29(1):183–193. doi: 10.1016/0092-8674(82)90102-7. [DOI] [PubMed] [Google Scholar]
- Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26(2):365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- Hoenen T, Biedenkopf N, Zielecki F, Jung S, Groseth A, Feldmann H, Becker S. Oligomerization of Ebola virus VP40 is essential for particle morphogenesis and regulation of viral transcription. J Virol. 2010;84(14):7053–7063. doi: 10.1128/JVI.00737-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou-Jong MH, Larsen SH, Roman A. Role of the agnoprotein in regulation of simian virus 40 replication and maturation pathways. J Virol. 1987;61(3):937–939. doi: 10.1128/jvi.61.3.937-939.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen M, Myhre MR, Dragset M, Tummler C, Moens U. Phosphorylation of human polyomavirus BK agnoprotein at Ser-11 is mediated by PKC and has an important regulative function. Virology. 2008;379(1):97–109. doi: 10.1016/j.virol.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Johannessen M, Walquist M, Gerits N, Dragset M, Spang A, Moens U. BKV agnoprotein interacts with alpha-soluble N-ethylmaleimide-sensitive fusion attachment protein, and negatively influences transport of VSVG-EGFP. PLoS One. 2011;6(9):e24489. doi: 10.1371/journal.pone.0024489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johne R, Buck CB, Allander T, Atwood WJ, Garcea RL, Imperiale MJ, Major EO, Ramqvist T, Norkin LC. Taxonomical developments in the family Polyomaviridae. Arch Virol. 2011;156(9):1627–1634. doi: 10.1007/s00705-011-1008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinschmidt-DeMasters BK, Tyler KL. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N Engl J Med. 2005;353(4):369–374. doi: 10.1056/NEJMoa051782. [DOI] [PubMed] [Google Scholar]
- Langer-Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med. 2005;353(4):375–381. doi: 10.1056/NEJMoa051847. [DOI] [PubMed] [Google Scholar]
- Lu JX, Sharpe S, Ghirlando R, Yau WM, Tycko R. Oligomerization state and supramolecular structure of the HIV-1 Vpu protein transmembrane segment in phospholipid bilayers. Protein Sci. 2010;19(10):1877–1896. doi: 10.1002/pro.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major EO. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annu Rev Med. 2010;61:35–47. doi: 10.1146/annurev.med.080708.082655. [DOI] [PubMed] [Google Scholar]
- Major EO, Amemiya K, Tornatore CS, Houff SA, Berger JR. Pathogenesis and molecular biology of progressive multifocal leukoencephalopathy, the JC virusinduced demyelinating disease of the human brain. Clin Microbiol Rev. 1992;5(1):49–73. doi: 10.1128/cmr.5.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major EO, Miller AE, Mourrain P, Traub RG, de Widt E, Sever J. Establishment of a line of human fetal glial cells that supports JC virus multiplication. Proc Nat Acad Sci U S A. 1985;82(4):1257–1261. doi: 10.1073/pnas.82.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolskee RF, Nathans D. Suppression of a VP1 mutant of simian virus 40 by missense mutations in serine codons of the viral agnogene. J Virol. 1983;48(2):405–409. doi: 10.1128/jvi.48.2.405-409.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell HK, Petersen NS, Buzin CH. Self-degradation of heat shock proteins. Proc Nat Acad Sci U S A. 1985;82(15):4969–4973. doi: 10.1073/pnas.82.15.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moens U, Ludvigsen M, Van Ghelue M. Human polyomaviruses in skin diseases. Pathol Res Int. 2011;2011:123491. doi: 10.4061/2011/123491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhre MR, Olsen GH, Gosert R, Hirsch HH, Rinaldo CH. Clinical polyomavirus BK variants with agnogene deletion are non-functional but rescued by trans-complementation. Virology. 2010;398(1):12–20. doi: 10.1016/j.virol.2009.11.029. [DOI] [PubMed] [Google Scholar]
- Ng SC, Mertz JE, Sanden-Will S, Bina M. Simian virus 40 maturation in cells harboring mutants deleted in the agnogene. J Biol Chem. 1985;260(2):1127–1132. [PubMed] [Google Scholar]
- Okada Y, Suzuki T, Sunden Y, Orba Y, Kose S, Imamoto N, Takahashi H, Tanaka S, Hall WW, Nagashima K, Sawa H. Dissociation of heterochromatin protein 1 from lamin B receptor induced by human polyomavirus agnoprotein: role in nuclear egress of viral particles. EMBO Rep. 2005;6(5):452–457. doi: 10.1038/sj.embor.7400406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou HD, Kwiatkowski W, Deerinck TJ, Noske A, Blain KY, Land HS, Soria C, Powers CJ, May AP, Shu X, Tsien RY, Fitzpatrick JA, Long JA, Ellisman MH, Choe S, O’Shea CC. A structural basis for the assembly and functions of a viral polymer that inactivates multiple tumor suppressors. Cell. 2012;151(2):304–319. doi: 10.1016/j.cell.2012.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5(4):725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowska B, Barrucco R, Khalili K, Safak M. Regulation of human polyomavirus JC virus gene transcription by AP-1 in glial cells. J Virol. 2003;77(1):665–672. doi: 10.1128/JVI.77.1.665-672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safak M, Barrucco R, Darbinyan A, Okada Y, Nagashima K, Khalili K. Interaction of JC virus agno protein with T antigen modulates transcription and replication of the viral genome in glial cells. J Virol. 2001;75(3):1476–1486. doi: 10.1128/JVI.75.3.1476-1486.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safak M, Khalili K. Physical and functional interaction between viral and cellular proteins modulate JCV gene transcription. J Neurovirol. 2001;7(4):288–292. doi: 10.1080/13550280152537111. [DOI] [PubMed] [Google Scholar]
- Safak M, Sadowska B, Barrucco R, Khalili K. Functional interaction between JC virus late regulatory agnoprotein and cellular Y-box binding transcription factor, YB-1. J Virol. 2002;76(8):3828–3838. doi: 10.1128/JVI.76.8.3828-3838.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, Arachea BT, White MK, Viola RE, Safak M. Human polyomavirus JC small regulatory agnoprotein forms highly stable dimers and oligomers: implications for their roles in agnoprotein function. Virology. 2011;420(1):51–65. doi: 10.1016/j.virol.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, Ozdemir A, Lam C, Safak M. JC virus-induced progressive multifocal leukoencephalopathy. Future Virol. 2010;5(3):313–323. doi: 10.2217/fvl.10.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, White MK, Safak M. JC virus agnoprotein enhances large T antigen binding to the origin of viral DNA replication: evidence for its involvement in viral DNA replication. Virology. 2012;433(1):12–26. doi: 10.1016/j.virol.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariyer IK, Akan I, Palermo V, Gordon J, Khalili K, Safak M. Phosphorylation mutants of JC virus agnoprotein are unable to sustain the viral infection cycle. J Virol. 2006;80(8):3893–3903. doi: 10.1128/JVI.80.8.3893-3903.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariyer IK, Khalili K, Safak M. Dephosphorylation of JC virus agnoprotein by protein phosphatase 2A: inhibition by small t antigen. Virology. 2008;375(2):464–479. doi: 10.1016/j.virol.2008.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariyer IK, Saribas AS, White MK, Safak M. Infection by agnoproteinnegative mutants of polyomavirus JC and SV40 results in the release of virions that are mostly deficient in DNA content. Virol J. 2011;8(1):255. doi: 10.1186/1743-422X-8-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418(6898):646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- Shishido-Hara Y, Hara Y, Larson T, Yasui K, Nagashima K, Stoner GL. Analysis of capsid formation by human polyomavirus JC(Tokyo-1 strain) by a eucaryotic expression system: Splicing of late RNAs, translation and nuclear transport of major capsid protein VP1, and capsid assembly. J Virol. 74:1840–53. doi: 10.1128/jvi.74.4.1840-1853.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Orba Y, Okada Y, Sunden Y, Kimura T, Tanaka S, Nagashima K, Hall WW, Sawa H. The human polyoma JC virus agnoprotein acts as a viroporin. PLoS Pathog. 2010;6(3):e1000801. doi: 10.1371/journal.ppat.1000801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterstab G, Gosert R, Leuenberger D, Lorentz P, Rinaldo CH, Hirsch HH. The polyomavirus BK agnoprotein co-localizes with lipid droplets. Virology. 2010;399(2):322–331. doi: 10.1016/j.virol.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Uversky VN, Oldfield CJ, Dunker AK. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recogn. 2005;18(5):343–384. doi: 10.1002/jmr.747. [DOI] [PubMed] [Google Scholar]
- Van Assche G, Van Ranst M, Sciot R, Dubois B, Vermeire S, Noman M, Verbeeck J, Geboes K, Robberecht W, Rutgeerts P. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N Engl J Med. 2005;353(4):362–368. doi: 10.1056/NEJMoa051586. [DOI] [PubMed] [Google Scholar]
- Ziegler K, Bui T, Frisque RJ, Grandinetti A, Nerurkar VR. A rapid in vitro polyomavirus DNA replication assay. J Virol Methods. 2004;122(1):123–127. doi: 10.1016/j.jviromet.2004.08.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.