

Abstract
The mechanistic target of rapamycin (mTOR) is recognized as a sensor of mitochondrial dysfunction and effector of T-cell lineage development, however, its role in autoimmunity, including systemic lupus erythematosus, remains unclear. Here, we prospectively evaluated mitochondrial dysfunction and mTOR activation in PBL relative to SLE disease activity index (SLEDAI) during 274 visits of 59 patients and 54 matched healthy subjects. Partial least square-discriminant analysis identified 15 of 212 parameters that accounted for 70.2% of the total variance and discriminated lupus and control samples (p<0.0005); increased mitochondrial mass of CD3+/CD4−/CD8− double-negative (DN) T cells (p=1.1×10−22) and FoxP3 depletion in CD4+/CD25+ T cells were top contributors (p=6.7×10−7). Prominent necrosis and mTOR activation were noted in DN T cells during 15 visits characterized by flares (SLEDAI increase ≥4) relative to 61 visits of remission (SLEDAI decrease ≥4). mTOR activation in DN T cells was also noted at pre-flare visits of SLE patients relative to those of stable disease or healthy controls. DN lupus T cells showed increased production of IL-4, which correlated with depletion of CD25+/CD19+B cells. Rapamycin treatment in vivo blocked the IL-4 production and necrosis of DN T cells, increased the expression of FoxP3 in CD25+/CD4+T cells, and expanded CD25+/CD19+ B cells. These results identify mTOR activation to be a trigger of IL-4 production and necrotic death of DN T cells in patients with SLE.
INTRODUCTION
Systemic lupus erythematosus (SLE) is an autoimmune inflammatory disease of unknown etiology characterized by T-cell and B-cell dysfunction and anti-nuclear antibody production (1). Abnormal death signal processing plays a key role in driving anti-nuclear antibody production through the release of immunogenic nuclear materials from apoptotic (2) and necrotic cells (3,4).
Mitochondria play critical roles in activation and death pathway selection in T lymphocytes (5). Lupus T cells exhibit mitochondrial dysfunction, which is characterized by elevated mitochondrial transmembrane potential (ΔΨm) or persistent mitochondrial hyperpolarization (MHP) and results in ATP depletion, diminished activation-induced apoptosis and predisposition of T cells for necrosis (6). The increased release of necrotic materials from T cells could drive disease pathogenesis by enhancing the capacity of macrophages and dendritic cells (DC) to produce nitric oxide (NO) and interferon α (IFN-α) in SLE (4). Along this line, DCs exposed to necrotic, but not apoptotic, cells induce lupus-like disease in MRL mice and accelerate the disease of MRL/lpr mice (7).
The mammalian target of rapamycin (mTOR) is located in the outer mitochondrial membrane and serves as a sensor of mitochondrial dysfunction and ATP depletion in T cells (8). mTOR activity is increased in lupus T cells (9). Treatment with rapamycin markedly decreased disease activity in lupus-prone mice (10) and SLE patients resistant or intolerant to conventional immunosuppressants (11). MHP persisted while CD3/CD28-induced Ca2+ fluxing was normalized in T cells of rapamycin-treated patients, suggesting that altered Ca2+ fluxing is downstream of mitochondrial dysfunction (11). Without moderating MHP, blockade of mTOR by N-acetylcysteine (NAC) also improved disease activity in patients with SLE (12). The activation of mTOR was inducible by NO (9), a key trigger of MHP and mitochondrial biogenesis (13). mTOR is also activated by oxidative stress (14) which is detectable in lupus T cells via increased production of reactive oxygen intermediates (ROI) and GSH depletion (6,15). Increased mTOR activity may cause the apoptosis resistance (16), promote necrosis (17), and suppress the expression of the FoxP3 transcription factor (18–21) and deplete CD4+/CD25+/FoxP3+ Tregs (22) which are deficient in patients with active SLE (23,24). Depletion of C3 and C4 (25) and increased anti-DNA antibodies have long been associated with disease activity, particularly with renal flares (26). However, neither hypocomplementemia (27) nor changes in anti-DNA (28) predict future flares (27,28). Therefore, we evaluated checkpoints of mitochondrial dysfunction, that may drive abnormal death signaling and anti-DNA production, as measures of disease activity in SLE. The present study reveals that mTOR activation causes increased production of IL-4 and necrosis of DN T cells, mediates lineage skewing in T-cell and B-cell compartments, predicts flares, and thus serves as mechanistically relevant target for treatment of SLE.
MATERIALS AND METHODS
Human subjects
Peripheral blood lymphocytes (PBL) were isolated during 274 visits of 59 SLE patients and evaluated by flow cytometry in parallel with 214 PBL samples from 54 healthy controls. The mean (±SEM) age of patients was 43.1±1.6 years, ranging between 20–65 years. 56 patients were females including 49 Caucasians, six African-Americans, and one Hispanic. 3 patients were Caucasian males. 54 healthy subjects were individually matched for each patient blood donation for age within ten years, gender, and ethnic background and their freshly isolated cells were studied in parallel as controls for flow cytometry studies. The mean (±SEM) age of controls was 39.1±1.8 years, ranging between 20–62 years. 47 controls were females including 40 Caucasians, five African-Americans, and two Hispanic. 7 controls were Caucasian males. SLE disease activity was assessed by the British Isles Lupus Assessment Group (BILAG) (29) and SLE Disease Activity Index (SLEDAI) (30,31). Pre-flare visits were defined as visits change of SLEDAI by < 4 relative to preceding visit and followed by a flare characterized by ≥4 increase of SLEDAI on subsequent visit in 129 ± 29 days (n=15). Stable disease was defined as visits with SLEDAI change < 4 relative to preceding and follow-up visit (n=124). Remission visits were defined as decrease of SLEDAI ≥ 4 relative to preceding visit occurred after a follow-up interval of 228 ± 24 days (n=61). We documented concurrent use and dosage of medications. Routine blood tests included complete blood count, liver and kidney function test, urinalysis and traditional lupus-relevant serological biomarkers, such as anti-double-stranded DNA, C3, and C4. 14 patients enrolled into the prospective Study of Rapamycin for the Treatment of SLE (ClinicalTrials.gov Identifier:NCT00779194) were also investigated. The mean (±SEM) age of these patients was 44.3±4.2 years, ranging between 18–65 years. 12 patients were Caucasian females and 2 patients were Caucasian males. 17 healthy subjects were individually matched for each patient blood donation for age within ten years, gender, and ethnic background and their freshly isolated cells were studied in parallel as controls for flow cytometry studies. The mean (±SEM) age of controls was 37.4±3.4 years, ranging between 20–63 years. 14 controls were Caucasian females and 3 controls were Caucasian males.
Assessment of metabolic biomarkers in live cells by flow cytometry
We examined unstimulated cells and cells stimulated with CD3/CD28 for 16 h (9). T-cell subsets were analyzed by staining with antibodies to CD4, CD8, and CD25. B cell subsets were identified by CD19 and CD25 staining. Cell death pathway selection was monitored with annexin V-FITC, annexin V-PE, or annexin V-AlexaFluor-647 matched with emission spectra of propidium iodide (PI) to detect Annexin V+/PI− apoptotic cells and PI+ necrotic cells (6). Mitochondrial transmembrane potential (ΔΨm) was assessed with positively charged cationic dyes (DiOC6, 40 nM, excitation: 488 nm, emission: 525 nm recorded in FL-1; TMRM, 100 nM, excitation: 543 nm, emission: 567 nm recorded in FL-2). Mitochondrial mass was evaluated with potential-insensitive mitochondrial dyes MitoTracker Green-FM (MTG, 100 nM; excitation: 490 nm, emission: 516 nm recorded in FL-1) or nonyl acridine orange (NAO, 50 nM; excitation: 490 nm, emission: 540 nm recorded in FL-1). Reactive oxygen intermediates (ROI) were assessed with superoxide-sensing hydroethidine (HE, 1 μM) and H2O2-sensing dichlorofluorescein diacetate (DCF-DA, 1 μM), nitric oxide (NO) sensor 4-amino-5-methylamino-2′,7′-difluoroflourescein diacetate (DAF-FM, 1 μM, excitation: 495, emission: 515 nm recorded in FL-1). Cytosolic Ca2+was assessed with Fluo-3 (1 μM, excitation: 506 nm, emission: 526 nm recorded in FL-1) and mitochondrial Ca2+ was assessed with Rhod-2 (1 μM; excitation: 552 nm, emission: 581 nm recorded in FL-2), respectively. All metabolic and mitochondrial sensor dyes were obtained from Invitrogen (Carlsbad, CA) and used as earlier described (9,13,32,33). We recorded up to 12 parameters simultaneously using a Becton Dickinson LSRII flow cytometer equipped with 20 mW solid-state Nd-YAG (emission at 355 nm), 20 mW argon (emission at 488 nm), 10 mW diode pumped solid state yellow-green (emission 561 nm) and 16 mW helium-neon lasers (emission at 634 nm). Necrostatin-1 (34), necrostatin-5(35), and necrostatin-7 were obtained from Biovision (Milpitas, CA) and used to inhibit necrosis, as earlier described (36). Each patient’s cells were freshly isolated, stained and analyzed in parallel with a matched control. Mean channel fluorescence (MFI) values of patient samples were normalized to controls set at 1.0 for each analysis and expressed as fold changes. Frequencies of cell populations were compared as absolute values.
Assessment of mTOR activity, FoxP3 expression, and cytokine production by flow cytometry
We examined unstimulated cells and cells stimulated with CD3/CD28 for 16 h. For detection of mTOR activity and FoxP3 expression, cells were permeabilized with Cytofix/CytopermPlus (eBiosciences) and stained with AlexaFluor-488 or AlexaFluor-647-conjugated antibody to pS6RP (Cell Signaling; Beverly, MA; Cat. No. 4851) and AlexaFluor-647-conjugated antibody to FoxP3 (BioLegend, San Diego, CA; Cat No 320014), as earlier described (12). Intracellular cytokine production was measured after additional in vitro stimulation for 3 h with 50 ng/ml phorbol myristyl acetate (PMA) and 1 μg/ml ionomycin in the presence of 10 μg/ml Brefeldin-A. PMA, ionomycin and Brefeldin-A were purchased from Sigma-Aldrich (St. Louis, MO), followed by fixation and permeabilization and staining with antibodies from BD Biosciences: FITC-conjugated anti-IFN-γ (Cat.No. 554700), APC-conjugated anti-IL-4 (Cat.No. 560671), and PE-conjugated anti-IL-17a (Cat.No. 560436). Relative fluorescence intensity (RFI) was calculated by comparison of mean channel fluorescence intensity (MFI) values of patients’ cells to healthy subjects’ cells, which were analyzed in parallel and normalized to 1.0.
Statistics
Pearson’s correlations between disease activity and biomarkers, partial least square-discriminant analysis (PLS-DA), principal component analysis (PCA), factor analysis, paired or unpaired t-test with Welch’s correction, and chi-square and Fischer’s exact tests were performed with the Statistical Package for the Social Sciences (SPSS Chicago, Ill), Stata (StataCorp, College Station, TX) and Prism softwares (GraphPad, San Diego, CA). Multivariate analyses of biomarkers capable of discriminating between SLE and control subjects were performed by partial least squares - discriminant analysis (PLS-DA). PLS is a supervised method that uses multi-variate regression technique to extract via linear combination of biomarkers (X) the information that can predict the subject group membership (Y). The classification and cross validation were performed using the wrapper function offered by the caret package in Metaboanalyst software (37). In order to assess that the class discrimination is statistically significant, a permutation test was performed. In each permutation, a PLS-DA model was built between the data (X) and the permuted class labels (Y) using the optimal number of components determined by cross validation for the model based on the original class assignment. The ratio of the between sum of the squares and the within sum of squares (B/W-ratio) for the class assignment prediction of each model was calculated. PLS-DA model of 212 metabolic biomarkers was validated by permutation tests’ p value < 0.001, which generated components 1 and 2 accounting for 24.7% and 21% of the total variance in lupus and control PBL. Validity of the PLS-DA model was retained at permutation tests’ p value < 0.0005 when using the top 15 contributors, which generated components 1, 2, and 3 accounting for 28.3%, 29.2%, and 12.7% of the total variance in 274 lupus and 214 control visits. False discovery rate (FDR) p values were determined for each contributing biomarker and considered significant at p<0.000236 with correction for multiple comparisons (0.05/212). Comparison of lupus and control PBL with PCA and factor analysis revealed top differentiating biomarker sets similar to those identified by PLS-DA. Individual biomarkers were compared between control and lupus PBL by paired or unpaired t-test with Welch’s correction using Prism. Medication use was compared between patient groups exhibiting flare and remission with chi-square and Fischer’s exact tests using Prism.
Supplemental materials include Figs. S1–3 and Table S1.
RESULTS
Prominent MHP and accumulation of mitochondrial mass in CD3+/CD4+/CD8− double-negative (DN) T cells of patients with SLE
We observed overall depletion of CD3+ T cells and expansion of CD19+ B cells in SLE patients (Fig. 1A). The prevalence of necrotic cells was moderately increased within CD3+, CD4+, CD8+, and, most noticeably, in DN T cells (control: 2.96±0.22%, SLE: 4.14±0.32%, p=0.0026; data not shown). In accordance with earlier findings, elevated ΔΨm or MHP was confirmed in lupus T cells using potentiometric dyes DiOC6 and TMRM (Fig. S1 and Fig. 1). Interestingly, the extent of MHP varied among CD3+/CD4+ (DiOC6: 1.3-fold; p=5.9×10−19), CD3+/CD8+ (DiOC6: 1.5-fold; p=2.9×10−15), and DN T cells subsets (DiOC6: 1.80-fold; p=1.3×10−20). MHP of CD4+ T cells was significantly exceeded by CD8+ T cells (p=0.045) and, particularly, DN T cells (p=0.000215).
Fig. 1.

Expansion of DN T cells with MHP and increased mitochondrial mass in PBL of patients with SLE. Frequency of annexin V− cell subsets (panel A) and subpopulations with increased ΔΨm (panel B: DiOC6 fluorescence; panel C: TMRM fluorescence) and mitochondrial mass (panel D, MTG fluorescence) were assessed in PBL during 274 visits of 59 patients relative to 214 PBL samples of 54 healthy subjects. p < 0.05 reflects unpaired two-tailed t-test.
Mitochondrial potentiometric and mass-sensing dyes detected discrete cell populations exhibiting MHP (DiOC6 and TMRM) and increased mitochondrial mass (MTG; Fig. S1). MHP (%DiOC6hi:1.9-fold, p=5.9×10−22, Fig. 1B; %TMRMhi:1.8-fold, p=2.1×10−22, Fig. 1C) and mitochondrial mass were most prominently increased in DN lupus T cells (%MTGhi:1.6-fold, p=3.9×10−26; Fig. 1D). Of note, B cells exhibited a modest decline of TMRM fluorescence, as cells exhibiting high TMRM fluorescence were depleted in the B-cell compartment of SLE patients (Fig. 1C).
The population of cells with high mitochondrial mass was overall increased in the CD3+ T-cell compartment (p=1.1×10−8), most considerably in DN T cells of SLE patients (35.3±1.0%) relative to controls (21.8±0.7%, p=3.9×10−26). PLS-DA model of 212 metabolic biomarkers identified 15 top contributors and generated three component matrices that accounted for 28.3%, 29.2%, and 12.7% of the total variance (Fig. 2A) and allowed discrimination between lupus and control PBL (Fig. 2B), as validated by permutation tests p value < 0.0005 (Fig. 2C). Increased mitochondrial mass (%MTGhi) of DN T cells (FDR p=1.1×10−22) and reduced FoxP3 expression within CD4+/CD25+ T cells (FDR p=6.7×10−7) were the most robust contributors to PLS-DA components 1–3 (Fig. 2A).
Fig. 2.

Partial Least Squares - Discriminant Analysis (PLS-DA) of 212 metabolic biomarkers in 274 lupus and 214 healthy control PBL samples. A, Coefficient-based importance measures of the top 15 contributors to components 1, 2, and 3. B, 3-dimensional score plot of PLS-DA using components 1, 2, and 3, accounting for 28.3% and 29.2%, and 12.7% of total variance, respectively. C, Validation of PLS-DA by permutation tests.
mTOR activation of DN T cells correlates with diminished FoxP3 expression of CD4+/CD25+ T cells in patients with SLE
Activation of mTOR, which serves as a sensor of ΔΨm in T lymphocytes (8), has been implicated in controlling the development of regulatory T cells (Tregs) dependent on expression of the transcription factor FoxP3 (38). Therefore, mTOR activity was evaluated by intracellular staining of its downstream target, the phosphorylated form of the S6 ribosomal protein (pS6RP, Fig. 3A). pS6RP+ cells were expanded within the DN T-cell subset (12.7±0.7%) relative to CD4+ (0.6±0.07%; p=4.1×10−33), CD8+ (1.0±0.1%; p=9.4×10−20) or all T cells in normal PBL (1.8±0.1%; p=2.3×10−42). The frequency of pS6RP+ DN T cells was increased in SLE patients (p=0.007; Fig. 3B). While the percentage of FoxP3+/CD25+ cells within the CD3+/CD4+ compartment was not diminished, a discordant expression FoxP3 and CD25 was observed between lupus and control T cells (Fig. 3C). The percentage of FoxP3+ cells within the CD3+/CD4+/CD25+ compartment was diminished in lupus patients relative to controls (p=2.7×10−7, Figs. 3A and C). In contrast, FoxP3 expression was significantly increased in CD4+/CD25−T cells of SLE patients (p=1.6 × 10−5, Figs. 3A and C). Increased mTOR activity in DN T cells showed moderate but significant correlation with diminished FoxP3 expression in CD4+/CD25+ T cells (r =−0.1319, p=0.030; Fig. 3D). mTOR activity in DN T cells did not correlate with FoxP3 expression in non-regulatory CD4+/CD25−T cells either in healthy (Pearson’s r=−0.037; p=0.59) or SLE donors (Pearson’s r=0.043; p=0.47). A lack of detectable mTOR hyperactivity in CD4+/CD25+ lupus T cells pointed to the involvement of indirect mechanisms driving diminished expression of FoxP3 in SLE.
Fig. 3.

mTOR activation in DN T cells correlates with contraction of Tregs in SLE. A, Upper panel: Detection of increased mTOR activity via phosphorylation of S6 ribosomal protein (pS6RP) in T-cell subsets from lupus and matched control donors. %pS6RPhi cells are indicated for control (blue histograms) and lupus T cells (red histograms), respectively. Lower panel: Dot plot and histogram of FoxP3 expression within CD25+ T cells, gating on CD3+/CD4+ T cells in control and lupus PBL. B, Cumulative analysis of mTOR activity in T-cell subsets in 274 lupus and 214 control PBL samples. C, Cumulative analysis of CD4+ T cells by expression of FoxP3 and CD25. D, Correlation of mTOR activity in DN T cells with expression of FoxP3 in CD4+/CD25+ T cells of 264 lupus PBL samples.
Correlation of metabolic biomarkers with disease activity in SLE
We evaluated 212 parameters reflecting ΔΨm, mitochondrial mass, NO and ROI production, [Ca2+]c and [Ca2+]m, mTOR and FoxP3 expression in T-cell and B-cell subsets for correlation with disease activity; p values <0.000236 were considered significant after correcting for multiple comparisons (0.05/212, representative correlations are shown in Fig. 4). The SLEDAI (6.2±0.3) and BILAG scores (24.5±0.6) showed significant correlation (r=0.450; p=1.4 × 10−14; Table S1). SLEDAI correlated with absolute values of traditional biomarker components C3 (r = −0.225; p=0.00037), C4 (r=−0.237; p=0.00016), and anti-DNA (r=0.237; p=0.00039; Fig. 4). C3 and C4 correlated with each other (r=0.695; p=1.4×10−38). Anti-DNA correlated with C3 (r=−0.191; p=0.004) and C4 (r=−0.323; p=6×10−7).
Fig. 4.

Correlation of SLEDAI and traditional biomarkers of disease activity, such as C3, C4, and anti-DNA, with 212 metabolic, cell surface, and gene expression biomarkers during 274 visits of 59 patients with SLE. Due to missing data, the actual number of available data pairs are indicated for each comparison (n = 228–255). Correlation r values were considered significant at p < 0.000236 when corrected for multiple comparisons (0.05/212). Correlation p values above and below 0.000236 are indicated in blue and red symbols, respectively.
As shown in Table S1 and for representative data in Fig. 4, SLEDAI was found to be correlated positively with the overall percentage of necrotic CD3+ (r=0.254; p=0.0001), CD4+ (r=0.260; p=0.00003) and DN T cells (r = 0.230; p=0.00019) as well as MHP (%DiOC6hi r=0.264; p=0.00022) and mitochondrial mass of DN T cells (%MTGhi r=0.254; p=0.00003). SLEDAI correlated negatively with FoxP3+/CD25+/CD4+ T cells after CD3/CD28 co-stimulation (r=−0.229; p= 0.00018).
Among the serological components of SLEDAI, C4 negatively correlated (%TMRMhi r=−0.316; p=2.0×10−7) while anti-DNA positively correlated with MHP of DN T cells (%TMRMhi r=0.410; p=1.6×10−10). C3 (r=0.249; p=0.00009) and C4 negatively correlated with oxidative stress in CD3+ T cells (DCF-DA r=0.235; p=0.00022; Table S1). Likewise, C3 (r=−0.294; p=3.3×10−7) and C4 negatively correlated with oxidative stress in CD8+ T cells (r=−0.239; p=0.00017). Along these lines, C4 positively correlated with CD4+ (r=0.268; p=0.00001) and negatively correlated with CD8+ T cell frequencies (r=−0.234; p=0.00014), MHP of DN T cells (%DiOC6hi r=−0.295; p=0.00004; %TMRMhi r=−0.316; p=2.6×10−7), mitochondrial mass of DN T cells (%MTGhi r=−0.264; p=0.00002) and B cells (%MTGhi r=−0.274; p=0.00001). Anti-DNA positively correlated with MHP (%DiOC6hi r=0.380, p=3.2×10−7; %TMRMhi r=0.410, p=1.6×10−10; TMRM MFI r=0.517, p=9.2×10−17) and mitochondrial mass of DN T cells (%MTGhi r=0.284, p=0.00001; MTG MFI r=0.357, p=3.6×10−8) and oxidative stress in CD8+ T cells (r=0.300; p=0.00001; Table S1).
Increased necrosis, mTOR activation in DN T cells and depletion of Tregs distinguish SLE patients in flare
To further evaluate the relationship of metabolic biomarkers with disease activity, we compared their performance during 15 patient visits with ≥ 4 increase of SLEDAI (+5.3±1.5) relative to 61 patient visits with ≥ 4 decrease of SLEDAI (−7.8±0.5, p=3.9×10−22). During the remaining 198 patient visits SLEDAI changes were < 4. While anti-DNA was increased at 605±181 U/ml relative to 174±47 U/ml (p=0.003), unexpectedly, C3 and C4 levels were not different between flaring and remitting patients, possibly due to the relatively modest number of flaring patients. The individual doses and numbers of patients taking prednisone were similar between the groups in flare and remission (data not shown). Flaring SLE patients exhibited increased necrosis of CD3+ (SLE: 1.4±0.3 %; control: 0.5±0.07%, p=0.0196), CD4+ (SLE: 0.9±0.3 %; control: 0.2±0.2%, p=0.0413), and DN T cells (SLE: 9.8±2.7%; control: 3.4±0.5%, p=0.0247; Figs. 5A and B). Patients in remission did not show increased frequency of necrotic cells within the T cell compartment (Figs. 5A and B).
Fig. 5.

Activation of mTOR, depletion of Tregs, and expansion of necrotic DN T cells distinguish SLE patients in flare. A, Detection of necrotic cells by PI staining in healthy (Control), remitting (SLE remission) and flaring SLE donors (SLE flare). B, Cumulative analyses of necrotic T cells during 214 healthy subject visits, 61 remitting SLE patient visits, and 15 flaring SLE patient visits. C, ΔΨm (DiOC6 and TMRM), mitochondrial mass (MTG), and mTOR activity (pS6RP) in healthy subjects, remitting SLE patients, and flaring SLE patients. D, Frequency of FoxP3−/CD25+ cells within CD4+ T cells and frequency of FoxP3+ cells within CD25+/CD4+ T cell compartments of healthy subjects, remitting SLE patients, and flaring SLE patients. p < 0.05 reflect unpaired two-tailed t-test.
In keeping with their increased rate of necrosis, DN T cells of flaring patients exhibited MHP (%TMRMhi SLE: 25.6±5.1%, control: 11.2±0.4 %; p=0.014), increased mitochondrial mass (%MTGhi SLE: 43.1± 5.3%, control: 21.8±0.7 %; p=0.0013), and increased mTOR activity (%pS6RPhi SLE: 17.6±3.2%, control: 12.7±0.7 %; p=0.0247; Fig. 5C). Patients in flare had increased numbers of FoxP3−/CD25+/CD4+/CD3+ T cells (SLE:7.5±1.6%, control: 3.0±0.2 %; p=0.0015;) and decreased numbers of FoxP3+ cells within the CD3+/CD4+/CD25+ T cell compartment (SLE:33.8±5.5%, control: 49.8±1.2 %; p=0.0015; Fig. 5D).
Patients in remission did not show increased frequency of necrotic cells within the T cell compartment (Figs. 5A and 5B), however, they still exhibited MHP of CD3+ (%DiOC6hi SLE: 63.3±1.8%, control: 50.8±1.1 %; p=1.0×10−8), CD4+ (%DiOC6hi SLE: 75.4±1.4%, control: 61.7±1.2 %; p=2.5×10−8), CD8+ (%DiOC6hi SLE: 33.4±2.0%, control: 24.3±0.9%; p=0.0001), and DN T cells (%DiOC6hi SLE: 33.4±2.4%, control: 15.6±0.6 %; p= 7.9×10−10; Fig. 5C) as well as increased mitochondrial mass in CD3+ (MTG MFI 1.3±0.06-fold; p=4.6×10−6), CD4+ (MTG MFI 1.3±0.05-fold; p=3.8×10−6), CD8+ (MTG MFI 1.4±0.1-fold; p=0.001) and DN T cells (MTG MFI 1.8±0.1-fold, p=5.3×10−7; %MTGhi p=3.6×10−9, Fig. 5C). SLE patients in remission did not show increased mTOR activity in DN T cells relative to healthy controls (%pS6RPhi SLE: 12.3±1.8 %, control: 12.7±0.7%; p=0.83) but did show diminished mTOR activity in all CD3 T cells relative to all patients upon study entry (61 SLE patient visits in remission %pS6RPhi: 1.6±0.3%, all 59 SLE patients at baseline: 3.7±0.9%; p=0.037). FoxP3 expression within CD3+/CD4+/CD25+ T cells of SLE patients in remission remained low (42.7±3.2%) in comparison to healthy controls (49.8±1.3%; p=0.043; Fig. 5D). mTOR blockers were being used in three of the 15 flare visits (rapamycin in two patients and NAC in one patient) and 56 of 61 remission visits (rapamycin in 36 cases and NAC in 20 cases (Fischer’s exact test p value < 0.0001). No difference was found in the use of prednisone, mycophenolate, or hydroxychloroquine.
mTOR activation in DN T cells and discordant expression of FoxP3 and CD25 in CD4+ T cells predict flare in SLE
To identify metabolic biomarkers potentially predicting disease flares, we compared pre-flare parameters of 15 patient visits, which had preceded the SLEDAI elevation of ≥4 by 129±32 days, to those of healthy controls, 124 stable SLE patient visits defined as showing SLEDAI changes < 4 compared to previous and subsequent visits, and 61 remission visits defined by ≥4 decrease of SLEDAI relative to prior visits. Within the live compartment, reduced numbers of CD3+ T cells were noted in all SLE patients relative to healthy controls, while pre-flare patients had fewer CD4+ T cells and more CD8+ and DN T cells than stable patients treated without mTOR inhibitors (Fig. 6A). 3-dimensional PLS-DA showed clear distinction of pre-flare and flare visits largely dominated by parameters of increased T-cell necrosis (not shown). Necrosis of DN T cells was reduced in pre-flare (3.5±0.8 %) relative to flare visits (9.8±2.7 %, p=0.020 using paired t-test) but unaffected in comparison to stable visits (4.4±0.5 %, p=0.068; Fig. 6B). mTOR activity was remarkably increased in DN T cells at pre-flare visits (%pS6RPhi 30.5±6.1%), as compared to healthy controls (12.7±0.7%; p=0.0161) and stable (18.2±1.2%; p=0.032) or remission SLE visits (12.3±1.8%; p=0.0004; Fig. 6C). mTOR blockers were only used during two of 12 pre-flare visits relative 56/61 remission visits (p<0.0001) and 75/124 stable patient visits (p=0.0048). mTOR activity was lower in DN T cells of stable patient treated with mTOR inhibitors (11.0±1.2%) relative to those without (21.7±2.1%; p=7.5×10−6; Fig. 6C). However, mTOR activity was also increased in CD4+ and CD8+ T cells during pre-flare visits relative to stable visits without mTOR blockade (Fig. 6C). Upon CD3/CD28 co-stimulation FoxP3 expression was inappropriately increased in CD4+/CD25− T cells in SLE patients at pre-flare visits (2.9±0.65%) relative to matched controls (0.8±0.04%; p=0.024) and SLE patients with stable disease (1.4±0.12%; p=0.0016; Fig. 6D).
Fig. 6.

mTOR activation in DN T cells and discordant expression FoxP3 and CD25 in CD4+ T cells predict flare in SLE patients with stable disease. Pre-flare visits were defined as visits preceding flare characterized by ≥4 increase of SLEDAI on subsequent visit in 129 ± 29 days (n=15). Pre-flare visits were compared to visits of patients with stable disease, defined as having SLEDAI change < 4 relative to preceding and follow-up visit (n=124). Within the stable visit group, treatment regimens included mTOR inhibitors, rapamycin or NAC, in 75 cases; for the remaining 49 cases, patients were treated without mTOR blockade. A, Frequency of live (% AnnV−/PI−) cells in T-cell compartments. B, Assessment of necrosis (% PI+ cells) in CD3, CD4, CD8 and DN T-cell compartments. p values < 0.05 reflect comparison to flaring patients using two-tailed t-test. C, Assessment of mTOR activity (% pS6RPhi cells) in CD3, CD4, CD8 and DN T-cell compartments. p values < 0.05 reflect comparison to pre-flare patients using two-tailed t-test. D, Detection of FoxP3+CD25−CD4+ T cells following CD3/CD28 activation. p values < 0.05 reflect comparison to pre-flare patients using two-tailed t-test.
Increased IL-4 expression by DN T cells correlates with skewing of the B cell compartment and production of anti-DNA
As disease activity has been associated with B-cell activation and production of anti-DNA, we investigated the possible influence of DN T cells on skewing of the B-cell compartment via cytokine production. Within an expanded B-cell compartment (Fig. 1A), the frequency of CD25+/CD19+ B cells was diminished in SLE patients (4.8±0.3%) relative to controls (7.8±0.4%; p=2.7×10−10). T-cell activation markedly increased the frequency of CD25+/CD19+ B cells, however, they remained depleted in SLE (70.6±1.6%) relative to control subjects (78.4±0.4%; p=4.9×10−5). To understand the mechanism by which DN T cells may impact lupus disease activity, we investigated their production of Th1 (IFN-γ), Th2 (IL-4), and Th17 cytokines (IL17a) in 26 patients that have not been exposed to mTOR inhibitor treatment with rapamycin or NAC. DN T cells harbored the highest frequency of IL-4-producing cells relative to other T-cell subsets both in SLE and matched healthy subjects (p<0.0001; Fig. 7A). IL-4 production was detected in 11.2±1.7% of DN T cells in SLE patients relative to 6.9±0.8% of DN T cells in healthy subjects (p=0.007; Fig. 7A). Representative dot plots are shown in Fig. S2. Following CD3/CD28 co-stimulation, IL-4-producing cells remained increased in the DN T-cell compartment (p=0.045, data not shown). IFN-γ production was reduced in CD8+ lupus T cells (p=0.045, data not shown). While the frequencies of IL17+ T cells were not increased significantly in SLE (Fig. 7B), moderately enhanced production of IL-17 was observed when comparing MFI of T cells between lupus patients and matched controls using paired t-test (Fig. 7C). IL4production by DN T cells was found to be positively correlated with anti-DNA (r=0.601; p=0.006; Fig. 7D) and CD25−/CD19+ B cells (r=0.520; p=0.009; Fig. 7E) and negatively correlated with CD25+/CD19+ B cells (r=−0.446; p=0.029; Fig. 7F). In contrast, IL-17 production by DN T cells did not correlate with anti-DNA (Fig. 7G) or frequencies of CD25−/CD19+ (Fig. 7H) or CD25+/CD19+ B cells (Fig. 7I).
Fig. 7.

Increased IL-4 production by DN T cells correlates with skewing of B-cell subsets and anti-DNA in patients with SLE. A, Intracellular production of IL-4 by CD3+, CD4+, CD8+, and DN T cells in 26 patients with SLE and 26 matched healthy subjects. Percentage of IL-4-producing cells was determined by flow cytometry. %IL-4+ cells were increased among DN T cells relative to other T-cell subsets both in lupus and control PBL (<0.0001). p < 0.05 reflects paired two-tailed t-test comparing lupus and healthy subjects. B, Intracellular production of IL-17 by CD3+, CD4+, CD8+, and DN T cells in SLE and matched healthy subjects. %IL-17+ cells were increased among CD4+ T cells relative to other T-cell subsets both in lupus and control PBL. There was no difference in %IL-17+ cells between lupus and control subjects using paired t-test. C, Production of IL-4 and IL-17 by MFI of CD3+, CD4+ and DN T cells of lupus patients normalized to matched healthy controls set at 1.0 for each analysis and expressed as fold changes. p < 0.05 reflects comparison of lupus and matched healthy subjects with paired two-tailed t-test. D, Correlation of %IL-4+ DN T cells with anti-DNA levels in SLE. E, Positive correlation of IL-4+ DN T cell and CD25− B cell frequencies. F, Negative correlation of IL-4+ DN T cell and CD25+ B cell frequencies. G, Correlation analysis of %IL-17+ DN T cells and anti-DNA levels in SLE. H, Correlation analysis of IL-17+ DN T cell and CD25− B cell frequencies. I, Correlation analysis of IL-17+ DN T cell and CD25+ B cell frequencies.
Treatment with rapamycin reduces necrosis and IL-4 production of DN lupus T cells
To determine if mTOR has a controlling influence over lineage skewing and abnormal death signaling in SLE, we examined 14 patients, who have been enrolled in a prospective open-label study and achieved therapeutic plasma levels of rapamycin at 8.7±1.2 ng/ml after 126±18 days. Preliminary analysis of this ongoing study revealed a significant improvement of disease activity, as measured by the reduction of SLEDAI to 5.7 ±1.0 from 11.8 ±1.1 at baseline (p = 0.0028) and BILAG to 20.9 ± 2.4 from 29.7±3.2 at baseline (p = 0.027; Fig. 8A). mTOR activity, as measured by %S6RPhi cells, was reduced in all T cell subsets of rapamycin-treated SLE patients relative to matched controls studied in parallel (p < 0.03; data not shown). Rapamycin-treated lupus patients exhibited diminished necrosis of CD4+ T cells (Fig. 8B). Necrosis of DN T cells was also diminished following CD3/CD28 co-stimulation (Fig. 8C). Rapamycin inhibited IL-4 production by DN T cells of lupus patients as indicated by reduced IL-4 MFI from 2.2 ± 0.5-fold to 0.9 ± 0.1-fold relative to matched controls (p=0.034; Fig. 8D). The percentage of IL-4+ DN T cells was not reduced significantly by rapamycin from 15.4 ± 2.6% to 12.4 ±1.9% (p=0.26). Rapamycin did not significantly affect the frequency of IL-17+ DN T cells (pretreatment: 0.21±0.12%, rapamycin treatment: 0.11±0.03%; two-tailed paired t-test p=0.33) or the IL-17 MFI in DN T cells of lupus patients (p=0.59). In contrast, rapamycin increased the expression of FoxP3 in CD4+/CD25+ T cells (Fig. 8E). Within the B-cell compartment, the frequency of CD25+/CD19+ cells was increased from 1.9 ± 0.4% to 3.3 ±0.7% in rapamycin-treated patients (p=0.029; Fig. 8F). The role of programmed necrosis or necroptosis (39) was evaluated by using necrostatins (34–36). Necrostatin-7, but not necrostatin-1 or necrostatin-5, reduced CD3/CD28 costimulation-induced necrosis of DN T cells in PBL of healthy subjects from 4.3±0.7% to 2.2±0.3% (p=0.013). None of the necrostatins reduced necrosis or IL-4 production of DN T cells in PBL of 8 patients with SLE (data not shown).
Fig. 8.
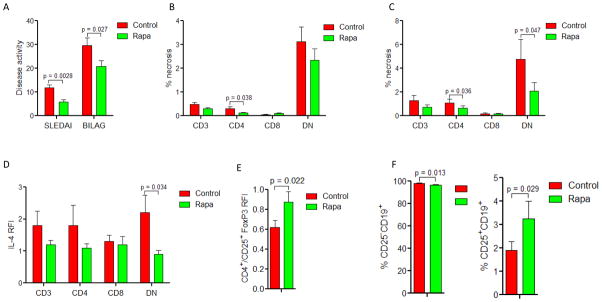
Effect of rapamycin on biomarkers of disease activity in patients with SLE. A, SLEDAI and BILAG disease activity scores in 14 SLE patients before and during rapamycin treatment of 126±18 days monitored by therapeutic plasma levels of 8.7±1.2 ng/ml; p values reflect paired t-test. B, Effect of rapamycin treatment on necrosis monitored by the prevalence of PI+ cells in CD3+, CD4+, CD8+, and DN T-cell subsets. C, Effect of rapamycin treatment on necrosis monitored by the prevalence of PI+ cells in CD3+, CD4+, CD8+, and DN T-cell subsets following CD3/CD28 co-stimulation. D, Effect of rapamycin on IL-4 expression in CD3+, CD4+, CD8+, and DN T-cell subsets assessed by relative fluorescence intensity (RFI) in comparison to matched healthy controls. E, Effect of rapamycin on FoxP3 expression in CD4+/CD25+ T cells assessed by relative fluorescence intensity (RFI) in comparison to matched healthy controls. F, Effect of rapamycin on the frequency of CD25−/CD19+ and CD25+/CD19+ B cells in SLE patients.
DISCUSSION
Lupus pathogenesis involves abnormal activation and death signaling in the immune system (1) that culminates in the release of pro-inflammatory nuclear debris, mostly from necrotic cells (4,6), driving the production of anti-nuclear auto-antibodies with over 100 known specificities (40). The present study provides evidence that metabolic biomarkers of T-cell dysfunction correlate with disease activity in SLE patients. The mitochondrial and cell death signal processing defects, such as MHP, increased mitochondrial mass, and necrosis, are most prominently exhibited by DN T cells and correlate with SLEDAI, contraction of Tregs, and activation of B cells. Necrotic cell death of DN T cells was strikingly increased in flaring SLE patients. mTOR, which is a sensor of ΔΨm and ATP depletion in T cells (8), is activated in DN T cells of flaring SLE patients. The correlation of SLEDAI with checkpoints of mitochondrial dysfunction in DN T cells reached or exceeded that with hypocomplementemia or increased anti-DNA. In contrast to mTOR activation, MHP and accumulation of mitochondria were observed in all T cells of SLE patients, even in remission, which suggested that mitochondrial dysfunction was a cause rather than a consequence of mTOR activation and disease flares. Indeed, mTOR activation is downstream of mitochondrial dysfunction as depicted in a schematic diagram (Fig. S3) and evidenced by 1) the induction of mTOR by NO (9), a trigger of MHP (13) and mitochondrial biogenesis (41), and 2) the persistence of MHP and accumulation of mitochondria during mTOR blockade in rapamycin-treated patients (9,11). The notion that altered mitochondrial homeostasis contributes to lupus pathogenesis has been supported by the recent identification of a lupus susceptibility gene as estrogen-related receptor gamma, which accounts for increased mitochondrial voltage-dependent anion channel protein levels in the spleen of lupus-prone Sle1c2 mice (42).
mTOR activation of DN T cells and expansion of FoxP3+/CD25−/CD4+T cells have been identified as predictors of flare. Of note, FoxP3+/CD25−/CD4+T cells are also expanded in new-onset SLE (43–45), suggesting a mechanistic role for this T-cell subset in triggering disease activation. The clinical improvement of SLE following mTOR blockade both in mice (10) and humans (11,12,46) supports the notion that mTOR activation represents a pathogenetically relevant biomarker, potentially causing as well as predicting flares and responsiveness to treatment. Measurement of MHP and mitochondrial mass, to a lesser extent, are dependent on cellular viability and require parallel analysis of healthy controls (47). In contrast, mTOR activity and FoxP3 expression can be measured in fixed and permeabilized cells that may be stored, shipped, and analyzed in batches, making these checkpoints of T-cell activation potentially useful biomarkers of disease activity in the clinical setting.
In addition to apoptosis and autophagy, necrosis is one of three fundamental forms of cell death, (48). Necrosis has long been considered a passive or accidental form of cell death, which is characterized by membrane damage and cellular swelling due to the depletion of ATP required for the activity of enzymes that maintain membrane integrity (4,49). However, it has lately become apparent that many different necrosis pathways exist (50). In fact, several necrosis pathways may also be actively regulated, which are termed necroptosis (39). While necroptosis has been initially implicated in mouse ischemic brain injury (34), neurodegeneration, and infection (39), it has become apparent that necroptosis also occurs in cells of the immune system (50). The best studied necroptosis pathway is triggered by tumor necrosis factor α (TNFα) and transduced by the kinase, receptor-interacting protein-1 (RIP1) (51). However, necroptosis inhibitor necrostatins failed to reduce necrosis or IL-4 production of DN T cells in PBL of patients with SLE. The lack of inhibition by necrostatins is in accordance with a central role for TNFα in necroptosis (52) and the diminished production of this cytokine in patients with SLE (53). Of note, mTOR activation has recently been found to promote necrotic cell death both in vivo (54) and in vitro (17). This is consistent with our findings shown in Fig. 8, indicating that in vivo treatment of SLE patients with rapamycin inhibited both necrosis and IL-4 production of DN T cells. As necrotic materials are more potent than apoptotic materials in triggering antinuclear antibody production in lupus-prone mice (7), inhibition of necrosis may have contributed to reduced anti-DNA production in rapamycin-treated SLE patients (11).
mTOR activation has been associated with the contraction of CD4+/CD25+/Foxp3+ Tregs in healthy subjects (22), type I diabetes patients (22), transplant recipients (55) and SLE patients (12,46). While inactivation of mTOR favors T-cell differentiation into Tregs (38), recent findings indicate that mTOR may not be completely dispensable for Treg development in mice (56). In our study, the inverse correlation between elevated mTOR in DN T cells and diminished frequency of FoxP3+/CD25+/CD4+ T cells pointed to the involvement of indirect mechanisms. In particular, the enhanced secretion of IL-4 by DN T cells may block the differentiation of FoxP3+ Tregs from naïve CD4+ T cells (57). The enhanced production of IL-4 by DN T cells may also promote the differentiation of Th2 cells and their production of IL-4, which are also mTOR dependent (58). The indirect effect of mTOR blockade may involve the expansion of CD25+/CD19+ B cells, which have been found to expand Tregs (59) and to be depleted in SLE (60). Increased IL-4 production by DN T cells has also been implicated in B-cell activation and anti-DNA production in SLE (61,62). Interim analysis of our ongoing prospective trial indicated that rapamycin treatment lowered the production of IL-4 by DN T cells and expanded CD25+/CD19+ B cells and Tregs in SLE patients. In summary, the present study has identified mTOR activation as a therapeutically targetable checkpoint of disease pathogenesis that may account for elevated production of IL-4 by DN T cells, thus causing Treg depletion, B-cell activation as well as increased anti-DNA production in SLE. mTOR activation warrants further characterization as a mechanism of pathogenesis and predictor of flares in SLE.
Supplementary Material
Acknowledgments
This work was supported in part by grant AI072648 from the National Institutes of Health, the Alliance for Lupus Research, and the Central New York Community Foundation.
LIST OF ABBREVIATIONS
- ΔΨm
mitochondrial transmembrane potential
- AnnV
Annexin V
- BILAG
British Isles Lupus Assessment Group
- Ca2+
calcium
- DAF-FM
4-amino-5-methylamino-2′,7′-difluoroflourescein diacetate; nitric oxide indicator
- DAR-4M
Diaminorhodamine-4M; peroxynitrite indicator
- DCF-DA
dichlorofluorescein diacetate, H2O2 sensor
- DiOC6
3,3′-dihexyloxacarbocyanine iodide; mitochondrial potential indicator
- DN T
CD3+CD4−CD8− double-negative T cell
- HE
hydroethidine; ROI sensor
- MFI
mean fluorescence intensity
- MHP
mitochondrial hyperpolarization
- MTG
MitoTracker Green
- mTOR
mammalian/mechanistic target of rapamycin
- NAC
N-acetylcysteine
- NAO
nonyl acridine orange
- NO
nitric oxide
- PBL
peripheral blood lymphocytes
- PBS
phosphate buffered saline
- PLS-DA
partial least square-discriminant analysis
- PI
propidium iodide
- ROI
reactive oxygen intermediates
- SLE
systemic lupus erythematosus
- SLEDAI
systemic lupus erythematosus disease activity index
- TMRM
tetramethylrhodamine methyl ester
- Treg
regulatory T cell; CD3+CD4+CD25+Foxp3+ T cell
References
- 1.Tsokos GC. Systemic Lupus Erythematosus. N Engl J Med. 2011;365:2110–2121. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- 2.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perl A. Systems biology of lupus: Mapping the impact of genomic and environmental factors on gene expression signatures, cellular signaling, metabolic pathways, hormonal and cytokine imbalance, and selecting targets for treatment. Autoimmunity. 2010;43:32–47. doi: 10.3109/08916930903374774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perl A, Gergely P, Jr, Nagy G, Koncz A, Banki K. Mitochondrial hyperpolarization: a checkpoint of T cell life, death, and autoimmunity. Trends Immunol. 2004;25:360–367. doi: 10.1016/j.it.2004.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandez D, Perl A. Metabolic control of T cell activation and death in SLE. Autoimmun Rev. 2009;8:184–189. doi: 10.1016/j.autrev.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gergely PJ, Grossman C, Niland B, Puskas F, Neupane H, Allam F, Banki K, Phillips PE, Perl A. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arth Rheum. 2002;46:175–190. doi: 10.1002/1529-0131(200201)46:1<175::AID-ART10015>3.0.CO;2-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma L, Chan KW, Trendell-Smith NJ, Wu A, Tian L, Lam AC, Chan AK, Lo CK, Chik S, Ko KH, To CK, Kam SK, Li XS, Yang CH, Leung SY, Ng MH, Stott DI, MacPherson GG, Huang FP. Systemic autoimmune disease induced by dendritic cells that have captured necrotic but not apoptotic cells in susceptible mouse strains. Eur J Immunol. 2005;35:3364–3375. doi: 10.1002/eji.200535192. [DOI] [PubMed] [Google Scholar]
- 8.Desai BN, Myers BR, Schreiber SL. FKBP12-rapamycin-associated protein associates with mitochondria and senses osmotic stress via mitochondrial dysfunction. Proc Natl Acad Sci USA. 2002;99:4319–4324. doi: 10.1073/pnas.261702698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandez DR, Telarico T, Bonilla E, Li Q, Banerjee S, Middleton FA, Phillips PE, Crow MK, Oess S, Muller-Esterl W, Perl A. Activation of mTOR controls the loss of TCR. in lupus T cells through HRES-1/Rab4-regulated lysosomal degradation. J Immunol. 2009;182:2063–2073. doi: 10.4049/jimmunol.0803600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warner LM, Adams LM, Sehgal SN. Rapamycin prolongs survival and arrests pathophysiologic changes in murine systemic lupus erythematosus. Arth Rheum. 1994;37:289–297. doi: 10.1002/art.1780370219. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez D, Bonilla E, Mirza N, Perl A. Rapamycin reduces disease activity and normalizes T-cell activation-induced calcium fluxing in patients with systemic lupus erythematosus. Arth Rheum. 2006;54:2983–2988. doi: 10.1002/art.22085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lai ZW, Hanczko R, Bonilla E, Caza TN, Clair B, Bartos A, Miklossy G, Jimah J, Doherty E, Tily H, Francis L, Garcia R, Dawood M, Yu J, Ramos I, Coman I, Faraone SV, Phillips PE, Perl A. N-acetylcysteine reduces disease activity by blocking mTOR in T cells of lupus patients. Arth Rheum. 2012;64:2937–2946. doi: 10.1002/art.34502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagy G, Koncz A, Perl A. T cell activation-induced mitochondrial hyperpolarization is mediated by Ca2+- and redox-dependent production of nitric oxide. J Immunol. 2003;171:5188–5197. doi: 10.4049/jimmunol.171.10.5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarbassov dD, Sabatini DM. Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. J Biol Chem. 2005;280:39505–39509. doi: 10.1074/jbc.M506096200. [DOI] [PubMed] [Google Scholar]
- 15.Gergely PJ, Niland B, Gonchoroff N, Pullmann R, Jr, Phillips PE, Perl A. Persistent mitochondrial hyperpolarization, increased reactive oxygen intermediate production, and cytoplasmic alkalinization characterize altered IL-10 signaling in patients with systemic lupus erythematosus. J Immunol. 2002;169:1092–1101. doi: 10.4049/jimmunol.169.2.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strauss L, Czystowska M, Szajnik M, Mandapathil M, Whiteside TL. Differential Responses of Human Regulatory T Cells (Treg) and Effector T Cells to Rapamycin. PLoS ONE. 2009;4:e5994. doi: 10.1371/journal.pone.0005994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu YT, Tan HL, Huang Q, Ong CN, Shen HM. Activation of the PI3K-Akt-mTOR signaling pathway promotes necrotic cell death via suppression of autophagy. Autophagy. 2009;5:824–834. doi: 10.4161/auto.9099. [DOI] [PubMed] [Google Scholar]
- 18.Liu G, Yang K, Burns S, Shrestha S, Chi H. The S1P1-mTOR axis directs the reciprocal differentiation of TH1 and Treg cells. Nat Immunol. 2010;11:1047–1056. doi: 10.1038/ni.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen C, Liu Y, Liu Y, Zheng P. Mammalian target of rapamycin activation underlies HSC defects in autoimmune disease and inflammation in mice. J Clin Invest. 2010;120:4091–4101. doi: 10.1172/JCI43873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merkenschlager M, Von Boehmer H. PI3 kinase signalling blocks Foxp3 expression by sequestering Foxo factors. J Exp Med. 2010;207:1347–1350. doi: 10.1084/jem.20101156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turnquist HR, Cardinal J, Macedo C, Rosborough BR, Sumpter TL, Geller DA, Metes D, Thomson AW. mTOR and GSK-3 shape the CD4+ T-cell stimulatory and differentiation capacity of myeloid DCs after exposure to LPS. Blood. 2010;115:4758–4769. doi: 10.1182/blood-2009-10-251488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin Promotes Expansion of Functional CD4+CD25+FOXP3+ Regulatory T Cells of Both Healthy Subjects and Type 1 Diabetic Patients. J Immunol. 2006;177:8338–8347. doi: 10.4049/jimmunol.177.12.8338. [DOI] [PubMed] [Google Scholar]
- 23.Crispin JC, Martinez A, Alcocer-Varela J. Quantification of regulatory T cells in patients with systemic lupus erythematosus. J Autoimmun. 2003;21:273–276. doi: 10.1016/s0896-8411(03)00121-5. [DOI] [PubMed] [Google Scholar]
- 24.Valencia X, Yarboro C, Illei G, Lipsky PE. Deficient CD4+CD25high T Regulatory Cell Function in Patients with Active Systemic Lupus Erythematosus. J Immunol. 2007;178:2579–2588. doi: 10.4049/jimmunol.178.4.2579. [DOI] [PubMed] [Google Scholar]
- 25.Petri M, Singh S, Tesfasyone H, Malik A. Prevalence of flare and influence of demographic and serologic factors on flare risk in systemic lupus erythematosus: A prospective study. J Rheumatol. 2009;36:2476–2480. doi: 10.3899/jrheum.090019. [DOI] [PubMed] [Google Scholar]
- 26.Linnik MD, Hu JZ, Heilbrunn KR, Strand V, Hurley FL, Joh D, arc+¦n-Segovia T, Appel G, Aranow C, Ballou S, Becker MA, Becker N, Belmont MH, Bohan A, Boling EP, Bombardieri S, Brelsford WG, Buyon JP, Cardiel MH, Carteron NL, Condemi JJ, Cronin ME, Cush JJ, DeHoratius R, Desir D, Edwards M, El-Shahawy MA, Emery P, Espinoza LR, Finkel TH, Fondal M, Fortin P, Furie RA, Genovese MC, Geppert T, Gil-Aguado A, Gilkeson GS, Ginzler E, Gluck OS, Granda JL, Grossman J, Hiepe F, Hill M, Howard P, Hura C, Ingelmo M, Jaffer A, Jakes J, Kalden JP, Kalunian KC, Kammer GM, Kanick K, Kaplan J, Kaplan S, Katz R, Kenney HM, Khamashta M, Kivitz AJ, Krishnan M, Kurtzman NA, Liebling M, Lindberg JS, Lourie SH, Loveless J, Manzi S, Martin K, McKay J, Merrill J, Moreland LW, Neuwelt MC, Petri M, Poque BC, Quinet RJ, Ramsey-Goldman R, Rosenblatt SG, Rothfield N, Scarpa N, Schneider M, Schousboe JT, Sewell LK, Shergy WJ, Sherrer Y, Sibilia J, Smith D, Spinowitz B, Spira M, Staud R, Stern S, Stevens MP, Sturfelt G, Surbeck W, Tindall EA, Torres A, Tumlin J, van Vollenhoven R, Varga J, Vijayan A, Vilardell-Tarres M, Wallace D, Weaver C, Zummer M. Relationship between anti-double-stranded DNA antibodies and exacerbation of renal disease in patients with systemic lupus erythematosus. Arth Rheum. 2005;52:1129–1137. doi: 10.1002/art.20980. [DOI] [PubMed] [Google Scholar]
- 27.Merrill JT, Buyon JP. The role of biomarkers in the assessment of lupus. Best Practice and Research: Clinical Rheumatology. 2005;19:709–726. doi: 10.1016/j.berh.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 28.van den Berg L, Nossent H, Rekvig O. Prior anti-dsDNA antibody status does not predict later disease manifestations in systemic lupus erythematosus. Clin Rheumatol. 2006;25:347–352. doi: 10.1007/s10067-005-0047-7. [DOI] [PubMed] [Google Scholar]
- 29.Isenberg DA, Rahman A, Allen E, Farewell V, Akil M, Bruce IN, D’Cruz D, Griffiths B, Khamashta M, Maddison P, McHugh N, Snaith M, Teh LS, Yee CS, Zoma A, Gordon C. BILAG 2004. Development and initial validation of an updated version of the British Isles Lupus Assessment Group’s disease activity index for patients with systemic lupus erythematosus. Rheumatology. 2005;44(7):902–6. doi: 10.1093/rheumatology/keh624. [DOI] [PubMed] [Google Scholar]
- 30.Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH and the committee on prognosis studies in SLE. . Derivation of the SLEDAI. A disease activity index for lupus patients. Arth Rheum. 1992;35:630–640. doi: 10.1002/art.1780350606. [DOI] [PubMed] [Google Scholar]
- 31.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arth Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 32.Banki K, Hutter E, Colombo E, Gonchoroff NJ, Perl A. Glutathione Levels and Sensitivity to Apoptosis Are Regulated by changes in Transaldolase expression. J Biol Chem. 1996;271:32994–33001. doi: 10.1074/jbc.271.51.32994. [DOI] [PubMed] [Google Scholar]
- 33.Banki K, Hutter E, Gonchoroff N, Perl A. Elevation of mitochondrial transmembrane potential and reactive oxygen intermediate levels are early events and occur independently from activation of caspases in Fas signaling. J Immunol. 1999;162:1466–1479. [PMC free article] [PubMed] [Google Scholar]
- 34.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 35.Wang K, Li J, Degterev A, Hsu E, Yuan J, Yuan C. Structure-activity relationship analysis of a novel necroptosis inhibitor, Necrostatin-5. Bioorg Med Chem Lett. 2007;17:1455–1465. doi: 10.1016/j.bmcl.2006.11.056. [DOI] [PubMed] [Google Scholar]
- 36.Zheng W, Degterev A, Hsu E, Yuan J, Yuan C. Structure-activity relationship study of a novel necroptosis inhibitor, necrostatin-7. Bioorg Med Chem Lett. 2008;18:4932–4935. doi: 10.1016/j.bmcl.2008.08.058. [DOI] [PubMed] [Google Scholar]
- 37.Xia J, Wishart DS. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat Protoc. 2011;6:743–760. doi: 10.1038/nprot.2011.319. [DOI] [PubMed] [Google Scholar]
- 38.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vanlangenakker N, Vanden Berghe T, Krysko DV, Festjens N, Vandenabeele P. Molecular mechanisms and pathophysiology of necrotic cell death. Current Molecular Medicine. 2008;8:207–220. doi: 10.2174/156652408784221306. [DOI] [PubMed] [Google Scholar]
- 40.Sherer Y, Gorstein A, Fritzler MJ, Shoenfeld Y. Autoantibody explosion in systemic lupus erythematosus: More than 100 different antibodies found in SLE patients. Semin Arth Rheum. 2004;34:501–537. doi: 10.1016/j.semarthrit.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 41.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide.[comment] Science. 2003;299:896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- 42.Perry DJ, Yin Y, Telarico T, Baker HV, Dozmorov I, Perl A, Morel L. Murine Lupus Susceptibility Locus Sle1c2 Mediates CD4+ T Cell Activation and Maps to Estrogen-Related Receptor gamma. J Immunol. 2012;189:793–803. doi: 10.4049/jimmunol.1200411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang B, Zhang X, Tang FL, Zhu LP, Liu Y, Lipsky PE. Clinical significance of increased CD4+CD25-Foxp3+ T cells in patients with new-onset systemic lupus erythematosus. Ann Rheum Dis. 2008;67:1037–1040. doi: 10.1136/ard.2007.083543. [DOI] [PubMed] [Google Scholar]
- 44.Yang HX, Zhang W, Zhao LD, Li Y, Zhang FC, Tang FL, He W, Zhang X. Are CD4+CD25-Foxp3+cells in untreated new-onset lupus patients regulatory T cells? Arth Res Ther. 2009:11. doi: 10.1186/ar2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Horwitz DA. Identity of mysterious CD4+CD25-Foxp3+ cells in SLE. Arth Res Ther. 2010;12:101. doi: 10.1186/ar2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lai Z, Telarico T, Bartos A, Miklossy G, Hanczko R, Jimah J, Clair B, Tily H, Francis L, Garcia R, Phillips PE, Ramos I, Perl A. Reversal of CD3/CD4/CD25/Foxp3 Treg Depletion in Active SLE Patients with Rapamycin. Suppl 10. 2010. p. 1196. [DOI] [Google Scholar]
- 47.Perl A, Hanczko R, Doherty E. Assessment of mitochondrial dysfunction in lymphocytes of patients with systemic lupus erythematosus. In: Perl A, editor. Meth Mol Med Autoimmunity: Methods and Protocols. Springer; Clifton, NJ: 2012. p. 61. [DOI] [PubMed] [Google Scholar]
- 48.Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci. 2007;32:37–43. doi: 10.1016/j.tibs.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 49.Cohen JJ, Duke RC, Fadok VA, Sellins KS. Apoptosis and programmed cell death in immunity. Ann Rev Immunol. 1992;10:267–293. doi: 10.1146/annurev.iy.10.040192.001411. [DOI] [PubMed] [Google Scholar]
- 50.Kaczmarek A, Vandenabeele P, Krysko D. Necroptosis: The Release of Damage-Associated Molecular Patterns and Its Physiological Relevance. Immunity. 2013;38:209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 51.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 52.Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, Duhadaway JB, Goossens V, Roelandt R, Van Hauwermeiren F, Libert C, Declercq W, Callewaert N, Prendergast GC, Degterev A, Yuan J, Vandenabeele P. Necrostatin-1 analogues: Critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012:3. doi: 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kovacs B, Vassilopoulos D, Vogelgesang SA, Tsokos GC. Defective CD3-mediated cell death in activated T cells from patients with systemic lupus erythematosus: role of decreased intracellular TNF-alpha. Clin Immunol Immunopathol. 1996;81:293–302. doi: 10.1006/clin.1996.0192. [DOI] [PubMed] [Google Scholar]
- 54.Carloni S, Buonocore G, Longini M, Proietti F, Balduini W. Inhibition of rapamycin-induced autophagy causes necrotic cell death associated with Bax/Bad mitochondrial translocation. Neuroscience. 2012;203:160–169. doi: 10.1016/j.neuroscience.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 55.Levitsky J, Mathew JM, Abecassis M, Tambur A, Leventhal J, Chandrasekaran D, Herrera N, Al-Saden P, Gallon Abdul-Nabi L, Yang GY, Kurian SM, Salomon DR, Miller J. Systemic immunoregulatory and proteogenomic effects of tacrolimus to sirolimus conversion in liver transplant recipients. Hepatology. 2013;57:239–248. doi: 10.1002/hep.25579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, Camirand G, Lin Y, Froicu M, Deng S, Shlomchik WD, Lakkis FG, Rothstein DM. Regulatory T cells require mammalian target of rapamycin signaling to maintain both homeostasis and alloantigen-driven proliferation in lymphocyte-replete mice. J Immunol. 2011;186:2809–2818. doi: 10.4049/jimmunol.0903805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wei J, Duramad O, Perng OA, Reiner SL, Liu YJ, Qin FXF. Antagonistic nature of T helper 1/2 developmental programs in opposing peripheral induction of Foxp3+ regulatory T cells. Proc Natl Acad Sci USA. 2007;104:18169–18174. doi: 10.1073/pnas.0703642104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cook KD, Miller J. TCR-Dependent Translational Control of GATA-3 Enhances Th2 Differentiation. J Immunol. 2010;185:3209–3216. doi: 10.4049/jimmunol.0902544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kessel A, Haj T, Peri R, Snir A, Melamed D, Sabo E, Toubi E. Human CD19+CD25high B regulatory cells suppress proliferation of CD4+ T cells and enhance Foxp3 and CTLA-4 expression in T-regulatory cells. Autoimmun Rev. 2012;11:670–677. doi: 10.1016/j.autrev.2011.11.018. [DOI] [PubMed] [Google Scholar]
- 60.Amu S, Tarkowski A, Dorner T, Bokarewa M, Brisslert M. The Human Immunomodulatory CD25+ B Cell Population belongs to the Memory B Cell Pool. Scand J Immunol. 2007;66:77–86. doi: 10.1111/j.1365-3083.2007.01946.x. [DOI] [PubMed] [Google Scholar]
- 61.Rajagopalan S, Zordan T, Tsokos GC, Datta SK. Pathogenic anti-DNA autoantibody-inducing T helper cell lines from patients with active lupus nephritis: isolation of CD4-8− T helper cell lines that express the gamma delta T-cell antigen receptor. Proc Natl Acad Sci USA. 1990;87:7020–7024. doi: 10.1073/pnas.87.18.7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sieling PA, Porcelli SA, Duong BT, Spada F, Bloom BR, Diamond B, Hahn BH. Human Double-Negative T Cells in Systemic Lupus Erythematosus Provide Help for IgG and Are Restricted by CD1c. J Immunol. 2000;165:5338–5344. doi: 10.4049/jimmunol.165.9.5338. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.