

Abstract
Articular cartilage is recalcitrant to endogenous repair and regeneration and thus a focus of tissue engineering and regenerative medicine strategies. A pre-requisite for articular cartilage tissue engineering is an understanding of the signal transduction pathways involved in mechanical compression during trauma or disease. We sought to explore the role of the extracellular signal-regulated kinase 1/2 (ERK 1/2) pathway in chondrocyte proliferation and proteoglycan synthesis following acute mechanical compression. Bovine articular cartilage explants were cultured with and without the ERK 1/2 pathway inhibitor PD98059. Cartilage explants were statically loaded to 40% strain at a strain rate of 1−sec for 5 seconds. Control explants were cultured under similar conditions but were not loaded. There were four experimental groups: 1) no load without inhibitor 2) no load with the inhibitor PD98059, 3) loaded without the inhibitor, and 4) loaded with the inhibitor PD98059. Explants were cultured for varying durations, from 5 minutes to 5 days. Explants were then analyzed by biochemical and immunohistochemical methods. Mechanical compression induced phosphorylation of ERK 1/2, and this was attenuated with the ERK 1/2 pathway inhibitor PD98059 in a dose-dependent manner. Chondrocyte proliferation was increased by mechanical compression. This effect was blocked by the inhibitor of the ERK 1/2 pathway. Mechanical compression also led to a decrease in proteoglycan synthesis that was reversed with inhibitor PD98059. In conclusion, the ERK 1/2 pathway is involved in the proliferative and biosynthetic response of chondrocytes following acute static mechanical compression.
Keywords: Articular cartilage, extracellular signal-regulated kinase 1/2 (ERK 1/2) pathway, chondrocyte proliferation, proteoglycan synthesis, mechanical compression, tissue engineering, regenerative medicine
Introduction
Osteoarthritis is the leading cause of arthritis in the United States, affecting an estimated 21 million people (Helmick et al., 2008). Traumatic joint injury is a significant cause of joint degeneration, leading to post-traumatic osteoarthritis, a significant cause of morbidity in an aging population. Numerous clinical studies relate acute joint injury and the subsequent development of post-traumatic arthritis (Gelber et al., 2000). Notably, individuals with a history of joint injury have a several fold increase in the development of hip and knee arthritis compared to individuals with no history of joint injury. Given this strong relationship, research over the past several decades has focused on the cartilage changes immediately following joint injury. Identified changes include the release and activation of proteases and cytokines (Cameron et al., 1997; Pickvance et al., 1993); increased collagen and proteoglycan degradation (Borrelli and Ricci, 2004; Chen et al., 2003; Patwari et al., 2003; Thibault et al., 2002); chondrocyte death by both necrosis and apoptosis (Borrelli et al., 2003; Chen et al., 2001; D'Lima et al., 2001a; Loening et al., 2000; Repo and Finlay, 1977); decreased proteoglycan synthesis (Jeffrey et al., 1997; Kurz et al., 2001); and lastly, chondrocyte proliferation (Mankin, 1962; Tew et al., 2001; Tew et al., 2000). Despite significant work in the area of cartilage injury, the molecular mechanisms underlying these biochemical changes remain poorly understood.
Studies investigating the molecular cascades promoting the above biochemical and structural changes in articular cartilage remain sparse. Recent studies have focused on the molecular pathways set into motion following physiologic compressive loads. Loads of this magnitude were found to induce activation of extracellular signal-regulated kinases 1/2 (ERK 1/2), p38 mitogen-activated protein kinase (p38), and c-Jun N-terminal kinase (JNK) (Fanning et al., 2003). These pathways are responsible for a large range of biologic responses. Additionally, studies on porcine articular cartilage support the activation of the ERK 1/2 pathway following mechanical injury (Vincent et al., 2004). The role of the ERK 1/2 pathway following injury is of interest in this study.
Tissue engineering is the science of design and manufacture of functional tissues, such articular cartilage, damaged or lost to trauma or disease (Reddi, 1998; Reddi, 2000). The three key ingredients for tissue engineering are inductive morphogens or signal transduction pathways that provide signaling cues, responding stem or progenitor cells, and a scaffolding such as the extracellular matrix (Reddi, 1998; Reddi, 2000). Our investigation sought to explore the activation of the ERK 1/2 pathway and its relationship to both the chondrocyte proliferative response and proteoglycan synthesis during injury. We hypothesized that injurious mechanical compression would lead to activation of ERK 1/2 via phosphorylation. Furthermore, we also hypothesized that injurious mechanical compression would lead to an increase in chondrocyte proliferation and a decrease in proteoglycan synthesis. Finally, we hypothesized that inhibition of the ERK 1/2 pathway would alter the increase in chondrocyte proliferation and the decrease in proteoglycan synthesis observed following injurious mechanical compression.
2 Materials and Methods
2.1. Materials
Dulbecco's Modified Eagle Medium: Nutrient Mixture F-12 (Ham) 1:1 (DMEM/F-12) and antibiotic solution (10,000 U/ml penicillin, 10 mg/ml streptomycin) were from Invitrogen (Carlsbad, CA). Bovine Serum Albumin (BSA), ascorbic acid 2-phosphate, inhibitor PD98059, protease inhibitor cocktail, and Proteinase K were from Sigma-Aldrich (St. Louis, MO). Protein Assay kit, protein standards, PVDF membranes, and semi-dry transblot system were from Bio-Rad Laboratories, Inc. (Richmond, CA). Monoclonal mouse antibodies for pERK were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Polyclonal rabbit antibodies for ERK were obtained from Cell Signaling Technology (Beverly, MA). Monoclonal mouse antibodies for proliferating cell nuclear antigen (PCNA) were obtained from Chemicon International (Temecula, CA). Cy™2-conjugated AffiniPure Goat Anti-mouse IgG was from Jackson ImmunoReseacrch Laboratories, Inc. (West Grove, PA). Mounting media for immunoflourescence was Prolong Gold anti-fade reagent with DAPI from Molecular Probes, Inc. (Eugene, OR). DNA was quantitated using PicoGreen® dsDNA Quantitation Kit from Molecular Probes (Eugene, OR). Radioactivity was determined in the liquid scintillation counter with a solution of Scintisafe Econo1 (Fisher, Hampton, NH).
2.2. Cartilage Explant Preparation
Cartilage explant preparations were prepared as previously described by our laboratory (Khalafi et al., 2007). Briefly, bovine stifle joints from 2-3 month old calves were obtained from a local slaughterhouse (Petaluma, CA). Full-thickness osteochondral plugs were removed from the weight-bearing surface of the distal femoral condyles utilizing a cylindrical coring punch 5 mm in diameter. Four osteochondral plugs were removed from each condyle (8 plugs per stifle joint). Osteochondral plugs were placed into a custom designed slicing device composed of aluminum and acrylic plexiglass, and sliced into superficial, middle, and deep layers. Each layer of the explant was 1.25 mm thick and 5 mm in diameter. The middle layer of the articular cartilage was utilized for most of the experiments, as it consisted of a homogenous population of cells, and was of uniform size and shape. Immediately following slicing, explants were equilibrated in DMEM/F12 culture media supplemented with 1% BSA, 20 μg/ml ascorbic acid, 10% FBS, 0.1% DMSO, and antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin), at 37°C and 5% CO2 for 48-72 hours.
2.3. Explant Loading
Mechanical compression of the cartilage explants was performed with an Instron 8511.2 digital servo-hydraulic mechanical testing device. Explants were first measured for exact thickness and diameter with a digital micro-caliper, as explants are known to swell slightly during the 48-72 hour equilibration period. This measurement allowed for precise positioning of the loading device at the start position. Also, these measurements permitted exact calculation of the stress applied to each loaded cartilage explant. The cartilage explants were then gently transferred from their equilibration wells to the loading wells with a flat, stainless steel spatula to minimize trauma to the cartilage specimens. Explants from the experimental group were placed, one at time, into the center of a custom-designed stainless steel well that had been filled with culture medium. The loading wells were designed to allow for radially unconfined mechanical compression of explants. The loading apparatus, measuring 2 cm in diameter, was fabricated with stainless steel to provide an effectively infinitely stiff surface for compression. A control explant was placed in a parallel, but non-communicating well, which had also been filled with identical medium. Explants were then statically loaded to 40% strain at a strain rate of 1−sec. The load was held for 5 seconds and then released. The stress generated at these loads was consistently ∼35 MPa. These loading parameters were chosen to provide a significant load to the articular cartilage discs (Ewers et al., 2002a; Ewers et al., 2001). Following compression, the macroscopic appearance of the cartilage explants was noted by visual inspection. All loaded explants were then transferred to fresh culture media and returned to an incubator at 37°C and 5% CO2 until the termination of the experiment. The elapsed time from initial removal from media to return to media following compression was less than 1 minute per explant.
2.4. Inhibitor Studies
PD98059, a selective MEK (MAPKK) activation inhibitor, was utilized for this study. PD98059 has been shown to prevent the activation of MEK by c-Raf or MEK kinase by blocking the MEK activation site (Alessi et al., 1995; Dudley et al., 1995). PD98059 was dissolved in DMSO and placed into culture media at two final concentrations, 50 μM and 200 μM. The maximum concentration of DMSO in any experiment was no more than 0.1%. Explants assigned to inhibitor groups were placed into PD98059 supplemented media immediately following cartilage plug extraction and remained under these conditions for the duration of study, including explant equilibration, loading, and post-loading incubation. Control explants were maintained in culture media containing 0.1% DMSO without PD98059.
2.5. Protein Extraction and Western Blots
At 0, 5, and 30 minutes and 2, 4, 18, 24, 48, and 72 hours after loading explants were flash frozen in liquid nitrogen and stored at −80°C. Four explants from each time point were combined in a cryo-preservation vial to provide sufficient cartilage for adequate protein extraction. Explants were placed into RIPA protein extraction buffer [50 mM sodium fluoride, 0.5% Igepal CA-630(NP-40), 10 mM sodium phosphate, 150 mM sodium chloride, 25 mM Tris pH 8.0, 1mM phenylmethylsulfonyl fluoride, 2 mM ethylenediaminetetraacetic acid, 1.2 mM sodium vanadate] supplemented with a protease inhibitor cocktail. A Polytron tissue homogenizer was then used to pulverize the cartilage explants. Homogenization was performed for 30 seconds. Homogenized samples were then frozen/thawed in liquid nitrogen for 3 cycles. This was followed by centrifugation at 14,000 × g for 10 minutes. Supernatant was removed and stored at −80°C until all samples had been collected. Extracted protein was then thawed and quantitated using Protein Assay kit. 12.5 μg of extracted protein was then combined with 15 μl of protein loading buffer and boiled for 5 minutes. Samples were run on 10% SDS-polyacrylamide gels for 1 hour. Gels were then transferred to PVDF membrane using a semi-dry transblot system for 35 minutes. Membranes were rinsed in ddH20 for 1 minute followed by 5% milk/TBST blocking for 2 hours. Following blocking, membranes were incubated with monoclonal mouse antibodies for pERK overnight at 4°C. After 3 washes in TBST and incubation with goat anti-mouse IgG-horseradish peroxidase conjugates for 1 hour at room temperature, membranes were then washed 3 times in TBST and exposed to ECL reagents for signal detection on X-ray films. Membranes were then stripped and incubated with polyclonal rabbit antibodies for ERK as a loading control.
2.6. Immunofluorescent Staining for Evaluation of Cellular Proliferation
At 0, 24, 48, and 72 hours following loading, explants were fixed in fresh 4% paraformaldehyde in phosphate buffer, pH 7.4, solution for 24 hours. Explants were then washed and placed into 70% ethanol. Tissues were embedded in paraffin, cut into 5 μm-thick sections, and mounted onto slides. Three explants were mounted in duplicate on each slide. Mounted specimens were incubated at 55°C for one hour to prevent detachment of sections from the slides during subsequent staining. Specimens were serially re-hydrated with decreasing concentrations of ethanol and ddH20. Antigen sites were unmasked by incubation in 0.1 M sodium citrate at 90° C for 10 minutes. Sections were subsequently incubated for 75 minutes with monoclonal mouse antibodies against proliferating cell nuclear antigen (PCNA) in normal horse blocking serum at a 1:50 dilution. Proliferating cell nuclear antigen is known to be up-regulated in proliferating cells (Garcia et al., 1989). Specimens were washed in PBS for 5 minutes 3 times. Sections were then incubated with Cy™2-conjugated AffiniPure Goat Anti-mouse IgG secondary antibody in normal horse blocking serum for 1 hour. Sections were again washed in PBS for 5 minutes, twice. Cover slips were then mounted using Prolong Gold anti-fade reagent with DAPI. Care was taken to keep specimens protected from light during the immunoflourescent steps and during slide storage. Immunoflourescent images were obtained using a Zeiss confocal microscope. Images were captured at 200× magnification. Two images were obtained for each specimen. First, images were obtained under UV light to visualize nuclear DAPI positive chondrocytes. Next, images were obtained under blue light to visualize PCNA positive cells in the same field, which appeared fluorescent green under these conditions. Images were analyzed using a computer grid to count number of positive cells per high power field. Results are expressed as number of PCNA positive cells as a percentage of DAPI positive cells.
2.7. Sulfate Incorporation for Evaluation of Proteoglycan Synthesis
At 24, 48, 72, 96, and 120 hours after loading, 4 cartilage explants specimens were incubated with 40 μCi of radioactive sodium sulfate (35SO4) for 4 hours. Following this incubation, the radioactive medium was removed and the explants were washed twice with culture medium and buffer containing non-radioactive sodium sulfate (10mM EDTA, 10mM sodium sulfate, 0.1M sodium phosphate, pH=6.5). Explants were then digested overnight in 0.833 mg/ml Proteinase K at 60 °C. After digestion, aliquots were taken for scintillation counting and DNA concentration. To determine radioactivity a 200μl aliquot was loaded onto a 2.0 ml polystyrene column (Pierce, Rockford, IL), containing 1.5 ml of Sephadex G-25 (Sigma), and 0.5 ml fractions were eluted with a solution of 4M Guanidine-HCl, 50mM Sodium Acetate, and 0.5% Triton-X, at a pH of 6.0. The first 3 fractions eluted were pooled and 9.0 ml of Scintisafe Econo1 (Fisher, Hampton, NH) was added. Previous work in our lab indicates the first 3 factions are the proteoglycan containing factions and derived of free sulfate(Luyten et al., 1992). The radioactivity was determined on a Beckman LS6500 liquid scintillation counter (Beckman, Fullerton, CA). DNA concentration was determined using a PicoGreen® dsDNA Quantitation Kit and scanned on a Storm® 860 (Amersham Biosciences, Uppsala, Sweden) fluorescence scanning system. The results of 35SO4 incorporation were expressed as CPM/μg DNA.
2.8. Statistical Analysis
Data for cell proliferation and sulfate incorporation were analyzed utilizing StatView version 5.0.1 (SAS Institute, Inc.). An ANOVA with Fisher's PLSD was performed with significance level set at 5%.
3. Results
3.1. Activation of the ERK 1/2 pathway by mechanical compression
Bovine articular cartilage explants were mechanically compressed to 40% strain, at a strain rate of 1sec- and this load was held for 5 seconds. All loaded explants were noted to have significant gross morphologic alternation, including surface fissuring, cracking, and flattening. This is consistent with previous studies utilizing similar loading parameters. Activation of the ERK 1/2 pathway via phosphorylation was evaluated with western blot using anti-phospho-ERK 1/2 antibodies. Initial experiments were conducted in superficial, middle, and deep layers of articular cartilage. Mechanical compression induced activation of pERK 1/2 in a time-dependent manner (Fig. 1). ERK 1/2 activation began at 30 minutes after loading, maintained from 30 minutes to 18 hours, and trailed off to control levels by 48 hours. This activation was not seen in control explants. Furthermore, this pattern of ERK 1/2 activation was seen in the different layers of articular cartilage, namely the superficial, middle, and deep layers. The superficial layer and middle layers demonstrated the strongest signal, with ERK 1/2 activation present up to 24 hours. All subsequent experiments were carried out utilizing the middle layer of cartilage only as it provided the uniform level surface for compression.
Fig. 1. Effect of mechanical compression on ERK 1/2 pathway activation.
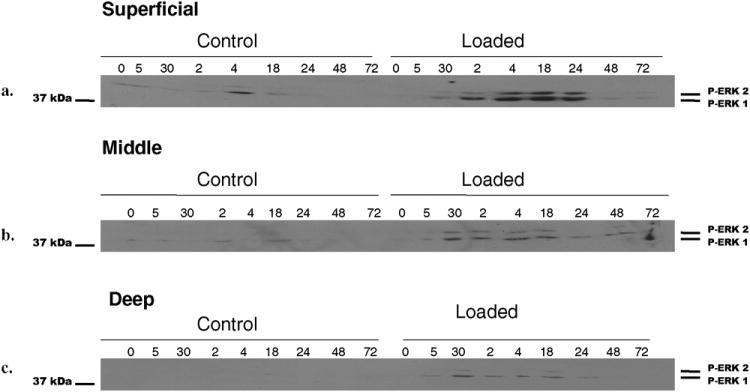
Cartilage explant discs from the superficial, middle, and deep zones were mechanically compressed to 40% strain based on the post-equilibration explant thickness following a 48-72 hour equilibration in culture medium. A strain rate of 1sec- was utilized. The load was held for 5 seconds. Compression was performed in the presence of culture medium. Control explants were placed in a parallel non-communicating well within the loading device. Following compression, explants were processed as described in Experimental Procedures and analyzed using standard Western blot technique for ERK 1/2 phosphorylation using a phospho-ERK 1/2 specific antibody (a-c). Each band represents four individually compressed and pooled explants.
3.2. Loading increased PCNA expression
Following injurious mechanical compression explants were evaluated immunohistochemically for cell proliferation. Proliferating cell nuclear antigen (PCNA) expression at 0, 24, 48, and 72 hours following loading was determined. Results were expressed as PCNA positive cells as a percentage of DAPI positive cells (Fig. 2a). With loading, a relative increase in PCNA expression was observed at 24, 48, and 72 hours (Fig. 2b). At 24 hours a 7% increase in chondrocyte proliferation was observed. At 48 hours a 10.8% increase in proliferation was observed, and at 72 hours a 10.5% increase in chondrocyte proliferation was observed. These differences were statistically significant above unloaded controls at all 3 time points (P < 0.05). These results suggest an increase in chondrocyte proliferation in cartilage explants following loading.
Fig. 2. Immunofluorescent staining.

5 μm-thick tissue sections were treated with monoclonal antibody for proliferating cell nuclear antigen (PCNA, 1:50), and subsequently incubated with Cy2-conjugated AffiniPure goat anti-mouse IgG secondary antibody (1:50) to allow for visualization of proliferating cells under blue light (a. right panel). Cover slips were then mounted using Prolong Gold anti-fade reagent with DAPI to allow for visualization of all cellular nuclei using UV light (a. left panel). Specimens were visualized at 200× magnification, and images were captured, saved, and then analyzed using a computer grid. Total cells (DAPI labeled as bright blue) were counted in control and loaded groups. PCNA positive cells (PCNA-Cy2 labeled as green) were then counted for the same field. Results were expressed as the percentage of PCNA positive cells per total DAPI positive cells (b). There were six explants for each experimental group at all the points. Note: The asterisk (*) represents statistical significance (P<0.05) compared to control. Error bars represent standard deviation.
3.3. Loading inhibited 35SO4 incorporation into proteoglycans
Explants were evaluated for sulfate incorporation into proteoglycans at 24, 48, 72, 96, and 120 hours following injurious mechanical compression. Following loading there was a decrease in sulfate incorporation at all time points (Fig. 3a). This decrease in sulfate incorporation was statistically significant at 24, 48, and 72 hours (P < 0.05); and the trend was continued at 96 and 120 hours. When sulfate incorporation in loaded explants was expressed as a percentage of sulfate incorporation in control explants (Fig. 3b) a relative decrease in sulfate incorporation ranging from 25% to 40% was observed. These data suggest compression of cartilage explants at injurious loads leads to a decrease in proteoglycan synthesis. These results are consistent with previous reports (Kurz et al., 2001).
Fig. 3. Sulfate incorporation following mechanical compression.
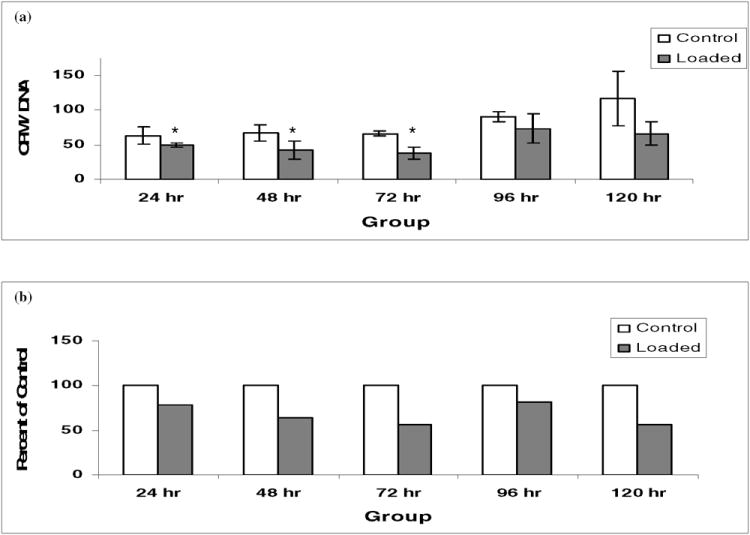
(a) Following loading, explants were treated and evaluated, as described in Experimental Procedures, at 24, 48, 72, 96, and 120 hours for biosynthetic activity. 35SO4 incorporation was utilized as a measure of proteoglycan production. Data presented are the average number of radioactive counts per minute (CPM) per microgram of DNA extracted from 2 pooled explants for n=3 experiments with 6 explants. ANOVA analysis revealed a significant difference existed between the groups (P < .0001). (b) Expressed as a percentage of 35SO4 incorporation in control explants, 35SO4 incorporation in loaded explants was decreased 25-40% versus controls. Note: The asterisk (*) represents statistical significance (P < 0.05) compared to control. Error bars represent standard deviation.
3.4. Inhibition of ERK 1/2 activation with PD98059
Bovine articular cartilage explants were cultured in media containing the MEK activation inhibitor PD98059. Explants were statically loaded to 40% strain, at a strain rate of 1sec-. This load was held for 5 seconds. Again, activation of the ERK 1/2 pathway via phosphorylation was evaluated with Western blot using anti-phospho-ERK 1/2 antibodies. As indicated previously, mechanical loading induced activation of ERK 1/2. With a concentration of PD98059 at 50 μM there was no inhibition of ERK 1/2 phosphorylation (Fig. 4a and b). However, a concentration of PD98059 at 200 μM effectively inhibited phosphorylation at nearly all time points (Fig. 4c). An example of loading control with ERK 1/2 is demonstrated (Fig. 4d). Given these results, all subsequent inhibitor experiments were conducted with a PD98059 concentration of 200 μM.
Fig. 4. Effect of ERK 1/2 pathway inhibitor PD98059 on ERK 1/2 activation following static mechanical compression.

Control and loaded explants were incubated with PD98059 at 50μm and 200μm final concentrations as described in Experimental Procedures. Immunoblot analysis with anti-phospho-ERK 1/2 of protein extracted from explants for both the control and loaded group at time points from 0-72 hours was performed for the no inhibitor (a), 50μm (b), and the 200μm (c) treated groups. Membranes were then stripped and probed with anti-ERK 1/2 antibody to verify that equivalent protein was present in each lane, for which one representative blot was shown (d).
3.5. Inhibition of ERK activation decreases PCNA expression
Explants were incubated with PD98059 at 200μM. Incubation with inhibitor occurred at all steps following explant preparation in an attempt to eliminate all possible ERK 1/2 activation. Immunohistochemical analysis for PCNA expression was performed at 0, 24, 48, and 72 hours after loading. Again, results were expressed as PCNA positive cells as a percentage of DAPI positive cells. With the addition of PD98059 to the culture media of the loaded specimens, PCNA expression did not differ significantly from unloaded controls at 0 and 24 hours. However, at the 48 and 72 hour time points a decrease in PCNA expression in the loaded, PD98059 supplemented specimens, was noted as compared to the amount of PCNA expression observed in the loaded specimens not supplemented with inhibitor (Fig. 5). At 48 hours the increase in PCNA expression following loading decreased from 10.8% in the uninhibited specimens to 3.8% in the PD98059 supplemented explants, and at 72 hours a decrease from 10.5% to 6.2% was observed. These differences were statistically significant at both time points (P < 0.05). These results suggest that with inhibition of ERK 1/2 activation, chondrocyte proliferation following cartilage compression is decreased, indicating a role of the ERK 1/2 pathway in the chondrocyte proliferative response.
Fig. 5. Effect of inhibition of ERK 1/2 activation on PCNA expression following mechanical compression.
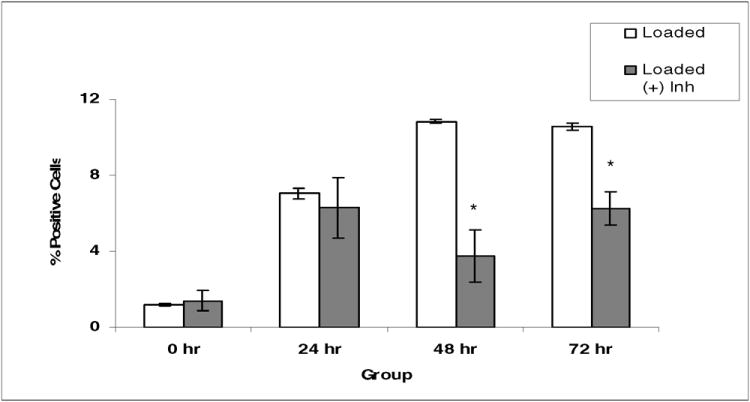
There were six explants per group. Explants were treated with PD98059, an ERK 1/2 pathway specific inhibitor, at a final concentration of 200μM. PCNA immunofluorescent staining was done as described in Fig. 2. Note: The asterisk (*) represents statistical significance compared to control. Error bars represent standard deviation.
3.6. Inhibition of ERK phosphorylation restores 35SO4 incorporation
Explants were incubated with PD98059 at 200μM. Explants were evaluated for sulfate incorporation at 24, 48, 96, and 120 hours following mechanical compression as previously described. With addition of PD98059 to the loaded explants, sulfate incorporation into proteoglycans was increased relative to the amount of sulfate incorporation observed in loaded, uninhibited explants (Fig. 6a). These differences were statistically significant at 24, 96, and 120 hours (P < 0.05). When results were expressed as loaded explants supplemented with inhibitor as a percentage of loaded explants without inhibitor (Fig. 6b), there was a 183% increase in sulfate incorporation at 24 hours, an 87% increase at 96 hours, and a 228% increase at 120 hours. These results suggest inhibition of ERK phosphorylation by PD98059 reduces the decrease in proteoglycan synthesis observed following injurious mechanical compression. These results affirm that the ERK 1/2 pathway may be a component of the proteoglycan biosynthetic response in articular cartilage following acute mechanical compression.
Fig. 6. Effect of inhibition of ERK 1/2 activation on sulfate incorporation following static mechanical compression.
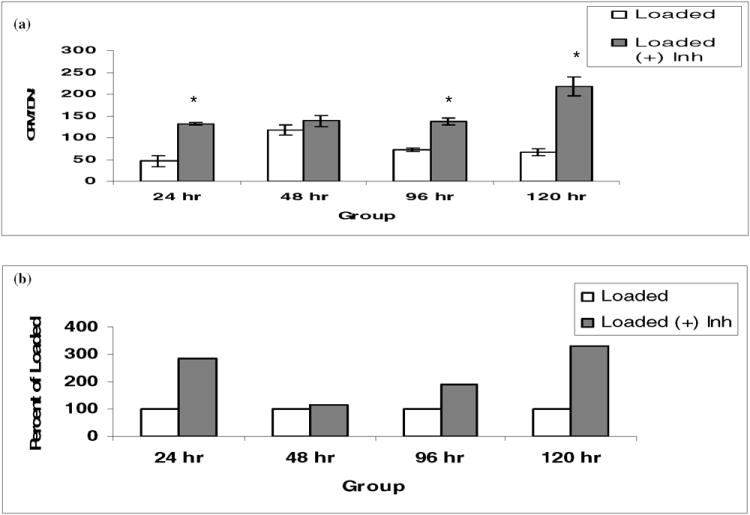
There were six explants per group. Explants were treated with PD98059, an ERK 1/2 pathway specific inhibitor, at a final concentration of 200μM, and 35SO4 incorporation assay was performed as described in Fig.3. Note: The asterisk (*) represents statistical significance compared to control. Error bars represent standard deviation.
Discussion
Articular cartilage responds to mechanical compression and injury through a number of homeostatic mechanisms. These cellular and matrix changes that occur following mechanical compression rely on activation of molecular cascades involving both latent and active proteins, which ultimately leads to genetic transcription and finally altered chondrocyte morphology and function. These changes can alter the normal cartilage architecture, resulting in abnormal wear characteristics, decreased structural integrity, and overall accelerated degeneration.
Significant work describing the structural, functional, and morphologic changes of both chondrocytes and their surrounding extracellular cartilage matrix has been accomplished. However, identification of the molecular mechanisms underlying these biochemical and structural changes remains elusive. Recent studies suggest the mitogen-activated protein (MAP) kinase pathways may have a role in articular cartilage homeostasis under physiologic mechanical loads. Fanning et al.(Fanning et al., 2003) reported activation of ERK 1/2, p38, and JNK under physiologic load conditions. These pathways are known to be activated in numerous cell types and by numerous modes of stimulation, including cytokines, growth factors, local chemical environmental changes, structural alterations, and mechanical stimulation (Johnson and Lapadat, 2002; Kolch, 2000; O'Neill and Kolch, 2004; Pouyssegur et al., 2002; Stork and Schmitt, 2002; Vincent et al., 2004; Watanabe et al., 2001). Activation under physiologic loads likely represents activation of processes responsible for cellular and matrix integrity needed under physiologic wear. However, activation of the ERK pathway generated under injurious loads represents a stress response. Recent studies by Vincent et al.(Vincent et al., 2002; Vincent et al., 2004) showed that acute injury to porcine articular cartilage resulted in activation of the ERK 1/2 pathway, and that this response was mediated by basic fibroblast growth factor (bFGF).
The ERK 1/2 pathway is among the mitogen-activated protein kinase (MAPK) cellular signaling pathways (Kolch, 2000). It exists in virtually all eukaryotic cells and controls fundamental cellular functions such as cell proliferation, differentiation, survival and apoptosis. Many different stimuli are known activators of this pathway, including growth factors, cytokines, carcinogens, and mechanical deformation (Johnson and Lapadat, 2002). This pathway is known to interact extensively with other MAPK pathways and its regulation and modulation is complex (O'Neill and Kolch, 2004; Pouyssegur et al., 2002; Stork and Schmitt, 2002; Watanabe et al., 2001). Investigation of the full breadth of stimuli, functions, and interactions of the ERK 1/2 pathway in articular cartilage homeostasis constitute ongoing studies in our laboratory.
The present investigation focused on chondrocyte proliferation and proteoglycan synthesis following acute mechanical compression. In this study, injurious loads of 40% strain, at a strain rate 1 −sec, were applied. The stress generated at these loads was consistently ∼35 MPa. The forces generated in our model most likely represent a scenario similar to that seen in an acute injurious event. In addition, previous work has indicated that these loading parameters significantly disrupt articular cartilage, yet do not generate conditions associated with fracture of the subchondral bone (Ewers et al., 2001; Ewers et al., 2002b).
To evaluate the role of the ERK 1/2 pathway, explants were incubated in the inhibitor PD98059 at all times following explantation. The vehicle dimethyl sulfoxide (DMSO) was utilized to dissolve this inhibitor and was also added to control specimens. Our data indicated that baseline chondrocyte proliferation and proteoglycan synthesis did not differ in the DMSO treated controls as compared to the non-DMSO treated controls utilized in the initial explants loading studies. PD98059 prevents the activation of MEK, the immediate upstream activator of ERK, by c-Raf or MEK kinase, thus effectively inhibiting the ERK 1/2 pathway (Alessi et al., 1995; Davies et al., 2000; Dudley et al., 1995). Previous investigators have utilized PD98059 final concentrations of 0.2 μM – 100 μM to evaluate the role of the ERK 1/2 pathway in chondrocytes in culture (Berenbaum et al., 2003; Bobick and Kulyk, 2004; Wang et al., 2004). To our knowledge, only one previous investigator has treated intact cartilage explants with PD98059 prior to tissue processing. However, this study failed to quantify the level of ERK 1/2 pathway inhibition (Pelletier et al., 2003). Our study is the first to measure ERK 1/2 activity following treatment of an intact cartilage explant with PD98059. Our goal was complete inhibition of the ERK 1/2 pathway as demonstrated by Western blot. This level of inhibition was deemed necessary to asses the role of the ERK pathway in light of the fact that it is known to interact with several other signaling cascades within the chondrocyte cell body. By minimizing the level of ERK activity, the effect of the ERK pathway on these additional pathways would be minimized. Many proteins, such as Ser/Thr kinases, protein Thr kinases, and PI-3 kinase, have been tested for activity in the presence of PD98059 at concentrations that provide near complete inhibition of ERK 1/2. Under these conditions, these protein kinases have been shown to have unchanged activity and are comparable to controls, despite the presence of PD98059 (Alessi et al., 1995). Therefore, experiments were conducted to determine the minimum concentration of PD98059 necessary to inhibit the phosphorylation of ERK 1/2. At a final concentration of 50 μM, PD98059 did not inhibit activation of ERK 1/2 following mechanical compression. However, at a concentration of 200 μM, near complete inhibition of the phosphorylation of ERK 1/2 was achieved. Presumably, the dense cartilage matrix surrounding the chondrocytes necessitated the utilization of a higher concentration of inhibitor than has been described by previous authors working with chondrocytes in culture.
With the addition of PD98059, the chondrocyte proliferative response following loading was significantly attenuated. This was observed at the 48 and 72 hours time points, thus supporting the role of the ERK 1/2 pathway in chondrocyte proliferation following an acute mechanical compressive load. Additionally, the decrease in proteoglycan synthesis seen following acute loading was effectively reversed with the addition of PD98059. This supports the hypothesis that the ERK 1/2 pathway is involved in the decrease in proteoglycan synthesis following acute mechanical compressive injury. Taken together, these results support the role of the ERK 1/2 pathway in the cellular changes which occur following injury.
Given the broad spectrum of responses with which the ERK 1/2 pathway has been demonstrated to be involved, including the up-regulation of MMP-13 by RUNX2 and FGF2(Wang et al., 2004) and the up-regulation of microsomal prostaglandin E synthase 1 by interleukin-1β (Masuko-Hongo et al., 2004), it is not surprising that our data support yet another role for the ERK 1/2 pathway in the cartilage stress response. In addition, the ERK 1/2 pathway has been demonstrated to promote de-differentiation and protect against apoptosis in response to nitric oxide stimulation, a substance know to promote articular cartilage damage (Kim et al., 2002). This role of the ERK 1/2 as a promoter of de-differentiation is consistent with our finding of decreased proteoglycan synthesis and increased proliferation in response to chondrocyte stress.
We did not investigate chondrocyte death by apoptosis or necrosis in response to injurious mechanical compression or the addition of the ERK 1/2 pathway inhibitor PD98059. Previous authors have demonstrated increased chondrocyte death by apoptosis following acute injurious mechanical compression (D'Lima et al., 2001b; D'Lima et al., 2001c; D'Lima et al., 2001d). Given this variable was kept constant in both PD98059 treated and untreated explants, we chose to explore relative changes in chondrocyte proliferation and proteoglycan synthesis. Our results indicate a relative decrease in the chondrocyte proliferative response following loading with addition of the ERK 1/2 pathway inhibitor, as well as relative reversal of decreased proteoglycan synthesis. Additionally, the role of ERK1/2 in cellular apoptosis is unclear. Previous investigators have shown that strong activation of ERK in CCL39 fibroblasts is necessary to protect cells from apoptosis, whereas moderate activation is required to permit apoptosis in response to stress (Le Gall et al., 2000; Pouyssegur et al., 2002; Zugasti et al., 2001). We are unaware of similar studies carried out in articular cartilage chondrocytes. Investigation into chondrocyte death by apoptosis which was augmented with addition of PD98059 would help explain the decrease in chondrocyte proliferation observed with addition of the ERK 1/2 pathway inhibitor, as there would be a relative decrease in the number of living cells available for mitosis. These would not be reflected in our total number of cells given the nuclear stain DAPI does not discriminate between living and dead cells to our knowledge. Investigation into this relationship would provide insight into the interpretation of our results. Further, investigation into the role of the ERK 1/2 pathway and its relationship to an apoptotic stress response would be of interest. However, an increase in chondrocyte apoptosis resulting from addition of ERK pathway inhibitor would not explain the relative increase in proteoglycan synthesis following loading observed after addition of PD98059. The decrease in number of cells capable of creating proteoglycans would be lower than those incubated without the ERK 1/2 pathway inhibitor rather than greater. Certainly the articular cartilage cellular stress response is a combination of complex regulatory mechanisms with which the ERK pathway is involved and further investigation into this complex pathway is needed.
While previous findings indicate the role of the ERK signaling pathway in chondrocyte differentiation and biosynthetic function (Bobick and Kulyk, 2004), our study extends these findings by showing evidence that ERK signaling pathway is a component of these additional processes. Given the broad spectrum of roles with which the ERK 1/2 pathway is involved; it is possible that it is associated with other cartilage stress responses, including the release and activation of catabolic cytokines and growth factors. Work by Gruber et al. (Gruber et al., 2004) has shown that explant slicing results in an immediate increase in IL-1, a known inflammatory cytokine. The cartilage stress response is a combination of complex regulatory mechanisms with which the ERK pathway is intimately involved.
The role of increased chondrocyte proliferation and decreased proteoglycan synthesis to the subsequent progression to post-traumatic arthritis is unknown. However, these alterations indicate a disruption of the normal cartilage homeostatic balance necessary for proper tissue maintenance. With traumatic joint injury as a significant cause of joint degeneration, our study, together with the others, highlights the importance of the ERK signaling pathway in the pathogenesis of early changes to articular cartilage following acute injury and may generate clues for selecting physiologically relevant signals towards tissue engineering of articular cartilage.
Acknowledgments
We thank Oscar Yeh, Ph.D., for his generous assistance with computer programming and mechanical testing, Maggie Chiu for her excellent assistance with histology, and Michelle Martin, M.D. and Lisa Mejia, M.D., for their assistance with manuscript editing. Dr. James A. Ryan was supported by a resident research fellowship from the Orthopaedic Research and Education Foundation. This study was in part supported by NIH grant (RO1 AR47345-02) and the Lawrence Ellison Endowed Chair to A. Hari Reddi.
This study was in part supported by NIH grant (1RO1-AR47345) and the Lawrence J. Ellison Endowed Chair to A. Hari Reddi.
References
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–94. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Berenbaum F, Humbert L, Bereziat G, Thirion S. Concomitant recruitment of ERK1/2 and p38 MAPK signalling pathway is required for activation of cytoplasmic phospholipase A2 via ATP in articular chondrocytes. J Biol Chem. 2003;278:13680–7. doi: 10.1074/jbc.M211570200. [DOI] [PubMed] [Google Scholar]
- Bobick BE, Kulyk WM. The MEK-ERK signaling pathway is a negative regulator of cartilage-specific gene expression in embryonic limb mesenchyme. J Biol Chem. 2004;279:4588–95. doi: 10.1074/jbc.M309805200. [DOI] [PubMed] [Google Scholar]
- Borrelli J, Jr, Ricci WM. Acute effects of cartilage impact. Clin Orthop. 2004:33–9. doi: 10.1097/01.blo.0000132627.13539.02. [DOI] [PubMed] [Google Scholar]
- Borrelli J, Jr, Tinsley K, Ricci WM, Burns M, Karl IE, Hotchkiss R. Induction of chondrocyte apoptosis following impact load. J Orthop Trauma. 2003;17:635–41. doi: 10.1097/00005131-200310000-00006. [DOI] [PubMed] [Google Scholar]
- Cameron M, Buchgraber A, Passler H, Vogt M, Thonar E, Fu F, Evans CH. The natural history of the anterior cruciate ligament-deficient knee. Changes in synovial fluid cytokine and keratan sulfate concentrations. Am J Sports Med. 1997;25:751–4. doi: 10.1177/036354659702500605. [DOI] [PubMed] [Google Scholar]
- Chen CT, Bhargava M, Lin PM, Torzilli PA. Time, stress, and location dependent chondrocyte death and collagen damage in cyclically loaded articular cartilage. J Orthop Res. 2003;21:888–98. doi: 10.1016/S0736-0266(03)00050-0. [DOI] [PubMed] [Google Scholar]
- Chen CT, Burton-Wurster N, Borden C, Hueffer K, Bloom SE, Lust G. Chondrocyte necrosis and apoptosis in impact damaged articular cartilage. J Orthop Res. 2001;19:703–11. doi: 10.1016/S0736-0266(00)00066-8. [DOI] [PubMed] [Google Scholar]
- D'Lima D, Hashimoto S, Chen P, Colwell C, Lotz M. Cartilage injury: impact of mechanical trauma on matrix and cells. Clinical Orthopaedics and Related Research. 2001a;391S:S90–S99. doi: 10.1097/00003086-200110001-00009. [DOI] [PubMed] [Google Scholar]
- D'Lima DD, Hashimoto S, Chen PC, Colwell CW, Jr, Lotz MK. Human chondrocyte apoptosis in response to mechanical injury. Osteoarthritis Cartilage. 2001b;9:712–9. doi: 10.1053/joca.2001.0468. [DOI] [PubMed] [Google Scholar]
- D'Lima DD, Hashimoto S, Chen PC, Colwell CW, Jr, Lotz MK. Impact of mechanical trauma on matrix and cells. Clin Orthop Relat Res. 2001c:S90–9. doi: 10.1097/00003086-200110001-00009. [DOI] [PubMed] [Google Scholar]
- D'Lima DD, Hashimoto S, Chen PC, Lotz MK, Colwell CW., Jr Cartilage injury induces chondrocyte apoptosis. J Bone Joint Surg Am. 2001d;83-A(2):19–21. doi: 10.2106/00004623-200100021-00004. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci U S A. 1995;92:7686–9. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewers B, Weaver B, ET S, Haut R. Chronic changes in rabbit retro-patellar cartilage and subchondral bone after blunt impact loading of the patellofemoral joint. Journal of Orthopaedic Research. 2002a;20:545–550. doi: 10.1016/S0736-0266(01)00135-8. [DOI] [PubMed] [Google Scholar]
- Ewers BJ, Dvoracek-Driksna D, Orth MW, Haut RC. The extent of matrix damage and chondrocyte death in mechanically traumatized articular cartilage explants depends on rate of loading. J Orthop Res. 2001;19:779–84. doi: 10.1016/S0736-0266(01)00006-7. [DOI] [PubMed] [Google Scholar]
- Ewers BJ, Jayaraman VM, Banglmaier RF, Haut RC. Rate of blunt impact loading affects changes in retropatellar cartilage and underlying bone in the rabbit patella. J Biomech. 2002b;35:747–55. doi: 10.1016/s0021-9290(02)00019-2. [DOI] [PubMed] [Google Scholar]
- Fanning PJ, Emkey G, Smith RJ, Grodzinsky AJ, Szasz N, Trippel SB. Mechanical regulation of mitogen-activated protein kinase signaling in articular cartilage. J Biol Chem. 2003;278:50940–8. doi: 10.1074/jbc.M305107200. [DOI] [PubMed] [Google Scholar]
- Garcia RL, Coltrera MD, Gown AM. Analysis of proliferative grade using anti-PCNA/cyclin monoclonal antibodies in fixed, embedded tissues. Comparison with flow cytometric analysis. Am J Pathol. 1989;134:733–9. [PMC free article] [PubMed] [Google Scholar]
- Gelber AC, Hochberg MC, Mead LA, Wang NY, Wigley FM, Klag MJ. Joint injury in young adults and risk for subsequent knee and hip osteoarthritis. Ann Intern Med. 2000;133:321–8. doi: 10.7326/0003-4819-133-5-200009050-00007. [DOI] [PubMed] [Google Scholar]
- Gruber J, Vincent TL, Hermansson M, Bolton M, Wait R, Saklatvala J. Induction of interleukin-1 in articular cartilage by explantation and cutting. Arthritis Rheum. 2004;50:2539–46. doi: 10.1002/art.20369. [DOI] [PubMed] [Google Scholar]
- Helmick CG, Felson DT, Lawrence RC, Gabriel S, Hirsch R, Kwoh CK, Liang MH, Kremers HM, Mayes MD, Merkel PA, Pillemer SR, Reveille JD, Stone JH. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I. Arthritis Rheum. 2008;58:15–25. doi: 10.1002/art.23177. [DOI] [PubMed] [Google Scholar]
- Jeffrey JE, Thomson LA, Aspden RM. Matrix loss and synthesis following a single impact load on articular cartilage in vitro. Biochim Biophys Acta. 1997;1334:223–32. doi: 10.1016/s0304-4165(96)00097-9. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–2. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Khalafi A, Schmid TM, Neu C, Reddi AH. Increased accumulation of superficial zone protein (SZP) in articular cartilage in response to bone morphogenetic protein-7 and growth factors. J Orthop Res. 2007;25:293–303. doi: 10.1002/jor.20329. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Ju JW, Oh CD, Yoon YM, Song WK, Kim JH, Yoo YJ, Bang OS, Kang SS, Chun JS. ERK-1/2 and p38 kinase oppositely regulate nitric oxide-induced apoptosis of chondrocytes in association with p53, caspase-3, and differentiation status. J Biol Chem. 2002;277:1332–9. doi: 10.1074/jbc.M107231200. [DOI] [PubMed] [Google Scholar]
- Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J. 2000;351(Pt 2):289–305. [PMC free article] [PubMed] [Google Scholar]
- Kurz B, Jin M, Patwari P, Cheng DM, Lark MW, Grodzinsky AJ. Biosynthetic response and mechanical properties of articular cartilage after injurious compression. J Orthop Res. 2001;19:1140–6. doi: 10.1016/S0736-0266(01)00033-X. [DOI] [PubMed] [Google Scholar]
- Le Gall M, Chambard JC, Breittmayer JP, Grall D, Pouyssegur J, Van Obberghen-Schilling E. The p42/p44 MAP kinase pathway prevents apoptosis induced by anchorage and serum removal. Mol Biol Cell. 2000;11:1103–12. doi: 10.1091/mbc.11.3.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loening AM, James IE, Levenston ME, Badger AM, Frank EH, Kurz B, Nuttall ME, Hung HH, Blake SM, Grodzinsky AJ, Lark MW. Injurious mechanical compression of bovine articular cartilage induces chondrocyte apoptosis. Arch Biochem Biophys. 2000;381:205–12. doi: 10.1006/abbi.2000.1988. [DOI] [PubMed] [Google Scholar]
- Luyten FP, Yu YM, Yanagishita M, Vukicevic S, Hammonds RG, Reddi AH. Natural bovine osteogenin and recombinant human bone morphogenetic protein-2B are equipotent in the maintenance of proteoglycans in bovine articular cartilage explant cultures. J Biol Chem. 1992;267:3691–5. [PubMed] [Google Scholar]
- Mankin H. Localization of tritiated thymidine in articular cartilage of rabbits. J Bone Joint Surg Am. 1962;44:688–698. [Google Scholar]
- Masuko-Hongo K, Berenbaum F, Humbert L, Salvat C, Goldring MB, Thirion S. Up-regulation of microsomal prostaglandin E synthase 1 in osteoarthritic human cartilage: critical roles of the ERK-1/2 and p38 signaling pathways. Arthritis Rheum. 2004;50:2829–38. doi: 10.1002/art.20437. [DOI] [PubMed] [Google Scholar]
- O'Neill E, Kolch W. Conferring specificity on the ubiquitous Raf/MEK signalling pathway. Br J Cancer. 2004;90:283–8. doi: 10.1038/sj.bjc.6601488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwari P, Cook MN, DiMicco MA, Blake SM, James IE, Kumar S, Cole AA, Lark MW, Grodzinsky AJ. Proteoglycan degradation after injurious compression of bovine and human articular cartilage in vitro: interaction with exogenous cytokines. Arthritis Rheum. 2003;48:1292–301. doi: 10.1002/art.10892. [DOI] [PubMed] [Google Scholar]
- Pelletier JP, Fernandes JC, Brunet J, Moldovan F, Schrier D, Flory C, Martel-Pelletier J. In vivo selective inhibition of mitogen-activated protein kinase kinase 1/2 in rabbit experimental osteoarthritis is associated with a reduction in the development of structural changes. Arthritis Rheum. 2003;48:1582–93. doi: 10.1002/art.11014. [DOI] [PubMed] [Google Scholar]
- Pickvance EA, Oegema TR, Jr, Thompson RC., Jr Immunolocalization of selected cytokines and proteases in canine articular cartilage after transarticular loading. J Orthop Res. 1993;11:313–23. doi: 10.1002/jor.1100110302. [DOI] [PubMed] [Google Scholar]
- Pouyssegur J, Volmat V, Lenormand P. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Biochem Pharmacol. 2002;64:755–63. doi: 10.1016/s0006-2952(02)01135-8. [DOI] [PubMed] [Google Scholar]
- Reddi AH. Role of morphogenetic proteins in skeletal tissue engineering and regeneration. Nat Biotechnol. 1998;16:247–52. doi: 10.1038/nbt0398-247. [DOI] [PubMed] [Google Scholar]
- Reddi AH. Morphogenesis and tissue engineering of bone and cartilage: inductive signals, stem cells, and biomimetic biomaterials. Tissue Eng. 2000;6:351–9. doi: 10.1089/107632700418074. [DOI] [PubMed] [Google Scholar]
- Repo RU, Finlay JB. Survival of articular cartilage after controlled impact. J Bone Joint Surg Am. 1977;59:1068–76. [PubMed] [Google Scholar]
- Stork PJ, Schmitt JM. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002;12:258–66. doi: 10.1016/s0962-8924(02)02294-8. [DOI] [PubMed] [Google Scholar]
- Tew S, Redman S, Kwan A, Walker E, Khan I, Dowthwaite G, Thomson B, Archer CW. Differences in repair responses between immature and mature cartilage. Clin Orthop. 2001:S142–52. doi: 10.1097/00003086-200110001-00014. [DOI] [PubMed] [Google Scholar]
- Tew SR, Kwan AP, Hann A, Thomson BM, Archer CW. The reactions of articular cartilage to experimental wounding: role of apoptosis. Arthritis Rheum. 2000;43:215–25. doi: 10.1002/1529-0131(200001)43:1<215::AID-ANR26>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Thibault M, Poole AR, Buschmann MD. Cyclic compression of cartilage/bone explants in vitro leads to physical weakening, mechanical breakdown of collagen and release of matrix fragments. J Orthop Res. 2002;20:1265–73. doi: 10.1016/S0736-0266(02)00070-0. [DOI] [PubMed] [Google Scholar]
- Vincent T, Hermansson M, Bolton M, Wait R, Saklatvala J. Basic FGF mediates an immediate response of articular cartilage to mechanical injury. Proc Natl Acad Sci U S A. 2002;99:8259–64. doi: 10.1073/pnas.122033199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent TL, Hermansson MA, Hansen UN, Amis AA, Saklatvala J. Basic fibroblast growth factor mediates transduction of mechanical signals when articular cartilage is loaded. Arthritis Rheum. 2004;50:526–33. doi: 10.1002/art.20047. [DOI] [PubMed] [Google Scholar]
- Wang X, Manner PA, Horner A, Shum L, Tuan RS, Nuckolls GH. Regulation of MMP-13 expression by RUNX2 and FGF2 in osteoarthritic cartilage. Osteoarthritis Cartilage. 2004;12:963–73. doi: 10.1016/j.joca.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Watanabe H, de Caestecker MP, Yamada Y. Transcriptional cross-talk between Smad, ERK1/2, and p38 mitogen-activated protein kinase pathways regulates transforming growth factor-beta-induced aggrecan gene expression in chondrogenic ATDC5 cells. J Biol Chem. 2001;276:14466–73. doi: 10.1074/jbc.M005724200. [DOI] [PubMed] [Google Scholar]
- Zugasti O, Rul W, Roux P, Peyssonnaux C, Eychene A, Franke TF, Fort P, Hibner U. Raf-MEK-Erk cascade in anoikis is controlled by Rac1 and Cdc42 via Akt. Mol Cell Biol. 2001;21:6706–17. doi: 10.1128/MCB.21.19.6706-6717.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]