

Abstract
Designers of clinical trials for Alzheimer's disease (AD) and mild cognitive impairment (MCI) are actively considering structural and functional neuroimaging, cerebrospinal fluid and genetic biomarkers to reduce the sample sizes needed to detect therapeutic effects. Genetic pre-selection, however, has been limited to Apolipoprotein E (ApoE). Recently discovered polymorphisms in the CLU, CR1 and PICALM genes are also moderate risk factors for AD; each affects lifetime AD risk by ~ 10–20%. Here, we tested the hypothesis that pre-selecting subjects based on these variants along with ApoE genotype would further boost clinical trial power, relative to considering ApoE alone, using an MRI-derived 2-year atrophy rate as our outcome measure. We ranked subjects from the Alzheimer's Disease Neuroimaging Initiative (ADNI) based on their cumulative risk from these four genes. We obtained sample size estimates in cohorts enriched in subjects with greater aggregate genetic risk. Enriching for additional genetic biomarkers reduced the required sample sizes by up to 50%, for MCI trials. Thus, AD drug trial enrichment with multiple genotypes may have potential implications for the timeliness, cost, and power of trials.
Keywords: Alzheimer's disease, Neuroimaging, Brain atrophy, Genetics, Genetic risk score, Clinical trial enrichment
Highlights
-
•
ApoE genotype status helps enrich MCI trials, using a structural MRI outcome measure.
-
•
CLU, PICALM and CR1 risk genes boost potential MCI trial power beyond ApoE alone.
-
•
CLU, PICALM and CR1 show significant, aggregate effects on TBM maps of brain atrophy.
1. Introduction
Dementia is a devastating disease, with costs exceeding those of cancer and heart disease (Hurd et al., 2013). There is a crucial need for Alzheimer's disease treatments that go beyond merely alleviating symptoms of this devastating disease. Powerful clinical trial designs are vital to novel drug development. In recent years, multiple approaches using neuroimaging, biochemical and genetic biomarkers have been proposed for enrichment of AD trials, i.e., reducing the number of subjects needed to detect potential drug effects. By enriching a sample in subjects who are more likely to cognitively decline, there is a greater change for the treatments to resist, and an expected gain in power. Structural MRI measures, for instance, are useful not only for measuring brain atrophy (Frisoni et al., 2010; Hua et al., 2011; Leow et al., 2006; McEvoy et al., 2010) but also for predicting brain atrophy, which can be used to pre-select subjects who will decline in a clinical trial (Lorenzi et al., 2010; McEvoy et al., 2010). PET scanning with FDG or PiB, cerebrospinal fluid (CSF) measures of Abeta42, tau and phosphorylated tau, as well as genotyping for the Apolipoprotein E (ApoE) ε4 allele (ApoE4) have also been proposed to pre-select people for clinical trials, reducing sample size requirements (Lorenzi et al., 2010; McEvoy et al., 2010). Multivariate methods can also combine multiple biomarkers to select subjects more likely to show detectable change in hypothetical AD trials (Kohannim et al., 2010).
ApoE is the greatest known genetic risk factor for late-onset AD (Bertram et al., 2007) and ApoE genotyping has already been studied for pre-selecting individuals for potential AD trial enrichment (Beckett et al., 2010; Kohannim et al., 2010; McEvoy et al., 2010; Nestor et al., 2008; Risacher et al., 2010). Carriers of at least one copy of the ApoE4 variant make up ~ 20% of the normal elderly population (e.g., Crivello et al., 2010), and each E4 allele carried roughly triples a person's lifetime risk of developing AD. Even so, ApoE does not account for all of the inherited risk for AD. As other risk genes are discovered and validated, genotyping should even further reduce sample size requirements. Two recent genome-wide association (GWA) studies recently implicated several highly prevalent genetic variants in the clusterin (CLU), phosphatidylinositol binding clathrin assembly protein (PICALM) and the complement component receptor 1 (CR1) genes, beyond the well-known ApoE gene; carriers of risk-conferring variants in any of these genes have an additional 10–20% increased risk for Alzheimer's disease, if other risk factors are equal (CLU and CR1; Lambert et al., 2009, CLU and PICALM; Harold et al., 2009). Since then, several studies, including meta-analyses, have replicated these findings in large cohorts of healthy controls and pathologically confirmed, late-onset AD patients (Carrasquillo et al., 2010; Corneveaux et al., 2010; Jun et al., 2010). Most recently, Ferrari et al. (2012) also verified the associations of these genetic variants with AD by sequencing the respective genes' coding regions. Though the precise role of these specific variants remains unknown, there are several links between the protein products of CLU, PICALM, and CR1 and AD pathogenesis: CLU with beta-amyloid aggregation, deposition, and clearance, PICALM with synaptic function, clathrin-mediated endocytosis, and amyloid precursor protein recycling, and CR1 with inflammation and complement-mediated beta-amyloid clearance (Holton et al., 2013; Jun et al., 2010; Sleegers et al., 2010).
Here, we assessed how the 3 recently discovered AD risk genes (i.e., PICALM, CLU and CR1), along with ApoE, combine to affect brain tissue loss rates in Alzheimer's Disease Neuroimaging Initiative (ADNI) subjects. We hypothesized that a genetic score derived from these top AD risk genes would enrich a hypothetical 2-year clinical trial beyond just selecting ApoE4 carriers. We focused on MCI and healthy controls, similarly to prior enrichment studies (Lorenzi et al., 2010; McEvoy et al., 2010), mainly due to the crucial need for early interventions in AD (Kozauer and Katz, 2013).
2. Methods
2.1. Subjects
Subjects considered for this study were participants in the first phase of the ADNI project, also known as “ADNI1” (Weiner et al., 2012). ADNI was launched as a public–private partnership in 2004 by the NIH, private pharmaceutical companies, and non-profit organizations, The goal of ADNI is to determine biological markers of Alzheimer's disease through neuroimaging, genetics, neuropsychological tests and other measures, to develop and monitor new therapies, and reduce the time of clinical trials. Subjects were recruited from 58 sites across North America. The study was conducted according to the Good Clinical Practice guidelines, the Declaration of Helsinki, and U.S. 21 CFR Part 50—Protection of Human Subjects, and Part 56—Institutional Review Boards. Written informed consent was obtained from all participants before protocols were performed. All data acquired as part of this study are publicly available (http://adni.loni.ucla.edu/).
All ADNI subjects underwent thorough clinical and cognitive assessment at the time of scan acquisition to establish diagnosis. In this study, all subjects with both baseline 1.5-Tesla scans and longitudinal follow-up scans at 24 months were assessed. The 24-month follow-up was picked, as 2 years is a fairly typical duration for an MCI or AD clinical trial. All subjects with available 24-month scans and genotypes were included. The mini-mental state exam (MMSE) was administered to provide a global measure of mental status (Cockrell and Folstein, 1988). The Clinical Dementia Rating (CDR) was used to assess dementia severity (Morris, 1993). Healthy volunteer status was determined if a subject had MMSE scores between 24 and 30 (inclusive), a CDR of 0, and was non-depressed, non-MCI, and non-demented. Mild cognitive impairment (MCI) diagnosis was determined if a subject had MMSE scores between 24 and 30 (inclusive), a memory complaint, objective memory loss measured by education adjusted scores on the Wechsler Memory Scale Logical Memory II, a CDR of 0.5, absence of significant levels of impairment in other cognitive domains, essentially preserved activities of daily living, and an absence of dementia. AD was diagnosed based on the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association (NINCDS–ADRDA) criteria for probable AD (McKhann et al., 1984), MMSE scores between 20 and 26 (inclusive), and CDR of 0.5 or 1.0. Definitive autopsy-based diagnosis of AD was not possible.
Here, we studied 500 ADNI subjects (mean age of 75.5 ± 6.5 SD; 296 males and 204 females; see Table 1 for full demographics) with available genotypes and scans at baseline and 24 months after diagnosis. All subjects are Caucasian, identified by self-report, and confirmed with multi-dimensional scaling analysis (Stein et al., 2010).
Table 1.
Number of subjects, distribution of sex and age (mean ± SD years), number of carriers (C) and non-carriers (NC) for the ApoE4 risk allele, and distribution of alleles for CLU (rs11136000), PICALM (rs3851179) and CR1 (rs3818361) AD risk-conferring polymorphisms are displayed for subjects with Alzheimer's disease (AD), mild cognitive impairment (MCI), healthy controls (CTL), and also for all 500 ADNI subjects combined. These 500 subjects were studied, as they had available 24-month as well as baseline MRI scans. Minor allele frequencies (MAFs) and Hardy–Weinberg Equilibrium (HWE) p-values for the candidate variants are also shown in the last column. The risk allele for CR1 is the minor allele (i.e., T), but the risk alleles for CLU and PICALM are the major alleles (i.e., C and G, respectively).
| AD | MCI | CTL | All | MAF/HWE p | |
|---|---|---|---|---|---|
| N | 106 | 234 | 160 | 500 | |
| Age | 75.5 ± 7.4 | 74.9 ± 6.9 | 76.3 ± 4.9 | 75.5 ± 6.5 | |
| Sex | 50 F | 79 F | 75 F | 204 M | |
| 56 M | 155 M | 85 M | 296 F | ||
| ApoE4 | 73 C | 128 C | 45 C | 246 C | |
| 33 NC | 106 NC | 115 NC | 254 NC | ||
| CLU | 36 C/C | 95 C/C | 61 C/C | 192 C/C | 0.38/0.84 |
| 54 C/T | 104 C/T | 76 C/T | 234 C/T | ||
| 16 T/T | 35 T/T | 23 T/T | 74 T/T | ||
| PICALM | 44 G/G | 102 G/G | 58 G/G | 204 G/G | 0.36/0.69 |
| 50 G/A | 102 G/A | 82 G/A | 234 G/A | ||
| 12 A/A | 30 A/A | 20 A/A | 62 A/A | ||
| CR1 | 64 C/C | 153 C/C | 119 C/C | 336 C/C | 0.18/0.76 |
| 38 C/T | 72 C/T | 39 C/T | 149 C/T | ||
| 4 T/T | 9 T/T | 2 T/T | 15 T/T |
The allele frequency distributions between AD, MCI, and CTL did not show any statistically significant differences in CLU (χ2 = 1.55, p = 0.82), PICALM (χ2 = 2.60, p = 0.63), or CR1 (χ2 = 7.43, p = 0.11).
2.2. Imaging
MRI scans of the brain were obtained from ADNI subjects with a standard 1.5 T protocol. A sagittal 3D MP-RAGE sequence was used, that had been optimized for consistency across sites (Jack et al., 2008; Leow et al., 2006; TR/TE = 2400/1000 ms; flip angle = 8°; FOV = 24 cm; final reconstructed voxel resolution = 0.9375 × 0.9375 × 1.2 mm3). Scans at 12 and 24 months were linearly registered to baseline scans and aligned to a template after standard corrections, which include Gradwarp (Jovicich et al., 2006), B1-correction (Jack et al., 2008), bias field correction (Sled et al., 1998), and phantom-based geometrical scaling (Gunter et al., 2006). Masks excluding non-brain tissues were obtained using a robust brain extraction tool (ROBEX; Iglesias et al., 2011). A bias-free tensor-based morphometry analysis (TBM; Hua et al., 2013; Leow et al., 2006) was used based on a non-linear inverse consistent elastic registration that treats the baseline and follow-up scans identically, and symmetrically (Leow et al., 2005). In voxelwise analysis, the volumetric change over time (over a 2-year follow-up) was regressed against the joint effect of genetic variants at each voxel, using multiple linear regression. Correction for multiple spatial comparisons was performed with a regional false discovery rate (FDR) method (Langers et al., 2007). In power analyses, a 2-year atrophy rate was computed from a statistically defined region-of-interest to summarize temporal lobe atrophy (Hua et al., 2011).
2.3. Genetic risk score calculation
The ADNI genotyping procedures are thoroughly described in Saykin et al. (2010). For this study, a genetic risk score was calculated from previously identified and replicated top SNPs in CLU (rs11136000), PICALM (rs3851179) and CR1 (rs3818361) using a linear model of aggregate disease risk, with weights for each variant equal to the logarithm of odds ratios reported in a recent meta-analysis by Jun et al. (2010), which is in agreement with odds ratios reported in other studies (Harold et al., 2009; Lambert et al., 2009). Odds ratios for the minor alleles of CLU, PICALM, and CR1 were therefore set to 0.9, 0.9 and 1.1, respectively. The odds ratio for carrying ApoE4 was set to 3. This is an underestimation as odds ratios are higher in ApoE e4/e4 subjects (Farrer et al., 1997) who are also included here, but any larger value would yield the same ranking of subjects as the effect of ApoE4 overrides that of all other candidate SNPs (in other words the score assigned to ApoE4 is so large that the exact value of the score does not affect the eventual ranking). The score calculation and ranking procedure is further detailed in the Supplementary material and Inline Supplementary Table S1.
Inline Supplementary Table S1.
Table S1.
| ApoE (carrier status) |
PICALM (rs3851179) |
CLU (rs11136000) |
CR1 (rs3818361) |
Genotype risk score |
|---|---|---|---|---|
| 1 | 0 | 0 | 1 | 1.19 |
| 1 | 0 | 1 | 2 | 1.18 |
| 1 | 1 | 0 | 2 | 1.18 |
| 1 | 0 | 0 | 0 | 1.10 |
| 1 | 1 | 0 | 1 | 1.09 |
| 1 | 0 | 1 | 1 | 1.09 |
| 1 | 1 | 1 | 2 | 1.08 |
| 1 | 0 | 2 | 2 | 1.08 |
| 1 | 1 | 0 | 0 | 0.99 |
| 1 | 0 | 1 | 0 | 0.99 |
| 1 | 1 | 1 | 1 | 0.98 |
| 1 | 2 | 0 | 1 | 0.98 |
| 1 | 0 | 2 | 1 | 0.98 |
| 1 | 1 | 1 | 0 | 0.89 |
| 1 | 2 | 0 | 0 | 0.89 |
| 1 | 0 | 2 | 0 | 0.89 |
| 1 | 1 | 2 | 1 | 0.88 |
| 1 | 1 | 2 | 0 | 0.78 |
| 1 | 2 | 1 | 0 | 0.78 |
| 1 | 2 | 2 | 1 | 0.77 |
| 1 | 2 | 2 | 0 | 0.68 |
| 0 | 0 | 0 | 2 | 0.19 |
| 0 | 0 | 0 | 1 | 0.10 |
| 0 | 1 | 0 | 2 | 0.09 |
| 0 | 0 | 0 | 0 | 0.00 |
| 0 | 1 | 0 | 1 | − 0.01 |
| 0 | 0 | 1 | 1 | − 0.01 |
| 0 | 1 | 1 | 2 | − 0.02 |
| 0 | 2 | 0 | 2 | − 0.02 |
| 0 | 1 | 0 | 0 | − 0.11 |
| 0 | 0 | 1 | 0 | − 0.11 |
| 0 | 1 | 1 | 1 | − 0.12 |
| 0 | 2 | 0 | 1 | − 0.12 |
| 0 | 0 | 2 | 1 | − 0.12 |
| 0 | 1 | 2 | 2 | − 0.13 |
| 0 | 1 | 1 | 0 | − 0.21 |
| 0 | 2 | 0 | 0 | − 0.21 |
| 0 | 0 | 2 | 0 | − 0.21 |
| 0 | 1 | 2 | 1 | − 0.22 |
| 0 | 2 | 1 | 1 | − 0.22 |
| 0 | 1 | 2 | 0 | − 0.32 |
| 0 | 2 | 1 | 0 | − 0.32 |
| 0 | 2 | 2 | 1 | − 0.33 |
| 0 | 2 | 2 | 0 | − 0.42 |
The ADNI genotyping procedures are thoroughly described in Saykin et al. (2010). For this study, a genetic risk score was calculated from previously identified and replicated top SNPs in CLU (rs11136000), PICALM (rs3851179) and CR1 (rs3818361) using a linear model of aggregate disease risk, with weights for each variant equal to the logarithm of odds ratios reported in a recent meta-analysis by Jun et al. (2010), which is in agreement with odds ratios reported in other studies (Harold et al., 2009; Lambert et al., 2009). Odds ratios for the minor alleles of CLU, PICALM, and CR1 were therefore set to 0.9, 0.9 and 1.1, respectively. The odds ratio for carrying ApoE4 was set to 3. This is an underestimation as odds ratios are higher in ApoE e4/e4 subjects (Farrer et al., 1997) who are also included here, but any larger value would yield the same ranking of subjects as the effect of ApoE4 overrides that of all other candidate SNPs (in other words the score assigned to ApoE4 is so large that the exact value of the score does not affect the eventual ranking). The score calculation and ranking procedure is further detailed in the Supplementary material and Inline Supplementary Table S1.
Inline Supplementary Table S1 can be found online at http://dx.doi.org/10.1016/j.nicl.2013.05.007.
In voxelwise analysis, multiple linear regression was used to predict volumetric differences at each voxel based on candidate genotypes, after adjusting for covariates (sex, age, and population structure parameters derived from multi-dimensional scaling analysis (MDS) as detailed in Hibar et al., 2011; Stein et al., 2010). This multiple regression analysis was more stable than regressing the same aggregate risk score against the signal at each voxel, as the variants' odds ratios from case–control studies may not necessarily correspond to the appropriate weights for predicting volume differences at each voxel.
2.4. Power analysis
As in prior studies (Hua et al., 2011; Kohannim et al., 2010), sample size estimates were calculated according to the following formula:
Here σ and β denote the mean and standard deviation of the atrophy rates respectively, α is set to be nominal 0.05, and the desired power is 80%. Subjects were ranked according to their composite genetic score to determine how much the sample size estimates would be reduced if the study was restricted to those with higher-risk scores. The ranking procedure was randomized 1000 times to compare observed power results with those obtainable by chance, i.e., when just selecting subsets of people from the overall sample without regard to their genotypes.
3. Results
We first studied the sample size enrichment effect using genotype scores for a mixed group of MCI and cognitively healthy control subjects, in the proportions they were enrolled in ADNI (i.e., roughly 2 MCI subjects for each healthy control subject). We ranked the 394 subjects by their cumulative risk score based on ApoE4 carrier status and their CLU, CR1 and PICALM genotypes. We related our risk ranking to the sample size for a potential clinical trial needed to detect a 25% reduction in atrophy rates within two years with 80% power. The temporal lobe atrophy rate we used for our sample size calculations was significantly correlated with 2-year change in sum-of-boxes Clinical Dementia Rating (CDR-SOB) scores (R2 = 0.25, p < 2 × 10− 16; see Inline Supplementary Fig. S1 for details). We observed boosting of trial power by selecting those at greater genetic risk (Fig. 1). Sample size estimates for a trial evaluating temporal lobe atrophy were reduced from 142 to 94 by selecting ApoE4 carriers, and to even smaller estimates in subjects with more risk alleles (69 when selecting only the top ~ 15% of the subjects, and 60 when selecting only the top ~ 10% of the subjects who had the greatest aggregate risk). These cutoffs correspond to risk scores of 1.08 and 1.10, respectively (see Inline Supplementary Table S1). We ensured this was not due to chance through 1000 permutations: all subjects were ranked randomly and sample size estimates were computed for random subsets (Fig. 1). When we coded ApoE as 0, 1 or 2 for the number of ε4 alleles (instead of 0 for carrying no copies of ε4 and 1 for carrying any number of copies of ε4), boosting was still observed in a similar way (Inline Supplementary Fig. S2).
Inline Supplementary Figure S1.
Fig. S1.
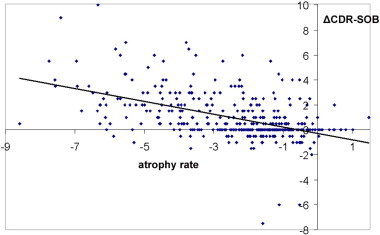
Two-year atrophy rates based on a statistically defined region-of-interest in the temporal lobes of a mixture of MCI and cognitively normal (N = 394) are negatively correlated with the 2-year change in sum-of-boxes Clinical Dementia Rating scores (ΔCDR-SOB) with R2 = 0.25; p < 2 × 10− 16. The scores are defined as the sum of memory, problem solving, orientation, community affairs, home and hobbies, and personal care. Negative values for atrophy rates reflect stronger atrophy, which correlate with change toward higher CDR-SOB scores (i.e., clinically worsening dementia). The correlation significance was unchanged after covarying for age and sex.
Fig. 1.
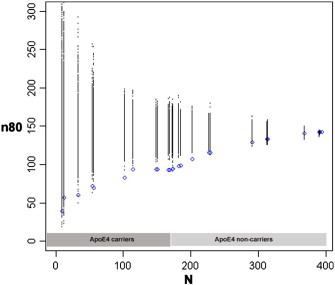
Sample size estimates (n80) are shown for a hypothetical clinical trial with a mixture of healthy control and mild cognitive impairment (MCI) subjects (in the proportions enrolled by ADNI), as a function of number of subjects (N) after ranking subjects according to a cumulative genetic risk score derived from ApoE4 (coded as 0 or 1 for carrier status) and single nucleotide polymorphisms in CLU, CR1 and PICALM (blue). The breakpoints represent different risk score values, as these are not continuous (see Inline Supplementary Table S1 for a list of risk scores and corresponding allele combinations). Permutations are performed by randomizing the ranking procedure 1000 times and calculating n80 estimates for each N (all permuted estimates are shown in black). ApoE4 reduces n80s from 142 to 94, based on data from a subset of 173 subjects (44% of all MCI and controls). Using the cumulative genetic risk score, this is further reduced to 69 when pre-selecting the top ~ 15%, and 60 when pre-selecting the top ~ 10% of the subjects.
Inline Supplementary Figure S2.
Fig. S2.
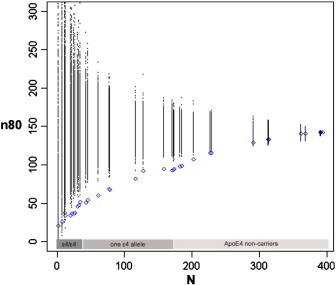
As in Fig. 1, sample size estimates (n80) are shown for a hypothetical clinical trial with a mixture of healthy control and mild cognitive impairment (MCI) subjects, as a function of number of subjects (N) after ranking subjects according to a cumulative genetic risk score derived from ApoE4 and single nucleotide polymorphisms in CLU, CR1 and PICALM (blue). Here, ApoE is coded as 0, 1 or 2 corresponding to the number of ε4 alleles. Permutations are performed by randomizing the ranking procedure 1000 times and calculating n80 estimates for each N (all permuted estimates are shown in black). Selection of ApoE ε4/ε4 subjects (a subset of 35 subjects) reduces n80s from 142 to 52. Although the number of subjects is limited, selecting fractions of ApoE ε4/ε4 individuals with higher cumulative genetic risk scores helps further reduce n80s to ~ 35.
We first studied the sample size enrichment effect using genotype scores for a mixed group of MCI and cognitively healthy control subjects, in the proportions they were enrolled in ADNI (i.e., roughly 2 MCI subjects for each healthy control subject). We ranked the 394 subjects by their cumulative risk score based on ApoE4 carrier status and their CLU, CR1 and PICALM genotypes. We related our risk ranking to the sample size for a potential clinical trial needed to detect a 25% reduction in atrophy rates within two years with 80% power. The temporal lobe atrophy rate we used for our sample size calculations was significantly correlated with 2-year change in sum-of-boxes Clinical Dementia Rating (CDR-SOB) scores (R2 = 0.25, p < 2 × 10− 16; see Inline Supplementary Fig. S1 for details). We observed boosting of trial power by selecting those at greater genetic risk (Fig. 1). Sample size estimates for a trial evaluating temporal lobe atrophy were reduced from 142 to 94 by selecting ApoE4 carriers, and to even smaller estimates in subjects with more risk alleles (69 when selecting only the top ~ 15% of the subjects, and 60 when selecting only the top ~ 10% of the subjects who had the greatest aggregate risk). These cutoffs correspond to risk scores of 1.08 and 1.10, respectively (see Inline Supplementary Table S1). We ensured this was not due to chance through 1000 permutations: all subjects were ranked randomly and sample size estimates were computed for random subsets (Fig. 1). When we coded ApoE as 0, 1 or 2 for the number of ε4 alleles (instead of 0 for carrying no copies of ε4 and 1 for carrying any number of copies of ε4), boosting was still observed in a similar way (Inline Supplementary Fig. S2).
Inline Supplementary Figs. S1 and S2 can be found online at http://dx.doi.org/10.1016/j.nicl.2013.05.007.
Next, we tested whether the boosting of trial power also applies to MCI subjects and cognitively healthy controls, when considered separately. Reduced sample sizes were observed in MCI subjects and were consistently lower than permuted estimates (Fig. 2A). In carriers of at least one copy of the risk allele, ApoE4, the sample size estimate was reduced to 67 (from 105 when considering all MCI subjects). Selecting the top ~ 20% and ~ 10% of high-risk MCI subjects yielded estimates of 58 and 50, respectively. These cut-offs correspond to the same risk score thresholds as those considered above, which led to reduced estimates of 69 and 60. In cognitively healthy subjects, a consistent pattern of reduced sample sizes from higher genetic risk scores was not observed (Fig. 2B). Selection of ApoE4 carriers reduced the sample size estimate only from 120 to 117, which was not significant. Selecting the top ~ 10% of control subjects with highest genetic risk reduces this further to 55, but this reduction still fell short of significance when compared to permutations (p ~ 0.11).
Fig. 2.
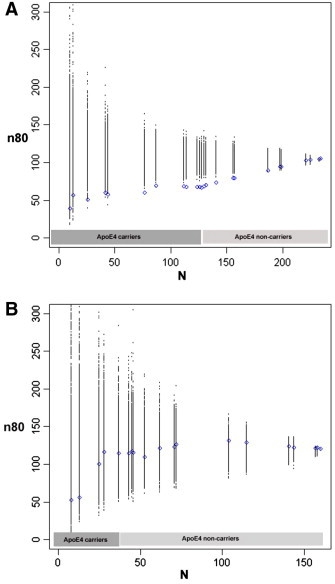
Sample size estimates (n80) are shown for hypothetical clinical trials as a function of number of subjects (N) after ranking subjects according to a cumulative genetic risk score derived from ApoE4 (coded as 0 or 1 for carrier status) and single nucleotide polymorphisms in CLU, CR1 and PICALM (blue). The breakpoints represent different risk score values, as these are not continuous (see Inline Supplementary Table S1 for a list of risk scores and corresponding allele combinations). Permutations are performed by randomizing the ranking procedure 1000 times and calculating n80 estimates for each N (all permuted estimates are shown in black). Panel A shows results for MCI subjects. Selection of subjects who carry at least one copy of the risk allele, ApoE4 (~ 55% of all MCI subjects), reduces n80s from 105 to 67. This is further reduced from the remaining three risk variants to 58 in the top ~ 20% and to 50 in the top ~ 10% of MCI subjects, respectively. Permutations consistently provide worse estimates. Panel B displays similar results for cognitively healthy subjects only. Selecting ApoE4 carriers (28% of all controls) reduces n80 minimally from 120 to 117. Selecting the top ~ 10% of control subjects with highest genetic risk reduces n80 to 55, still falling short of significance when compared to random permutations (p ~ 0.11).
We then sought to statistically explore how much CLU, PICALM, and CR1 contribute to the reduction in sample size, independently of ApoE. We performed 1000 permutations as above, but instead started with ApoE carriers and permuted within this subgroup only to see if further n80 reductions from the aggregate genotype risk score were statistically significant. In the mixture of MCI and cognitively normal ApoE carriers, boosting of n80 from 94 to 69 by selecting the subjects with risk scores above 1.08 (top ~ 15% in above analysis) was statistically significant (p = 0.035). Selecting those with scores above 1.10 (top ~ 10% above), which reduced n80 from 94 to 60 was suggestive, but did not reach significance (p = 0.070). When we considered MCI ApoE carriers alone, neither reductions from 67 to 58 and 50 were statistically significant (p = 0.20, and p = 0.14, respectively).
Although we were mostly interested in MCI and healthy control subjects due to the importance of early intervention in clinical trials, we also investigated a similar enrichment strategy within the whole cohort of 500 subjects, including those with AD. Similar results were obtained with the inclusion of AD subjects. Selecting the subset of 246 ApoE carriers reduced the sample size from 135 to 84. The sample sizes were further reduced to 65 and 59 by selecting subsets of subjects with high genetic risk scores with the same two score breakpoints used above. Permutations confirmed statistical significance. Boosting in the full cohort – and corresponding permutation results – are shown in Inline Supplementary Fig. S3.
Inline Supplementary Figure S3.
Fig. S3.
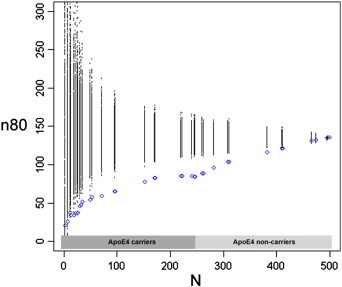
Similarly to Fig. 1, sample size estimates (n80) are displayed for a hypothetical clinical trial. Here, AD subjects are included in addition to the MCI and healthy control subjects (total N = 500). As in Fig. 1, ApoE is coded as 0 or 1, reflecting a subject's carrier status. Subjects are ranked based on their aggregate genetic risk score derived from ApoE, CLU, PICALM, and CR1 (shown in blue). Permutations are performed by randomizing the ranking procedure 1000 times and calculating n80 estimates for each N (all permuted estimates are shown in black). ApoE4 reduces n80s from 135 to 84. The cumulative genetic risk score further reduces n80 significantly to 65 and 59, using the 1.10 and 1.08 breakpoints, respectively (as done in the MCI and control groups).
Although we were mostly interested in MCI and healthy control subjects due to the importance of early intervention in clinical trials, we also investigated a similar enrichment strategy within the whole cohort of 500 subjects, including those with AD. Similar results were obtained with the inclusion of AD subjects. Selecting the subset of 246 ApoE carriers reduced the sample size from 135 to 84. The sample sizes were further reduced to 65 and 59 by selecting subsets of subjects with high genetic risk scores with the same two score breakpoints used above. Permutations confirmed statistical significance. Boosting in the full cohort – and corresponding permutation results – are shown in Inline Supplementary Fig. S3.
Inline Supplementary Fig. S3 can be found online at http://dx.doi.org/10.1016/j.nicl.2013.05.007.
When regressed against the 24-month atrophy rate based on the temporal lobe statistical ROI in all 500 ADNI subjects, our 4-gene risk score was statistically significant in the expected direction (β = − 1.43; p < 2 × 10− 16) after adjusting for age, sex, and population structure. When ApoE was not included in the score, but included as a covariate instead, the 3-gene risk score association was still in the expected direction, but no longer statistically significant (β = − 1.17; p = 0.11). In a post-hoc analysis, we assessed whether there were regions in our voxelwise atrophy maps where the 3-gene association was statistically significant. The cumulative effect of CLU, CR1 and PICALM SNPs was regressed against voxelwise measures of volumetric change over 2 years in maps of the temporal lobes. As explained in the Methods section, we used multiple linear regression with the 3 SNPs as predictors here, instead of a score based on the case–control odds ratios, which do not necessarily reflect the effect of each SNP on structural brain atrophy at each voxel. After adjusting for ApoE status and other covariates (i.e., sex, age and population structure) and correcting for multiple comparisons across voxels, we found statistically significant spatial effects in the temporal lobes for 500 subjects with the rate of volumetric brain change 24 months after the first scan, mostly in the entorhinal cortex and hippocampus (Fig. 3). In a separate analysis, we also added diagnostic status (AD, MCI, or control) as another covariate, and obtained similar, in fact stronger, results (Fig. 4).
Fig. 3.
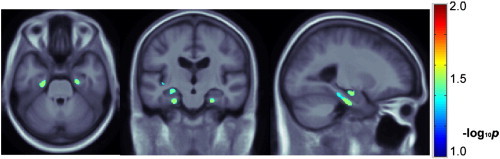
Single nucleotide polymorphisms in CLU, PICALM and CR1 are jointly regressed against voxelwise, 3D maps of 24-month temporal lobe atrophy in 500 ADNI subjects with available scans. Sex, age, ApoE status and population structure are adjusted for in each regression. Multiple comparisons across voxels are corrected with a regional FDR method; only voxels that survive this statistical correction are shown in color. Representative axial and sagittal slices are shown, which show the statistically significant additive, bilateral and rather symmetric effects of the variants in the hippocampus and entorhinal cortex. Warmer colors represent more significant effects. Images are in radiological convention. A total of 15,711 voxels survive the p < 0.05 threshold with a pmin of 1.04 × 10− 4. The regional (searchlight) FDR technique renders 3130 voxels significant (i.e., with corrected p-values below 0.05).
Fig. 4.
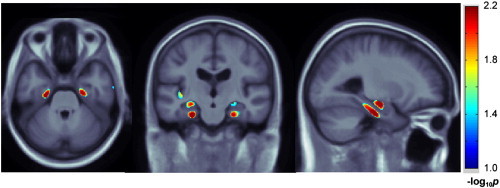
Single nucleotide polymorphisms in CLU, PICALM and CR1 are jointly regressed against voxelwise, 3D maps of 24-month temporal lobe atrophy in 500 ADNI subjects with available scans. Diagnostic status (i.e., either AD, MCI, or control) in addition to sex, age, ApoE and population structure is adjusted for in each regression. Multiple comparisons across voxels are corrected with a regional FDR method; only voxels that survive this statistical correction are shown in color. Representative axial and sagittal slices are shown, as in Fig. 3, indicating the statistically significant additive effects of the variants in the hippocampus and entorhinal cortex. Warmer colors represent ‘more significant’ effects. Images are in radiological convention. A total of 23,202 voxels survive the p < 0.05 threshold with a pmin of 2.51 × 10− 5. A subtotal of 6114 voxels survive the corrected p-value threshold of 0.05 with the regional (searchlight) FDR technique.
4. Discussion
In this study, we showed that an aggregate genotype risk score derived from variants in the CLU, PICALM and CR1 risk genes for Alzheimer's disease can be used to enrich a hypothetical clinical trial through genetic profiling. Prior studies show successful enrichment of AD and MCI trials with ApoE4 genotyping (Kohannim et al., 2010; McEvoy et al., 2010) and ApoE's significant effect on brain atrophy, independently of clinical diagnosis (Potkin et al., 2009). To our knowledge, no previous study has investigated whether we can further reduce sample sizes by including more risk variants to select the sample.
We first showed that selecting subjects who carry at least one copy of ApoE4 reduced the sample size required to detect an 80% atrophy rate in 2 years by about 34% in a group of MCI and healthy control subjects and 36% in MCI subjects only. This reduction is consistent with McEvoy et al.'s (2010) reported 10–43% reduction in sample size requirements when using ApoE4 to pre-select a sample. Enrichment was not explored for cognitively healthy controls in the McEvoy study, but we did not find a significant sample size reduction for ApoE carriers in this group. This is not surprising, as average atrophy maps show a low-grade and diffuse pattern of atrophy in cognitively healthy controls, whereas effects are stronger and more concentrated in regions such as the temporal lobes in AD and MCI subjects. Prior studies have uncovered significant differences in structural and functional imaging measures such as cortical thickness (Burggren et al., 2008) and beta amyloid (Morris et al., 2010) in cognitively healthy ApoE carriers, but the effects are either absent in classic AD brain areas such as the hippocampus, or they appear to be diffuse across several brain regions. Numerical summaries designed to capture localized atrophy in the classic AD brain areas (trained by AD subject scans) may, therefore, not work well to capture atrophy in healthy aging subjects (Hua et al., 2013).
In our study, selecting subjects who, in addition to carrying ApoE4, carried high-risk combinations of the CLU, CR1 and PICALM polymorphisms led to an additional reduction of 51–58% in sample size in a mixed group of MCI and controls and 45–52% in MCI subjects only. These were statistically significant when compared to a randomized subject selection procedure, though when considered strictly independently from ApoE, statistical significance was limited to the mixed group of MCI and controls with the 51% reduction. In cognitively healthy controls, a 56% reduction was observed through selecting the top 10% of high-risk subjects, but did not reach significance. A simple explanation may be that we need larger sample sizes for the healthy controls to demonstrate significant boosting of trial power, whereas we have fewer control subjects than MCI in our study. However, the discrepancy between the findings in the two diagnostic subgroups may also suggest that clinical trials might benefit from either including MCI subjects only, or enrolling a mixture of MCI and healthy controls (i.e., enrollment regardless of clinical diagnosis) who instead have higher genetic risk scores. Enrolling MCI subjects for AD trials is not without its limitations, as criteria for MCI may lead to missing AD converters or including controls (Visser et al., 2005). Nonetheless, this very limitation may be alleviated by further excluding subjects with lower genetic risk. In ADNI, as in most other neuroimaging studies, autopsy confirmation of AD diagnosis was not possible, and at least in principle, CSF biomarkers (for tau protein, etc.) could have been used to corroborate the clinical diagnosis, which was made according to the NINCDS–ADRDA criteria. CSF measures of Tau, P-Tau, and A-beta are available for only a subset of the ADNI subjects who have longitudinal data, so we did not use them here to further inform the diagnosis. Also, it must be conceded that some MCI participants might harbor AD pathology and might be considered as having AD in its initial stages.
We further reported regionally significant aggregate effects of CLU, PICALM and CR1 variants on TBM-derived three-dimensional maps of temporal lobe atrophy. The use of a cumulative genetic risk score has substantial practical consequences, beyond the standard practice of focusing on ApoE as the dominant risk gene for late-onset AD. The 3 more recently discovered AD risk genes have been previously associated with quantitative, neuroimaging summary measures (reviewed in Braskie et al., 2011a) in addition to their high degree of replication as AD risk genes in case–control studies (Carrasquillo et al., 2010; Ferrari et al., 2012; Harold et al., 2009; Jun et al., 2010; Lambert et al., 2009). Significant association of PICALM with MRI-derived hippocampal volume and entorhinal cortex thickness, and that of CR1 on entorhinal cortex thickness have been recently reported (Biffi et al., 2010). Another study found an association of CR1 with entorhinal cortex volume in young, healthy adults (Bralten et al., 2011). Although the association of our 3-SNP risk score with the average atrophy rate across the temporal lobe fell short of significance, our results provide a voxelwise complement to these studies, as we found spatially significant effects in temporal lobe regions that are in agreement with their findings. The rs11136000 variant in CLU, though not significant in Biffi et al.'s study, has been associated with microstructural white matter integrity using diffusion tensor imaging in young, healthy adults (Braskie et al., 2011b). Although the ADNI cohort was included in the Jun et al. (2010) meta-analysis, the three SNPs have been discovered and replicated in multiple, large cohorts that do not overlap with ADNI and there is, therefore, no circularity in our genetic results; in other words, the SNPs chosen were based on their association with AD in many other samples, and not because they were associated with brain atrophy in the ADNI dataset specifically.
The outcome measure we implemented here for performing our power analyses was based on numerical summaries of temporal lobe atrophy derived from tensor-based morphometry of MRI scans (Leow et al., 2006). Neuroimaging measures can boost power for clinical trials (Leung et al., 2010; McEvoy et al., 2010; Nestor et al., 2008; Risacher et al., 2010). As detailed in Hua et al. (2009), we used a statistically defined region-of-interest in the temporal lobes, which can be a more powerful outcome measure for tracking atrophy than a standard voxelwise average. This region-of-interest was obtained from a training set of 20 AD subjects. Since our enrichment analyses focused on MCI and control subjects, overfitting due to training and testing with overlapping subjects was avoided. Despite the use of a sophisticated imaging-based outcome measure, we demonstrate its correlation with clinical dementia scores, and expect that our findings with respect to sample size reduction using multiple AD risk genes should generalize to trials with purely clinical outcome measures as well, as these are top SNPs from case–control genome-wide association studies (Harold et al., 2009; Lambert et al., 2009).
A genotype-based enrichment approach may offer several potential advantages for AD drug trials. As discussed in Schork and Topol (2010), trials for both prevention and therapy of disease may benefit from such enrichment. For prevention, subjects with high-risk genotypes (e.g., ApoE4 carriers with multiple CLU, PICALM and CR1 risk alleles in our study) may be more likely to respond to a preventive agent, thereby making trials more cost-effective and efficient. The presence of subjects in a trial who are unlikely to decline at all makes it difficult to pick up drug effects. Though our multilocus enrichment approach is related to that of other AD enrichment studies, where neuroimaging and cerebrospinal fluid biomarkers are shown to help select at-risk subjects, it is particularly helpful as it may be less costly and can be obtained from subjects quickly and efficiently, and potentially avoid issues such as patients' willingness to undergo MRI or a lumbar puncture. Although several of the sample size reductions we obtained from our candidate genes were not significant after adjustment for ApoE alone, the general risk score approach, perhaps fine-tuned with additional genes and larger numbers of subjects, has the potential to offer such advantages.
A genotype profiling enrichment strategy is also promising for therapeutic trials, as risk genes often reflect molecular mechanisms of disease and, arguably, may affect subjects' degree of responsiveness to specific therapeutics. The protein products of CLU, CR1, and PICALM are linked to inflammatory, amyloid, lipid and chaperone pathways that are implicated in neurodegeneration (Sleegers et al., 2010). Genetic profiling based on these candidate genes may help with AD trial enrichment. This overall approach is related to pharmacogenetic sampling (i.e., selecting genetic subgroups of patients who are more likely to respond to treatment; Schork and Topol, 2010). However, the main premise is to avoid sampling people who will not decline during the trial, and preferentially sample those who will. This sampling may also be performed early, which may help in cases where therapeutics work better in the earlier stages of a disease. Such pharmacogenetic designs have already been explored with ApoE in the context of AD trials (Roses, 2009; Zamani et al., 2011) and hinted at in mouse model studies (Cramer et al., 2012).
Designing clinical trials based on genotypes also has some challenges and limitations. Screening out subjects with certain genotypes may even miss trial effects, particularly if it is uncertain how the risk gene affects responses to therapeutic agent. In theory at least, people at low genetic risk may respond better to a treatment; regardless of this, however, their response may be extremely challenging to detect if they decline less than others, when not treated. Similarly, our study is limited to four AD risk genes. The three candidate SNPs we included in addition to ApoE are top GWAS findings, with recent replications in very large cohorts (see Introduction). There are, however, other AD risk variants that might potentially be included in a risk score for trial enrichment, particularly once they are further validated. Genetic variants in BIN1 (Kingwell, 2013; Lambert et al., 2011) and those in ABCA7, EPHA1, CD33, CD2AP, and MS4A6A/MS4A4E (Hollingworth et al., 2011), for instance, were recently discovered in large AD GWAS studies, some of which may have roles in AD pathogenesis (Holton et al., 2013). Genetic variants in TOMM40 have also been linked to AD (Roses et al., 2010), though the proximity and linkage disequilibrium between this gene and ApoE led to recent investigations that failed to replicate this association and cast doubt on ApoE-independent risk signals in the chromosomal region (Cruchaga et al., 2011; Jun et al., 2012). In practice, our approach may be too selective for a clinical trial, but nonetheless serves as a proof-of-principle study to demonstrate enrichment using multiple genotypes.
Another limitation of our study is that we did not include gene–gene interactions in our model. As the first paper to consider genes beyond ApoE for clinical trial enrichment, we started with a linear, additive model, modeling SNP effects based on well-recognized, GWAS-validated odds ratios. Some interactions between our candidate genes, particularly those between PICALM and ApoE have been reported in prior work (Jun et al., 2010). However, they have not always been replicated. The first GWAS that identified PICALM in AD (Harold et al., 2009), in fact, failed to find such interaction, making effect size estimation for interaction terms more complex. In addition, we may be underpowered to pick up interaction effects, as they assume a main effect is present, and that modulatory effects on the main effect can be detected. Truly vast samples, as well as novel computational strategies, may be needed to detect interactions among all pairs of N SNPs. One effort in this regard is Hibar et al. (2013). Environmental factors may also modify genetic susceptibility in the context of complex traits and disorders (De Jager et al., 2009; Morrison et al., 2007; Qi et al., 2012). Nonetheless, the general approach proposed here could also be extended to include any known environmental risk factors that might modulate disease risk (such as educational level, exercise, and alcohol intake).
Allele frequencies in the general population may also raise some practical issues for implementing this approach. In our sample, the risk alleles for PICALM and CLU are relatively common (major allele frequencies of 0.74 and 0.72, respectively), but that of CR1 is lower (minor allele frequency of 0.18). Some studies report a higher MAF of 0.26 for the CR1 SNP (Jun et al., 2010), but our MAF estimate is consistent with those reported in initial GWAS studies in Caucasian subjects (Lambert et al., 2009). Uncommon genotypes may cause a trial to be more expensive if a large number of subjects need to be screened (Schork and Topol, 2010). The cost/benefit depends on the cost of screening procedures relative to neuroimaging and longitudinal assessment, which may be much greater. In addition, pharmacogenomic profiling introduces ethical dilemmas, which complicate its application to drug trials; specifically some people may not have access to a new treatment as quickly as others, if genetic tests are among the criteria required to obtain it (Issa, 2002). Nevertheless, as risks and benefits of using a multilocus genetic profiling are weighed for specific trials and more is understood about our candidate risk genes and their roles in AD pathology, our results may have important implications for the design of AD clinical trials. At the very least, genotyping could be done retrospectively, to see if effects would have been more powerful if obtained from a pre-selected sample.
Acknowledgments
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, and the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Abbott; Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Amorfix Life Sciences Ltd.; AstraZeneca; Bayer HealthCare; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals Inc.; Eli Lilly and Company; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech, Inc.; GE Healthcare; Innogenetics, N.V.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by NIH grants P30 AG010129 and K01 AG030514. Algorithm development and image analysis for this study was funded by grants to P.T. from the NIBIB (R01 EB007813, R01 EB008281, and R01 EB008432), NICHD (R01 HD050735), and NIA (R01 AG020098). O.K. was supported by NIH F30 AG041681 and the UCLA Dissertation Year Fellowship. D.H. is partially supported by NSF GRFP Grant DGE-0707424. N.J. was additionally supported by NIH NLM Grant T15 LM07356.
Disclosure statement
The authors have no financial disclosures and declare no competing financial interests, but some have received commercial funding unrelated to the topic of the paper. M. Weiner is on the following scientific advisory boards: Lilly, Araclon and Institut Catala de Neurociencies Aplicades, Gulf War Veterans Illnesses Advisory Committee, VACO, Biogen Idec, and Pfizer; received funding for consulting from: Astra Zeneca, Araclon, Medivation/Pfizer, Ipsen, TauRx Therapeutics LTD, Bayer HealthCare, Biogen Idec, Exonhit Therapeutics, SA, Servier, Synarc, Pfizer, and Janssen; received funding for travel from: NeuroVigil, Inc., CHRU-Hopital Roger Salengro, Siemens, AstraZeneca, Geneva University Hospitals, Lilly, University of California, San Diego-ADNI, Paris University, Institut Catala de Neurociencies Aplicades, University of New Mexico School of Medicine, Ipsen, CTAD (Clinical Trials on Alzheimer's Disease), Pfizer, AD PD Meeting, Paul Sabatier University, Novartis, and Tohoku University; received honoraria from: PMDA /Japanese Ministry of Health, Labour, and Welfare, Tohoku University, Neuro Vigil, Inc., and Insitut Catala de Neurociencies Aplicades; and received research support from: Merck, Avid, DoD, VA; Stock Options: Synarc, Elan; organizations contributing to the Foundation for NIH and thus to the NIA funded Alzheimer's Disease Neuroimaging Initiative: Abbott, Alzheimer's Association, Alzheimer's Drug Discovery Foundation, Anonymous Foundation, AstraZeneca, Bayer HealthCare, BioClinica, Inc. (ADNI 2), Bristol-Myers Squibb, Cure Alzheimer's Fund, Eisai, Elan, Gene Network Sciences, Genentech, GE Healthcare, GlaxoSmithKline, Innogenetics, Johnson & Johnson, Eli Lilly & Company, Medpace, Merck, Novartis, Pfizer Inc., Roche, Schering Plough, Synarc, and Wyeth.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supplementary data
Table S1: Cumulative genetic risk scores and corresponding allele combinations.
References
- Beckett L.A., Harvey D.J., Gamst A., Donohue M., Kornak J., Zhang H., Kuo J.H., Alzheimer's Disease Neuroimaging Initiative The Alzheimer's Disease Neuroimaging Initiative: annual change in biomarkers and clinical outcomes. Alzheimer's & Dementia. 2010;6(3):257–264. doi: 10.1016/j.jalz.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L., McQueen M.B., Mullin K., Blacker D., Tanzi R.E. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nature Genetics. 2007;39(1):17–23. doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- Biffi A., Anderson C.D., Desikan R.S., Sabuncu M., Cortellini L., Schmansky N., Salat D., Rosand J., Alzheimer's Disease Neuroimaging Initiative Genetic variation and neuroimaging measures in Alzheimer disease. Archives of Neurology. 2010;67(6):677–685. doi: 10.1001/archneurol.2010.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bralten J., Franke B., Arias-Vásquez A., Heister A., Brunner H.G., Fernández G., Rijpkema M. CR1 genotype is associated with entorhinal cortex volume in young healthy adults. Neurobiology of Aging. 2011;32(11):2106.e7–2106.e11. doi: 10.1016/j.neurobiolaging.2011.05.017. [DOI] [PubMed] [Google Scholar]
- Braskie M.N., Ringman J.M., Thompson P.M. Neuroimaging measures as endophenotypes in Alzheimer's disease. International Journal of Alzheimer's Disease. 2011;490140 doi: 10.4061/2011/490140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braskie M.N., Jahanshad N., Stein J.L., Barysheva M., McMahon K.L., de Zubicaray G.I., Martin N.G., Wright M.J., Ringman J.M., Toga A.W., Thompson P.M. Common Alzheimer's disease risk variant within the CLU gene affects white matter microstructure in young adults. Journal of Neuroscience. 2011;31(18):6764–6770. doi: 10.1523/JNEUROSCI.5794-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burggren A.C., Zeineh M.M., Ekstrom A.D., Braskie M.N., Thompson P.M., Small G.W., Bookheimer S.Y. Reduced cortical thickness in hippocampal subregions among cognitively normal apolipoprotein E e4 carriers. NeuroImage. 2008;41(4):1177–1183. doi: 10.1016/j.neuroimage.2008.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquillo M.M., Belbin O., Hunter T.A., Ma L., Bisceglio G.D., Zou F., Crook J.E., Pankratz V.S., Dickson D.W., Graff-Radford N.R., Petersen R.C., Morgan K., Younkin S.G. Replication of CLU, CR1, and PICALM associations with Alzheimer disease. Archives of Neurology. 2010;67(8):961–964. doi: 10.1001/archneurol.2010.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockrell J.R., Folstein M.F. Mini-Mental State Examination (MMSE) Psychopharmacology Bulletin. 1988;24(4):689–692. [PubMed] [Google Scholar]
- Corneveaux J.J., Myers A.J., Allen A.N., Pruzin J.J., Ramirez M., Engel A., Nalls M.A., Chen K., Lee W., Chewning K., Villa S.E., Meechoovet H.B., Gerber J.D., Frost D., Benson H.L., O'Reilly S., Chibnik L.B., Shulman J.M., Singleton A.B., Craig D.W., Van Keuren-Jensen K.R., Dunckley T., Bennett D.A., De Jager P.L., Heward C., Hardy J., Reiman E.M., Huentelman M.J. Association of CR1, CLU and PICALM with Alzheimer's disease in a cohort of clinically characterized and neuropathologically verified individuals. Human Molecular Genetics. 2010;19(16):3295–3301. doi: 10.1093/hmg/ddq221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer P.E., Cirrito J.R., Wesson D.W., Lee C.Y., Karlo J.C., Zinn A.E., Casali B.T., Restivo J.L., Goebel W.D., James M.J., Brunden K.R., Wilson D.A., Landreth G.E. ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science. 2012;335(6075):1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crivello F., Lemaître H., Dufouil C., Grassiot B., Delcroix N., Tzourio-Mazoyer N., Tzourio C., Mazoyer B. Effects of ApoE-epsilon4 allele load and age on the rates of grey matter and hippocampal volumes loss in a longitudinal cohort of 1186 healthy elderly persons. Neuroimage. 2010;53(3):1064–1069. doi: 10.1016/j.neuroimage.2009.12.116. [DOI] [PubMed] [Google Scholar]
- Cruchaga C., Nowotny P., Kauwe J.S., Ridge P.G., Bertelsen S., Hinrichs A., Fagan A.M., Holtzman D.M., Morris J.C., Goate A.M., Alzheimer's Disease Neuroimaging Initiative Association and expression analyses with single-nucleotide polymorphisms in TOMM40 in Alzheimer disease. Archives of Neurology. 2011;68(8):1013–1019. doi: 10.1001/archneurol.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jager P.L., Chibnik L.B., Cui J., Reischl J., Lehr S., Simon K.C., Aubin C., Bauer D., Heubach J.F., Sandbrink R., Tyblova M., Lelkova P., Steering committee of the BENEFIT study, Steering committee of the BEYOND study, Steering committee of the LTF study, Steering committee of the CCR1 study, Havrdova E., Pohl C., Horakova D., Ascherio A., Hafler D.A., Karlson E.W. Integration of genetic risk factors into a clinical algorithm for multiple sclerosis susceptibility: a weighted genetic risk score. Lancet Neurology. 2009;8(12):1111–1119. doi: 10.1016/S1474-4422(09)70275-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer L.A., Cupples L.A., Haines J.L., Hyman B., Kukull W.A., Mayeux R., Myers R.H., Pericak-Vance M.A., Risch N., van Duijn C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278(16):1349–1356. [PubMed] [Google Scholar]
- Ferrari R., Moreno J.H., Minhajuddin A.T., O'Bryant S.E., Reisch J.S., Barber R.C., Momeni P. Implication of common and disease specific variants in CLU, CR1 and PICALM. Neurobiology of Aging. 2012;33(8):1846.e7–1846.e18. doi: 10.1016/j.neurobiolaging.2012.01.110. [DOI] [PubMed] [Google Scholar]
- Frisoni G.B., Fox N.C., Jack C.R., Jr., Scheltens P., Thompson P.M. The clinical use of structural MRI in Alzheimer's disease. Nature Reviews Neurology. 2010;6(2):67–77. doi: 10.1038/nrneurol.2009.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter J., Bernstein M., Borowski B., Felmlee J., Blezek D., Mallozzi R. Validation testing of the MRI calibration phantom for the Alzheimer’s Disease Neuroimaging Initiative study. Proceedings on International Society for Magnetic Resonance in Medicine. 2006;14 [Google Scholar]
- Harold D., Abraham R., Hollingworth P., Sims R., Gerrish A., Hamshere M.L., Pahwa J.S., Moskvina V., Dowzell K., Williams A., Jones N., Thomas C., Stretton A., Morgan A.R., Lovestone S., Powell J., Proitsi P., Lupton M.K., Brayne C., Rubinsztein D.C., Gill M., Lawlor B., Lynch A., Morgan K., Brown K.S., Passmore P.A., Craig D., McGuinness B., Todd S., Holmes C., Mann D., Smith A.D., Love S., Kehoe P.G., Hardy J., Mead S., Fox N., Rossor M., Collinge J., Maier W., Jessen F., Schürmann B., van den Bussche H., Heuser I., Kornhuber J., Wiltfang J., Dichgans M., Frölich L., Hampel H., Hüll M., Rujescu D., Goate A.M., Kauwe J.S., Cruchaga C., Nowotny P., Morris J.C., Mayo K., Sleegers K., Bettens K., Engelborghs S., De Deyn P.P., Van Broeckhoven C., Livingston G., Bass N.J., Gurling H., McQuillin A., Gwilliam R., Deloukas P., Al-Chalabi A., Shaw C.E., Tsolaki M., Singleton A.B., Guerreiro R., Mühleisen T.W., Nöthen M.M., Moebus S., Jöckel K.H., Klopp N., Wichmann H.E., Carrasquillo M.M., Pankratz V.S., Younkin S.G., Holmans P.A., O'Donovan M., Owen M.J., Williams J. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nature Genetics. 2009;41(10):1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibar D.P., Stein J.L., Kohannim O., Jahanshad N., Saykin A.J., Shen L., Kim S., Pankratz N., Foroud T., Huentelman M.J., Potkin S.G., Jack C.R., Jr., Weiner M.W., Toga A.W., Thompson P.M., Alzheimer's Disease Neuroimaging Initiative Voxelwise gene-wide association study (vGeneWAS). Multivariate gene-based association testing in 731 elderly subjects. NeuroImage. 2011;56(4):1875–1891. doi: 10.1016/j.neuroimage.2011.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibar D.P., Stein J.L., Jahanshad N., Kohannim O., Toga A.W., McMahon K.L., de Zubicaray G.I., Montgomery G.W., Martin N.G., Wright M.J., Weiner M.W., Thompson P.M. Exhaustive Search of the SNP–SNP Interactome Identifies Replicated Epistatic Effects on Brain Volume, MICCAI 2013, Nagoya, Japan, Sept. 22–26. 2013. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingworth P., Harold D., Sims R., Gerrish A., Lambert J.-C., Carrasquillo M.M., Abraham R., Hamshere M.L., Singh Pahwa J., Moskvina V., Dowzell K., Jones N., Stretton A., Thomas C., Richards A., Ivanov D., Widdowson C., Chapman J., Lovestone S., Powell J., Proitsi P., Lupton M.K., Brayne C., Rubinsztein D.C., Gill M., Lawlor B., Lynch A., Brown K.S., Passmore P.A., Craig D., McGuinness B., Todd S., Holmes C., Mann D., Smith A.D., Beaumont H., Warden D., Wilcock G., Love S., Kehoe P.G., Hooper N.M., Vardy E.R.L.C., Hardy J., Mead S., Fox N.C., Rossor M., Collinge J., Maier W., Jessen F., Schürmann B., Rüther E., Heun R., Kölsch H., van den Bussche H., Heuser I., Kornhuber J., Wiltfang J., Dichgans M., Frölich L., Hampel H., Hüll M., Gallacher J., Rujescu D., Giegling I., Goate A.M., Kauwe J.S.K., Cruchaga C., Nowotny P., Morris J.C., Mayo K., Sleegers K., Bettens K., Engelborghs S., De Deyn P.P., Van Broeckhoven C., Livingston G., Bass N.J., Gurling H., McQuillin A., Gwilliam R., Deloukas P., Al-Chalabi A., Shaw C.E., Tsolaki M., Singleton A.B., Guerreiro R., Mühleisen T.W., Nöthen M.M., Moebus S., Jöckel K.-H., Klopp N., Wichmann H.-E., Pankratz V.S., Sando S.B., Aasly J.O., Barcikowska M., Wszolek Z.K., Dickson D.W., Graff-Radford N.R., Petersen R.C., the Alzheimer's Disease Neuroimaging Initiative, van Duijn C.M., Breteler M.M.B., Ikram M.A., DeStefano A.L., Fitzpatrick A.L., Lopez O., Launer L.J., Seshadri S., CHARGE consortium, Berr C., Campion D., Epelbaum J., Dartigues J.-F., Tzourio C., Alpérovitch A., Lathrop M., EADI1 consortium, Feulner T.M., Friedrich P., Riehle C., Krawczak M., Schreiber S., Mayhaus M., Nicolhaus S., Wagenpfeil S., Steinberg S., Stefansson H., Stefansson K., Snædal J., Björnsson S., Jonsson P.V., Chouraki V., Genier-Boley B., Hiltunen M., Soininen H., Combarros O., Zelenika D., Delepine M., Bullido M.J., Pasquier F., Mateo I., Frank-Garcia A., Porcellini E., Hanon O., Coto E., Alvarez V., Bosco P., Siciliano G., Mancuso M., Panza F., Solfrizzi V., Nacmias B., Sorbi S., Bossù P., Piccardi P., Arosio B., Annoni G., Seripa D., Pilotto A., Scarpini E., Galimberti D., Brice A., Hannequin D., Licastro F., Jones L., Holmans P.A., Jonsson T., Riemenschneider M., Morgan K., Younkin S.G., Owen M.J., O'Donovan M., Amouyel P., Williams J. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nature Genetics. 2011;43(5):429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holton P., Ryten M., Nalls M., Trabzuni D., Weale M.E., Hernandez D., Crehan H., Gibbs J.R., Mayeux R., Haines J.L., Farrer L.A., Pericak-Vance M.A., Schellenberg G.D., Alzheimer's Disease Genetics Consortium, Ramirez-Restrepo M., Engel A., Myers A.J., Corneveaux J.J., Huentelman M.J., Dillman A., Cookson M.R., Reiman E.M., Singleton A., Hardy J., Guerreiro R., Apostolova L.G., Arnold S.E., Baldwin C.T., Barber R., Barmada M.M., Beach T.G., Beecham G.W., Beekly D., Bennett D.A., Bigio E.H., Bird T.D., Blacker D., Boeve B.F., Bowen J.D., Boxer A., Burke J.R., Buros J., Buxbaum J.D., Cairns N.J., Cantwell L.B., Cao C., Carlson C.S., Carney R.M., Carrasquillo M.M., Carroll S.L., Chui H.C., Clark D.G., Cotman C.W., Crane P.K., Crocco E.A., Cruchaga C., Cummings J.L., De Jager P.L., DeCarli C., DeKosky S.T., Demirci F.Y., Diaz-Arrastia R., Dick M., Dickson D.W., Duara R., Ellis W.G., Ertekin-Taner N., Evans D., Faber K.M., Fallon K.B., Farlow M.R., Ferris S., Foroud T.M., Frosch M.P., Galasko D.R., Ganguli M., Gearing M., Geschwind D.H., Ghetti B., Gilbert J.R., Gilman S., Giordani B., Glass J.D., Goate A.M., Graff-Radford N.R., Green R.C., Growdon J.H., Hakonarson H., Hamilton R.L., Harrell L.E., Head E., Honig L.S., Hulette C.M., Hyman B.T., Jarvik G.P., Jicha G.A., Jin L.W., Jun G., Kamboh M.I., Karlawish J., Karydas A., Kauwe J.S., Kaye J.A., Kim R., Koo E.H., Kowall N.W., Kramer P., Kukull W.A., Lah J.J., Larson E.B., Levey A.I., Lieberman A.P., Lopez O.L., Lunetta K.L., Mack W.J., Marson D.C., Martin E.R., Martiniuk F., Mash D.C., Masliah E., McCormick W.C., McCurry S.M., McDavid A.N., McKee A.C., Mesulam M., Miller B.L., Miller C.A., Miller J.W., Montine T.J., Morris J.C., Naj A.C., Nowotny P., Parisi J.E., Peskind E., Petersen R.C., Poon W.W., Potter H., Quinn J.F., Raj A., Rajbhandary R.A., Raskind M., Reisberg B., Reitz C., Ringman J.M., Roberson E.D., Rogaeva E., Rosenberg R.N., Sano M., Saykin A.J., Schneider J.A., Schneider L.S., Seeley W.W., Shelanski M.L., Smith C.D., Sonnen J.A., Spina S., St George-Hyslop P., Stern R.A., Tanzi R.E., Trojanowski J.Q., Troncoso J.C., Tsuang D.W., Valladares O., Van Deerlin V.M., Vardarajan B.N., Vinters H.V., Vonsattel J.P., Wang L.S., Weintraub S., Welsh-Bohmer K.A., Williamson J., Woltjer R.L., Wright C.B., Younkin S.G. Initial assessment of the pathogenic mechanisms of the recently identified Alzheimer risk loci. Annals of Human Genetics. 2013;77(2):85–105. doi: 10.1111/ahg.12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X., Gutman B., Boyle C.P., Rajagopalan P., Leow A.D., Yanovsky I., Kumar A.R., Toga A.W., Jack C.R., Jr., Schuff N., Alexander G.E., Chen K., Reiman E.M., Weiner M.W., Thompson P.M., Alzheimer's Disease Neuroimaging Initiative Accurate measurement of brain changes in longitudinal MRI scans using tensor-based morphometry. NeuroImage. 2011;57(1):5–14. doi: 10.1016/j.neuroimage.2011.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X., Hibar D.P., Ching C.R., Boyle C.P., Rajagopalan P., Gutman B.A., Leow A.D., Toga A.W., Jack C.R., Jr., Harvey D., Weiner M.W., Thompson P.M., the Alzheimer's Disease Neuroimaging Initiative Unbiased tensor-based morphometry: improved robustness and sample size estimates for Alzheimer's disease clinical trials. NeuroImage. 2013;66C:648–661. doi: 10.1016/j.neuroimage.2012.10.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X., Lee S., Yanovsky I., Leow A.D., Chou Y.Y., Ho A.J., Gutman B., Toga A.W., Jack C.R., Jr., Bernstein M.A., Reiman E.M., Harvey D.J., Kornak J., Schuff N., Alexander G.E., Weiner M.W., Thompson P.M. Alzheimer's Disease Neuroimaging Initiative, Optimizing power to track brain degeneration in Alzheimer's disease and mild cognitive impairment with tensor-based morphometry: an ADNI study of 515 subjects. Neuroimage. 2009;48(4):668–681. doi: 10.1016/j.neuroimage.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd M.D., Martorell P., Delavande A., Mullen K.J., Langa K.M. Monetary costs of dementia in the United States. The New England Journal of Medicine. 2013;368(14):1326–1334. doi: 10.1056/NEJMsa1204629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias J.E., Liu C.Y., Thompson P.M., Tu Z. Robust brain extraction across datasets and comparison with publicly available methods. IEEE Transactions on Medical Imaging. 2011;30(9):1617–1634. doi: 10.1109/TMI.2011.2138152. [DOI] [PubMed] [Google Scholar]
- Issa A.M. Ethical perspectives on pharmacogenomic profiling in the drug development process. Nature Reviews. Drug Discovery. 2002;1(4):300–308. doi: 10.1038/nrd771. [DOI] [PubMed] [Google Scholar]
- Jack C.R., Jr., Bernstein M.A., Fox N.C., Thompson P., Alexander G., Harvey D., Borowski B., Britson P.J., Whitwell J.L., Ward C., Dale A.M., Felmlee J.P., Gunter J.L., Hill D.L., Killiany R., Schuff N., Fox-Bosetti S., Lin C., Studholme C., DeCarli C.S., Krueger G., Ward H.A., Metzger G.J., Scott K.T., Mallozzi R., Blezek D., Levy J., Debbins J.P., Fleisher A.S., Albert M., Green R., Bartzokis G., Glover G., Mugler J., Weiner M.W. The Alzheimer's Disease Neuroimaging Initiative (ADNI) MRI methods. Journal of Magnetic Resonance Imaging. 2008;27(4):685–691. doi: 10.1002/jmri.21049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovicich J., Czanner S., Greve D., Haley E., van der Kouwe A., Gollub R., Kennedy D., Schmitt F., Brown G., Macfall J., Fischl B., Dale A. Reliability in multi-site structural MRI studies: effects of gradient non-linearity correction on phantom and human data. NeuroImage. 2006;30(2):436–443. doi: 10.1016/j.neuroimage.2005.09.046. [DOI] [PubMed] [Google Scholar]
- Jun G., Naj A.C., Beecham G.W., Wang L.S., Buros J., Gallins P.J., Buxbaum J.D., Ertekin-Taner N., Fallin M.D., Friedland R., Inzelberg R., Kramer P., Rogaeva E., St George-Hyslop P., Alzheimer's Disease Genetics Consortium, Cantwell L.B., Dombroski B.A., Saykin A.J., Reiman E.M., Bennett D.A., Morris J.C., Lunetta K.L., Martin E.R., Montine T.J., Goate A.M., Blacker D., Tsuang D.W., Beekly D., Cupples L.A., Hakonarson H., Kukull W., Foroud T.M., Haines J., Mayeux R., Farrer L.A., Pericak-Vance M.A., Schellenberg G.D. Meta-analysis confirms CR1, CLU, and PICALM as Alzheimer disease risk loci and reveals interactions with APOE genotypes. Archives of Neurology. 2010;67(12):1473–1484. doi: 10.1001/archneurol.2010.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun G., Vardarajan B.N., Buros J., Yu C.E., Hawk M.V., Dombroski B.A., Crane P.K., Larson E.B., Alzheimer's Disease Genetics Consortium, Mayeux R., Haines J.L., Lunetta K.L., Pericak-Vance M.A., Schellenberg G.D., Farrer L.A. Comprehensive search for Alzheimer disease susceptibility loci in the APOE region. Archives of Neurology. 2012;69(10):1270–1279. doi: 10.1001/archneurol.2012.2052. (Oct.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingwell K. Alzheimer disease: BIN1 variant increases risk of Alzheimer disease through tau. Nature Reviews. Neurology. 2013;9(4):184. doi: 10.1038/nrneurol.2013.34. [DOI] [PubMed] [Google Scholar]
- Kohannim O., Hua X., Hibar D.P., Lee S., Chou Y.Y., Toga A.W., Jack C.R., Jr., Weiner M.W., Thompson P.M., Alzheimer's Disease Neuroimaging Initiative Boosting power for clinical trials using classifiers based on multiple biomarkers. Neurobiology of Aging. 2010;31(8):1429–1442. doi: 10.1016/j.neurobiolaging.2010.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozauer N., Katz R. Regulatory innovation and drug development for early-stage Alzheimer's disease. The New England Journal of Medicine. 2013;368(13):1169–1171. doi: 10.1056/NEJMp1302513. [DOI] [PubMed] [Google Scholar]
- Lambert J.C., Heath S., Even G., Campion D., Sleegers K., Hiltunen M., Combarros O., Zelenika D., Bullido M.J., Tavernier B., Letenneur L., Bettens K., Berr C., Pasquier F., Fiévet N., Barberger-Gateau P., Engelborghs S., De Deyn P., Mateo I., Franck A., Helisalmi S., Porcellini E., Hanon O., European Alzheimer's Disease Initiative Investigators, de Pancorbo M.M., Lendon C., Dufouil C., Jaillard C., Leveillard T., Alvarez V., Bosco P., Mancuso M., Panza F., Nacmias B., Bossù P., Piccardi P., Annoni G., Seripa D., Galimberti D., Hannequin D., Licastro F., Soininen H., Ritchie K., Blanché H., Dartigues J.F., Tzourio C., Gut I., Van Broeckhoven C., Alpérovitch A., Lathrop M., Amouyel P. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nature Genetics. 2009;41(10):1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- Lambert J.C., Zelenika D., Hiltunen M., Chouraki V., Combarros O., Bullido M.J., Tognoni G., Fiévet N., Boland A., Arosio B., Coto E., Del Zompo M., Mateo I., Frank-Garcia A., Helisalmi S., Porcellini E., Pilotto A., Forti P., Ferri R., Delepine M., Scarpini E., Siciliano G., Solfrizzi V., Sorbi S., Spalletta G., Ravaglia G., Valdivieso F., Alvarez V., Bosco P., Mancuso M., Panza F., Nacmias B., Bossù P., Piccardi P., Annoni G., Seripa D., Galimberti D., Licastro F., Lathrop M., Soininen H., Amouyel P. Evidence of the association of BIN1 and PICALM with the AD risk in contrasting European populations. Neurobiol Aging. 2011;32(4):756.e11-5. doi: 10.1016/j.neurobiolaging.2010.11.022. [DOI] [PubMed] [Google Scholar]
- Langers D.R.M., Jansen J.F.A., Backes W.H. Enhanced signal detection in neuroimaging by means of regional control of the global false discovery rate. NeuroImage. 2007;38(1):43–56. doi: 10.1016/j.neuroimage.2007.07.031. [DOI] [PubMed] [Google Scholar]
- Leow A., Huang S.C., Geng A., Becker J., Davis S., Toga A., Thompson P.M. Inverse consistent mapping in 3D deformable image registration: its construction and statistical properties. Information Processing in Medical Imaging. 2005;19:493–503. doi: 10.1007/11505730_41. [DOI] [PubMed] [Google Scholar]
- Leow A.D., Klunder A.D., Jack C.R., Jr., Toga A.W., Dale A.M., Bernstein M.A., Britson P.J., Gunter J.L., Ward C.P., Whitwell J.L., Borowski B.J., Fleisher A.S., Fox N.C., Harvey D., Kornak J., Schuff N., Studholme C., Alexander G.E., Weiner M.W., Thompson P.M., ADNI Preparatory Phase Study Longitudinal stability of MRI for mapping brain change using tensor-based morphometry. NeuroImage. 2006;31(2):627–640. doi: 10.1016/j.neuroimage.2005.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung K.K., Barnes J., Ridgway G.R., Bartlett J.W., Clarkson M.J., Macdonald K., Schuff N., Fox N.C., Ourselin S., for the Alzheimer's Disease Neuroimaging Initiative Automated cross-sectional and longitudinal hippocampal volume measurement in mild cognitive impairment and Alzheimer's disease. NeuroImage. 2010;51(4):1345–1359. doi: 10.1016/j.neuroimage.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzi M., Donohue M., Paternicò D., Scarpazza C., Ostrowitzki S., Blin O., Irving E., Frisoni G.B., for the Alzheimer's Disease Neuroimaging Initiative Enrichment through biomarkers in clinical trials of Alzheimer's drugs in patients with mild cognitive impairment. Neurobiology of Aging. 2010;31(8):1443–1451. doi: 10.1016/j.neurobiolaging.2010.04.036. [DOI] [PubMed] [Google Scholar]
- McEvoy L.K., Edland S.D., Holland D., Hagler D.J., Jr., Roddey J.C., Fennema-Notestine C., Salmon D.P., Koyama A.K., Aisen P.S., Brewer J.B., Dale A.M., for the Alzheimer's Disease Neuroimaging Initiative Neuroimaging enrichment strategy for secondary prevention trials in Alzheimer disease. Alzheimer Disease and Associated Disorders. 2010;24(3):269–277. doi: 10.1097/WAD.0b013e3181d1b814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann G., Drachman D., Folstein M., Katzman R., Price D., Stadlan E.M. Clinical diagnosis of Alzheimer's disease: report of the NINCDS–ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34(7):939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Morris J.C. The Clinical Dementia Rating (CDR). Current version and scoring rules. Neurology. 1993;43(11):2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- Morris J.C., Roe C.M., Xiong C., Fagan A.M., Goate A.M., Holtzman D.M., Mintun M.A. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Annals of Neurology. 2010;67(1):122–131. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison A.C., Bare L.A., Chambless L.E., Ellis S.G., Malloy M., Kane J.P., Pankow J.S., Devlin J.J., Willerson J.T., Boerwinkle E. Prediction of coronary heart disease risk using a genetic risk score: the Atherosclerosis Risk in Communities study. American Journal of Epidemiology. 2007;166(1):28–35. doi: 10.1093/aje/kwm060. [DOI] [PubMed] [Google Scholar]
- Nestor S.M., Rupsingh R., Borrie M., Smith M., Accomazzi V., Wells J.L., Fogarty J., Bartha R., Alzheimer's Disease Neuroimaging Initiative Ventricular enlargement as a possible measure of Alzheimer's disease progression validated using the Alzheimer's disease neuroimaging initiative database. Brain. 2008;131(9):2443–2454. doi: 10.1093/brain/awn146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potkin S.G., Guffanti G., Lakatos A., Turner J.A., Kruggel F., Fallon J.H., Saykin A.J., Orro A., Lupoli S., Salvi E., Weiner M., Macciardi F., Alzheimer's Disease Neuroimaging Initiative Hippocampal atrophy as a quantitative trait in a genome-wide association study identifying novel susceptibility genes for Alzheimer's disease. PLoS One. 2009;4(8):e6501. doi: 10.1371/journal.pone.0006501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Q., Li Y., Chomistek A.K., Kang J.H., Curhan G.C., Pasquale L.R., Willett W.C., Rimm E.B., Hu F.B., Qi L. Epidemiology and Prevention/Nutrition, Physical Activity and Metabolism Scientific Sessions. 2012. Physical activity, television watching and genetic predisposition in relation to body mass index in women and men. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risacher S.L., Shen L., West J.D., Kim S., McDonald B.C., Beckett L.A., Harvey D.J., Jack C.R., Jr., Weiner M.W., Saykin A.J., Alzheimer's Disease Neuroimaging Initiative Longitudinal MRI atrophy biomarkers: relationship to conversion in the ADNI cohort. Neurobiology of Aging. 2010;31(8):1401–1418. doi: 10.1016/j.neurobiolaging.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roses A.D. The medical and economic roles of pipeline pharmacogenetics: Alzheimer's disease as a model of efficacy and HLA-B(*)5701 as a model of safety. Neuropsychopharmacology. 2009;34(1):6–17. doi: 10.1038/npp.2008.153. [DOI] [PubMed] [Google Scholar]
- Roses A.D., Lutz M.W., Amrine-Madsen H., Saunders A.M., Crenshaw D.G., Sundseth S.S., Huentelman M.J., Welsh-Bohmer K.A., Reiman E.M. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer's disease. The Pharmacogenomics Journal. 2010;10(5):375–384. doi: 10.1038/tpj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saykin A.J., Shen L., Foroud T.M., Potkin S.G., Swaminathan S., Kim S., Risacher S.L., Nho K., Huentelman M.J., Craig D.W., Thompson P.M., Stein J.L., Moore J.H., Farrer L.A., Green R.C., Bertram L., Jack C.R., Jr., Weiner M.W., Alzheimer's Disease Neuroimaging Initiative Alzheimer’s Disease Neuroimaging Initiative biomarkers as quantitative phenotypes: genetics core aims progress and plans. Alzheimer's & Dementia. 2010;6(3):265–273. doi: 10.1016/j.jalz.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schork N.J., Topol E.J. Genotype-based risk and pharmacogenetic sampling in clinical trials. Journal of Biopharmaceutical Statistics. 2010;20(2):315–333. doi: 10.1080/10543400903572779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sled J.G., Zijdenbos A.P., Evans A.C. A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Transactions on Medical Imaging. 1998;17(1):87–97. doi: 10.1109/42.668698. [DOI] [PubMed] [Google Scholar]
- Sleegers K., Lambert J.C., Bertram L., Cruts M., Amouyel P., Van Broeckhoven C. The pursuit of susceptibility genes for Alzheimer's disease: progress and prospects. Trends in Genetics. 2010;26(2):84–93. doi: 10.1016/j.tig.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Stein J.L., Hua X., Morra J.H., Lee S., Hibar D.P., Ho A.J., Leow A.D., Toga A.W., Sul J.H., Kang H.M., Eskin E., Saykin A.J., Shen L., Foroud T., Pankratz N., Huentelman M.J., Craig D.W., Gerber J.D., Allen A.N., Corneveaux J.J., Stephan D.A., Webster J., DeChairo B.M., Potkin S.G., Jack C.R., Jr., Weiner M.W., Thompson P.M., Alzheimer's Disease Neuroimaging Initiative Genome-wide analysis reveals novel genes influencing temporal lobe structure with relevance to neurodegeneration in Alzheimer's disease. NeuroImage. 2010;51(2):542–554. doi: 10.1016/j.neuroimage.2010.02.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser P.J., Scheltens P., Verhey F.R. Do MCI criteria in drug trials accurately identify subjects with predementia Alzheimer's disease? J Neurol Neurosurg Psychiatry. 2005;76(10):1348–1354. doi: 10.1136/jnnp.2004.047720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner M.W., Veitch D.P., Aisen P.S., Beckett L.A., Cairns N.J., Green R.C., Harvey D., Jack C.R., Jagust W., Liu E., Morris J.C., Petersen R.C., Saykin A.J., Schmidt M.E., Shaw L., Siuciak J.A., Soares H., Toga A.W., Trojanowski J.Q. Alzheimer’s Disease Neuroimaging Initiative, The Alzheimer's Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers Dement. 2012;8(1 Suppl):S1–68. doi: 10.1016/j.jalz.2011.09.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamani M., Mehri M., Kollaee A., Yenki P., Ghaffarpor M., Harirchian M.H., Shahbazi M. Pharmacogenetic study on the effect of rivastigmine on PS2 and APOE genes in Iranian Alzheimer patients. Dementia and Geriatric Cognitive Disorders Extra. 2011;1(1):180–189. doi: 10.1159/000329514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Cumulative genetic risk scores and corresponding allele combinations.