

Abstract
Meckel’s cartilage is a transient supporting tissue of the embryonic mandible in mammals, and disappears by taking different ultimate cell fate along the distal-proximal axis, with the majority (middle portion) undergoing degeneration and chondroclastic resorption. While a number of factors have been implicated in the degeneration and resorption processes, signaling pathways that trigger this degradation are currently unknown. BMP signaling has been implicated in almost every step of chondrogenesis. In this study, we used Noggin mutant mice as a model for gain-of-BMP signaling function to investigate the function of BMP signaling in Meckel’s cartilage development, with a focus on the middle portion. We showed that Bmp2 and Bmp7 are expressed in early developing Meckels’ cartilage, but their expression disappears thereafter. In contrast, Noggin is expressed constantly in Meckel’s cartilage throughout the entire gestation period. In the absence of Noggin, Meckel’s cartilage is significantly thickened attributing to dramatically elevated cell proliferation rate associated with enhanced phosphorylated Smad1/5/8 expression. Interestingly, instead of taking a degeneration fate, the middle portion of Meckel’s cartilage in Noggin mutants undergoes chondrogenic differentiation and endochondral ossification contributing to the forming mandible. Chondrocyte-specific expression of a constitutively active form of BMPRIa but not BMPRIa leads enlargement of Meckel’s cartilage, phenocopying the consequence of Noggin deficiency. Our results demonstrate that elevated BMP signaling prevents degeneration and leads to endochondral ossification of Meckel’s cartilage, and support the idea that withdrawal of BMP signaling is required for normal Meckel’s cartilage development and ultimate cell fate.
Keywords: BMP signaling, Noggin, BmprIa, Meckel’s cartilage, development, differentiation
Introduction
In mammals, Meckel’s cartilage acts as a transient support tissue in mandibular arch during early embryogenesis and disappears at late gestation or neonatal stage of development (Dixon, 1997). In mice, the development of Meckel’s cartilage, which derives from cranial neural crest (CNC) cells, begins at the first molar bud region at embryonic day 11 (E11), and continues with extension at both the anterior and posterior ends with addition of CNC cells at the chondrogenic front, forming a pair of rod-like hyaline cartilage (Ito et al., 2002). Previous studies have established different ultimate fates of Meckel’s cartilage in rodents (Bhaskar et al., 1953; Bernick and Patek, 1969; Savostin-Asling and Asling, 1973; Harada and Ishizeki, 1998; Ishizeki et al., 2001). The distal region of Meckel’s cartilage undergoes endochondral ossification contributing to the symphysis of the mandible, whereas the most proximal portion (caudal end) gives rise to malleus and incus through endochondral ossification as well. However, in the middle portion, which counts for the majority of Meckel’s cartilage, the chondrocytes become hypertrophic, but never further differentiate and undergo degeneration subsequently, except that the posterior part of this portion is replaced by the sphenomandibular ligament (Harada and Ishizeki, 1998).
Similar to the development of other fetal cartilaginous skeleton, chondrogenesis of Meckel’s cartilage progresses via mesenchymal cell condensation, proliferation, and differentiation of chondrocytes. Despite its CNC-origin, the initial chondrogenesis of Meckel’s cartilage shares many similar mechanisms with those mesoderm-derived cartilaginous elements. For example, Sox9 is required for the determination of chondrogenic lineage in axial and appendicular skeletal elements as well as CNC-derived cartilages and endochondral bones (Bi et al., 1999; Mori-Akiyama et al., 2003). In addition, a number of signaling molecules, including bone morphogenetic protein (BMP), connective tissue growth factor (CTGF), fibroblast growth factor (FGF), transforming growth factor β (TGFβ), and Wnt that are known to regulate chondrogenesis of appendicular skeletons, have also been implicated in Meckel’s cartilage development (Chai et al., 1994; Nonaka et al., 1999; Ito et al., 2002; Shimo et al., 2004; Terao et al., 2011; Zhang et al., 2011). However, unlike the fate of those mesoderm-derived cartilaginous elements and other CNC-derived cartilages such as cranial base, the majority of Meckel’s cartilage does not develop further and becomes degenerated. While previous studies have revealed potential contributions of several factors and cellular processes to the disappearance/resorption (Trichilis and Wroblewski, 1997; Harada and Ishizeki, 1998; Sakakura et al., 2005, 2007a, 2007b; Tsuzurahara et al., 2011; Yang et al., 2012), signaling pathways that prevent further differentiation and trigger degeneration of Meckel’s cartilage remain elusive.
BMP signaling acts via binding to heterotetrameric complexes of type I and II receptors, leading to activation of Smad-dependent canonical pathway and Smad-independent noncanonical MAPK pathways that regulate targeted gene expression (Massagué, 2012). BMP signaling plays important roles in multiple steps of chondrogenesis and endochondral bone formation. In the earliest stage of chondrogenesis, BMP signaling promotes mesenchymal cells to differentiate into chondrocytes and stimulates chondrocyte proliferation by inducing Sox9 expression (Denker et al., 1999; Zehentner et al., 1999; Yoon et al., 2005). BMP signaling also controls chondrocyte differentiation by promoting chondrocyte hypertrophy and is required for endochondral bone formation (Minina et al., 2001; Valcourt et al., 2002; Kobayashi et al., 2005; Retting et al., 2009).
Noggin is a potent BMP antagonist, binding preferentially to BMP2, BMP4, and BMP7 to prevent their signaling (Zimmerman et al., 1996; Groppe et al., 2002; Chen et al., 2004). Noggin deficiency in mice, a model of gain-of-BMP function, leads to overgrowth of skeletal elements including the mandible (Brunet et al., 1998; Stottmann et al., 2001). However, whether or not loss of Noggin alters the development of Meckel’s cartilage has not been reported. In our and other’s previous studies on the effects of overdosed BMP activity on the development of palate, tooth, and temporomandibular joint using Noggin mutant mice as a model, the formation of significantly enlarged Meckel’s cartilage was observed (He et al., 2010; Lana-Elola et al., 2011; Wang et al., 2011; Hu et al., 2012). In the present study, we followed up our previous observation by detailed analysis of the phenotype in Noggin mutants with focus on the middle portion of Meckel’s cartilage. We show here that Noggin is expressed constantly in Meckel’s cartilage throughout the entire embryonic stage, whereas Bmp2 and Bmp7 but not Bmp4 are expressed in developing Meckel’s cartilage only at the early stage. In the absence of Noggin, the expression of phosphorylated Smad1/5/8 is significantly elevated associated with enhanced cell proliferation rate in Meckel’s cartilage, leading to formation of enlarged Meckel’s cartilage. Instead becoming arrest of further differentiation and subsequent degeneration, Meckel’s cartilage in Noggin mutants undergoes endochondral differentiation, unifying with the developing mandibular bone to form a larger mandible. Forced expression of a constitutively active form of BMPRIa in chondrocyte lineage also produces enlarged Meckel’s cartilage, mimicking the consequence of Noggin deficiency. We conclude that withdrawal of BMP signaling represents an essential step for arrest of chondrogenic differentiation and subsequent degeneration of Meckel’s cartilage during development.
Materials and Methods
Animals
The generation of Noggin mutant (Nog+/−), Col2a1-CreERT2, Rosa26 reporter, pMes-caBmprIa, and pMes-caBmprIb mice have been described previously (Soriano, 1999; McMahon et al., 1998; Chen et al., 2007; He et al., 2010; Yu et al., 2010). Nog−/− embryos were harvested timed pregnant Nog+/− females mated with Nog+/− males. To achieve chondrocyte-specific expression of constitutively active form of BMPRIa or BMPRIb in developing embryos, Col2-CreERT2 transgenic mice were mated with either pMes-caBmprIa or pMes-caBmprIb conditional transgenic mice, and timed pregnant females were injected intraperitoneally once with tamoxifen (Sigma) at embryonic day 12.5 (E12.5). Each mouse received a single injection of 200-ul tamoxifen solution, dissolved in corn oil (Sigma), at the final concentration of 1 mg/20 g body weight. Tail sample of each harvested embryo was subjected to PCR-based genotyping. Primer information for PCR-based genotyping for these mouse lines is available upon request. Use of animals in this study was approved by the Institutional Animal Care and Use Committee (IACUC) of Tulane University, and was in strict accordance with the recommendations in the Guide for Care and Use of Laboratory Animals of the National Institutes of Health.
Skeletal preparation, histology, immunostaining, in situ hybridization, X-gal staining, and BrdU labeling assays
Skeletal staining was conducted by Alcian blue/Alizarin red staining for cartilage and bone, as described previously (Zhang et al., 2000). For histological analysis and section in situ hybridization assay, samples were fixed in 4% paraformaldehyde (PFA)/PBS at 4°C for overnight, dehydrated through graded ethanol, paraffin embedded, and sectioned at 10 μm. Sections were subjected to standard Hematoxylin/Eosin staining for histological examination, and to in situ hybridization for gene expression assay using non-radioactive riboprobes, as described previously (St. Amand et al., 2000). For immunohistochemical staining, PFA fixed samples were washed in 30% sucrose/PBS, embedded in O.C.T. compound (Tissue-Tek) and cryo-sectioned at 10 μm. Prior to the application of primary antibody (anti-pSmad1/5/8; from Cell Signaling), slides were treated with acetone for 10-min and air-dried. For immunostaining with antibodies against type II and type X collagens, samples were processed same as that for histological analysis, and paraffin-embedded sections were subjected to immunostaining, as described previously (Xiong et al., 2009). For X-gal staining, samples were fixed in 0.2% glutaraldehyde at 4°C overnight, washed in ice-cold PBS and subsequently with 30% sucrose/PBS, embedded in O.C.T., and cryo-sectioned at 10 μm. X-gal staining was conducted as described previously (Ito et al., 2003). All experiments were repeated at least three times. To determine cell proliferation rate, we performed Bromodeoxyuridine (BrdU) labeling experiment using BrdU Labeling and detection Kit (Roche Diagnostics Corp.), as described previously (Xiong et al., 2009). BrdU-positive cells and total cell number in a section of Meckel’s cartilage were counted on higher magnification image on computer. Data collected from nine sections of three samples (three continuous sections from each sample) of either control or mutant were subjected to statistical analysis. The outcome was presented as percentage of labeled cells among total cells in Meckel’s cartilage, and Student’s t-test was used to determine the significance of difference.
Results
Expression of Noggin and Bmp genes in developing Meckel’s cartilage
To investigate the role of Noggin and BMP signaling in Meckel’s cartilage development and degeneration, we examined the expression of Noggin and several Bmp genes including Bmp2, Bmp4, and Bmp7 in the middle portion of Meckel’s cartilage. We used the first molar germ as a landmark for our analyses, since the development of Meckel’s cartilage initiates at this region. We took advantage of Nog+/− mice in which the Noggin coding sequences were replaced with the LacZ reporter gene by targeted knock-in approach (McMahon et al., 1998). We assayed Noggin expression by X-gal staining from E11.5 at which time Meckel’s cartilage condensation appears, to the last day day of gestation (E18.5). As shown in Fig. 1, LacZ activity was detected, though relatively weaker, in forming Meckel’s cartilage at E11.5 (Fig. 1A). Strong X-gal staining was observed in Meckel’s cartilage at E12.5 (Fig. 1B), and remained there throughout the entire embryonic stage (Fig. 1C, 1D). In situ hybridization assay conformed Noggin expression in Meckel’s cartilage (insert in Fig. 1C). We performed in situ hybridization assay to examine Bmp gene expression. Since all these three Bmp genes are expressed in the developing tooth, their expression in tooth germ was therefore used as positive control for in situ hybridization assay. We found that Bmp2 is expressed in Meckel’s cartilage condensation at E11.5, and the expression becomes stronger at E12.5 (Fig. 2A, 2D). However, from E13.5 on, Bmp2 expression became undetectable in Meckel’s cartilage, though lower level of Bmp2 expression was observed in surrounding mandibular mesenchyme and mandibular bone (Fig. 2G, 2J, 2M). While Bmp4 expression was never observed in developing Meckel’s cartilage in all stages examined, we did detect Bmp4 transcripts in the surrounding mandibular mesenchyme and mandibular bone at E11.5 to E13.5 as well as E16.5 (Fig. 2B, 2E, 2H, 2K, and 2N). Bmp7 expression was found in Meckel’s cartilage and surrounding mandibular mesenchyme at relatively lower level at E11.5 and E12.5 (Fig. 2C, 2F). However, at E13.5, Bmp7 expression disappeared, but was detected in the adjacent forming mandibular bone (Fig. 2I). At following E14.5 and E16.5 stages, Bmp7 expression was not found in Meckel’s cartilage as well as surrounding tissues (Fig. 2L, 2O). These results reveal dynamic expression patterns of these Bmp genes and implicate potential role of BMP signaling in Meckel’s cartilage development.
Figure 1. Expression of Noggin in developing Meckel’s cartilage.
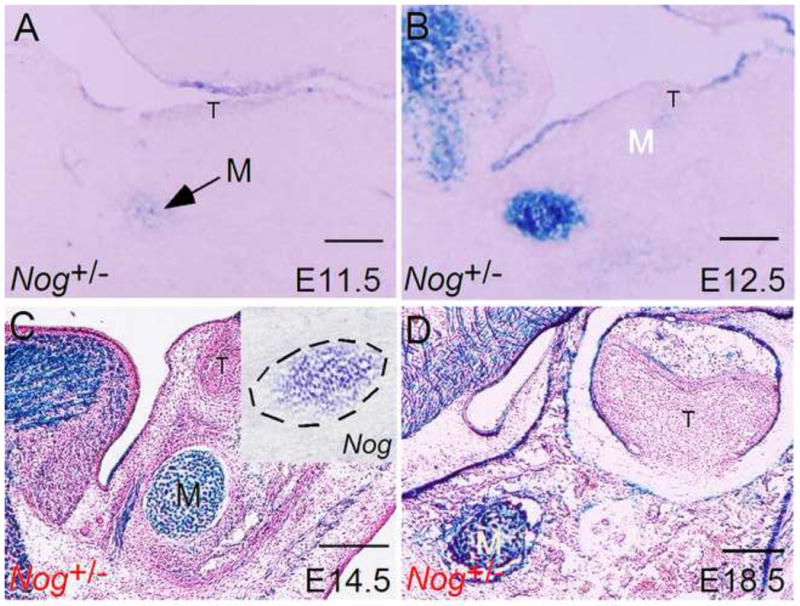
Noggin expression, as assessed by LacZ activity, is seen in the initial mesenchymal condensation of Meckel’s cartilage at E11.5 (A), and in Meckel’s cartilage through subsequent stages of embryogenesis (B–D). Insert in (C) shows in situ hybridization detection of Noggin expression in Meckel’s cartilage M, Meckel’s cartilage. Scale bar = 100-μm.
Figure 2. Expression of Bmp2, Bmp4, and Bmp7 in developing Meckel’s cartilage.

(A–C) In situ hybridization shows Bmp2 and Bmp7 but not Bmp4 expression in mesenchymal condensation of Meckel’s cartilage at E11.5. Note Bmp2 expression in the dental epithelium (arrow in panel A) and Bmp4 expression in the dental mesenchyme (arrow in panel B). (D–F) At E12.5, strong Bmp2 expression and relatively weaker Bmp7 expression but not Bmp4 is detected in Meckel’s cartilage. In addition, all three Bmp genes are expressed in the surrounding mesenchymal tissues. (G–O) At E13.5 and subsequently developmental stages at E14.5 and E16.5, these Bmp genes are no longer expressed in Meckels’ cartilage but are expressed at lower levels in the surrounding mesenchymal tissues and developing mandibular bone. M, Meckel’s cartilage; T, tooth; Mb, mandibular bone; Sek, secondary enamel knot. Scale bar = 100-μm.
Lack of Noggin leads to enlargement and endochondral ossification of Meckel’s cartilage
In our and other’s previous studies, the presence of significantly thickened Meckel’s cartilage in Noggin-deficient embryos was noticed (He et al., 2010; Lana-Elola et al., 2011; Wang et al., 2011). Indeed, skeleton preparations of embryonic heads revealed enlarged craniofacial cartilages in the mutants at E14.5 including Meckle’s cartilage, nasal cartilage, basisphenoid, and presphenoid, as compared to the controls (Fig. 3A, 3B). At E18.5, similar to the control, the enlarged causal end and symphysis of Meckel’s cartilage in the mutants remained an unossified status (Fig. 3C–F). To determine the developmental process and fate of the middle portion of Meckel’s cartilage in Noggin mutants, we made detailed histological analysis. We found that mesenchymal condensations of Meckel’s cartilage in Nog−/− embryos formed at the same time as that in the control at E11.5, evidenced by the expression of Sox9 (Fig. 4A, 4B). Comparing to the controls, the Sox9 expression domain in the mutants appeared larger at this stage, suggesting recruitment of more cells to the blastema of mutant Meckel’s cartilage. At E12.5, Meckel’s cartilage in both controls and mutants became discernibly larger in diameter (Fig. 4C, 4D). At E14.5, the size of Meckel’s cartilage increased only slightly in wild type controls (Fig. 4E), but became significantly larger in mutants (Fig. 4F). At E16.5 when chondrocytes had become hypertrophic and degeneration began in control Meckel’s cartilage (Harada and Ishizeki, 1998; Sakakura et al., 2005), the mutant counterpart had undergone extensive endochondral ossification at the first molar level (Fig. 4G, 4H). However, at the posterior region of the middle portion, endochondral ossification had not yet begun at this stage (Fig. 4J). At E18.5 when degeneration of Meckel’s cartilage appeared obviously in controls (Fig. 4K), Meckel’s cartilage in Nog−/− mutants had almost ossified and integrated into mandibular bone (Fig. 4L). At this time, the posterior middle portion had also begun to ossify (Fig. 4N), indicating delayed chondrogenic differentiation and endochondral ossification in the posterior domain. These observations demonstrate that in the absence of Noggin, Meckel’s cartilage becomes dramatically enlarged, and the middle portion of Meckel’s cartilage continues chondrogenic differentiation process and eventually undergoes endochondral ossification instead of degeneration.
Figure 3. Enlarged craniofacial cartilages in in Noggin mutants.

(A, B) Skeleton preparations of control (A) and Nog−/− (B) heads at E14.5 reveal generally enlarged craniofacial cartilages in the mutants. (C, D) Skeletal staining of mandibular arches of E18.5 control (C) and mutant (D) shows enlarged but unossified caudal end (black arrows) of Meckel’s cartilage in mutant. (E, F) Coronal sections of E18.5 control (E) and Nog−/− mutant (F) reveal enlarged and unossified symphysis in the mutant. I, incisor; S, symphysis; BS, basisphenoid; MC, Meckel’s cartilage; NC, nasal cartilage; PS, presphenoid; Dnt, dentary. Scale bar = 2-mm (A–D); 500-μm (E, F).
Figure 4. Lack of Noggin leads to formation of enlarged Meckel’s cartilage and endochondral ossification.
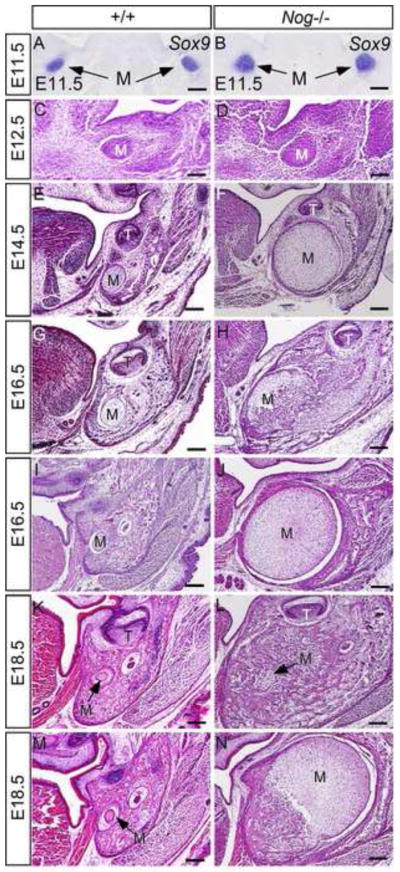
(A, B) In situ hybridization shows Sox9 expression in mesenchymal condensations of Meckel’s cartilage in control and Nog−/− embryos at E11.5. (C–F) Histological sections through the middle portion of Meckel’s cartilage of E12.5 and E14.5 control and Nog−/− embryos show enlarged Meckel’s cartilage in mutants. (G, H) Histological sections through the first molar level of mandible show Meckel’s cartilage with hypertrophic chondrocytes in E16.5 control mice (G) and Meckel’s cartilage in E16.5 Nog−/− mice that are undergoing endochondral ossification (H). (I, J) histological sections through the posterior part of middle portion of E16.5 control (I) and Nog−/− mutant (J) show enlarged Meckel’s cartilage in the mutant that has not yet started endochondral ossification process. (K–N) Coronal sections through the first molar region (K, L) and the posterior region (M, N) of the middle portion of Meckel’s cartilage of E18.5 control (K, M) and mutant (L, N) show degenerating Meckel’s cartilage in controls and almost completely ossified Meckel’s cartilage at the first molar level and Meckel’s cartilage undergoing endochondral ossification in the posterior region in mutants. M, Meckel’s cartilage; T, tooth; Scale bar = 100-μm.
To further confirm that Meckel’s cartilage in Nog−/− mice undergoes similar chondrogenic differentiation process as that in long bone formation, we examined the expression of chondrogenic differentiation markers at the first molar level: collagen type II (Col II), a marker for chondrocytes, Ihh, a marker for prehypertrophic chondrocytes, and collagen type X (Col X), a marker for hypertrophic chondrocytes. Our results showed that at E14.5, Col II expression was detected in Meckel’s cartilage in controls and Nog−/− mice (Fig. 5A, 5B). However, at this stage, expression of Ihh and Col X was also detected in Nog−/− mutants but was not in the controls (Fig. 5F, 5J), indicating that Meckel’s cartilage in the mutant had undergone chondrogenic differentiation. At E16.5, Col II expression became barely detectable in the controls (Fig. 5C), but was present in a few undifferentiated chondrocytes in the mutant (Fig. 5D). At this stage, Ihh and Col X expression was also absent in the control (Fig. 5G, 5K), despite the presence of hypertrophic chondrocytes (Harada and Ishizeki, 1998; Sakakura et al., 2005). This observation of lack of Col X expression in hypertrophic chondrocytes in the middle portion of Meckel’s cartilage is consistent with the previous report (Chung et al., 1995), suggesting that the cellular hypertrophy of Meckel’s cartilage is not necessarily associated with the expression of chondrogenic differentiation markers. In the mutant at this stage, while Ihh expression was no longer detectable, Col X was found in remaining chondrocytes that underwent final differentiation (Fig. 5J, 5L), consistent with histological evidence that the middle portion of Meckels’ cartilage in Nog−/− mutants has undergone extensive chondrogenic differentiation and endochondral ossification at this stage. To examine if there is an invasion of adjacent Noggin-negative mandibular mesenchymal cells into mutant Meckel’s cartilage that may function to resorb Meckel’s cartilage during its chondrogenic differentiation, we made LacZ staining on E14.5 mutant Meckel’s cartilage that had undergone chondrogenic differentiation. As shown in the insert in Fig. 5J, all cells in mutant Meckel’s cartilage are LacZ positive, suggesting a lack of invasion of cells from adjacent tissues.
Figure 5. Expression of chondrogenic differentiation markers developing Meckel’s cartilage of Nog−/− mutants.
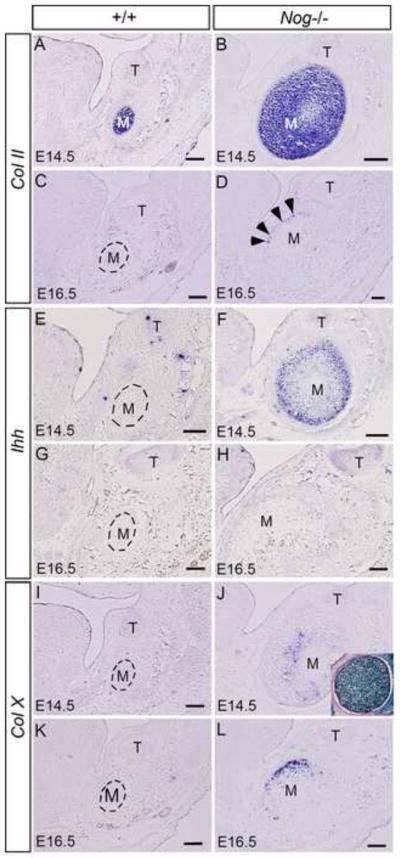
(A–D) Expression of Col II is seen in Meckel’s cartilage of wild type control and Nog−/− embryos at E14.5 (A, B), but is barely detectable in wild type Meckel’s cartilage at E16.5 (C). Col II expression is also observed in some undifferentiated chondrocytes (arrowheads) of Nog−/− Meckel’s cartilage at E16.5 (D). (E–H) The prehypertrophic chondrocyte marker Ihh is never expressed in control Meckel’s cartilage at E14.5 (E) and E16.5 (G). In contrast, Ihh is strongly expressed in Nog−/− Meckel’s cartilage at E14.5 (F) but not at E16.5 (H). (I–L) Col X expression is not detected in the middle portion of Meckel’s cartilage in wild type embryos at E14.5 (I) and E16.5 (K), but is seen in Nog−/− Meckel’s cartilage at both stages (J, L). Insert in (J) shows that all cells in E14.5 Nog−/− Meckel’s cartilage are LacZ-positive. M, Meckel’s cartilage; T, tooth. Scale bar = 100-μm.
Absence of Noggin leads to upregulated BMP signaling and elevated cell proliferation in Meckel’s cartilage
To determine if the absence of Noggin alters BMP signaling activity, we performed immunohistochemical staining to examine the expression of phosphorylated Smad1/5/8 (pSmad1/5/8). In wild type controls, at E12.5 when Bmp2 is strongly expressed in Meckel’s cartilage (Fig. 2), relatively abundant pSmad1/5/8 positive cells were detected (Fig. 6A). However, at E13.5 when Bmp genes are no longer expressed in Meckel’s cartilage (Fig. 2), the number of pSmad1/5/8 positive cells accordingly decreased dramatically (Fig. 6C). In contrast, as we expected, the number of pSmad1/5/8 positive cells increased significantly in Nog−/− Meckel’s cartilage compared to the controls at both stages (Fig. 6B, 6D). At E14.5, wild type Meckel’s cartilage remained a level of pSmad1/5/8 positive cells similar to that at E13.5, the amount of pSmad1/5/8 positive cells began to reduce (Fig. 6E, 6F).
Figure 6. Elevated pSmad activity in Nog−/− Meckel’s cartilage.

(A–F) Immunohistochemical staining show pSmad1/5/8 positive cells in control and Nog−/− Meckel’s cartilage. At E12.5, control Meckel’s cartilage has relatively high number of pSmad1/5/8 positive cells (A), but at E13.5 and E14.5, it contains only a few positive cells (C, E). In contrast, the number of pSmad1/5/8 positive cells increases significantly in Nog−/− Meckel’s cartilage at E12.5 (B) and E13.5 (D). However, at E14.5, the number of pSmad1/5/8 positive cells is reduced in Nog−/− Meckel’s cartilage (F). Scale bar = 100-μm.
We next conducted BrdU labeling experiment to determine if the enlarged Meckel’s cartilage is a consequence of increased cell proliferation rate. We found that cell proliferation rate was indeed increased significantly (P < 0.001) in mutants as compared with that in controls at E12.5 (Fig. 7A, 7B, 7G). However, at E14.5, we did not detect a significant difference in cell proliferation rates between controls and mutants (Fig. 7C, 7D, 7G), consistent with the reduced amount of pSmad1/5/8 positive cells in mutant Meckel’s cartilage. This observation could be attributed to the fact that the chondrocytes in mutant Meckel’s cartilage had begun to differentiate at this stage, as assessed by the expression of chondrogenic differentiation markers shown above. Elevated cell proliferation rate was also found in other cranial cartilages such as presphenoid (Fig. 7E, 7F, 7H), suggesting that elevated cell proliferation represents at least one of the mechanisms underlying the enlargement of cranial cartilages in Noggin mutants.
Figure 7. Lack of Noggin leads to elevated cell proliferation rate in Meckel’s cartilage.
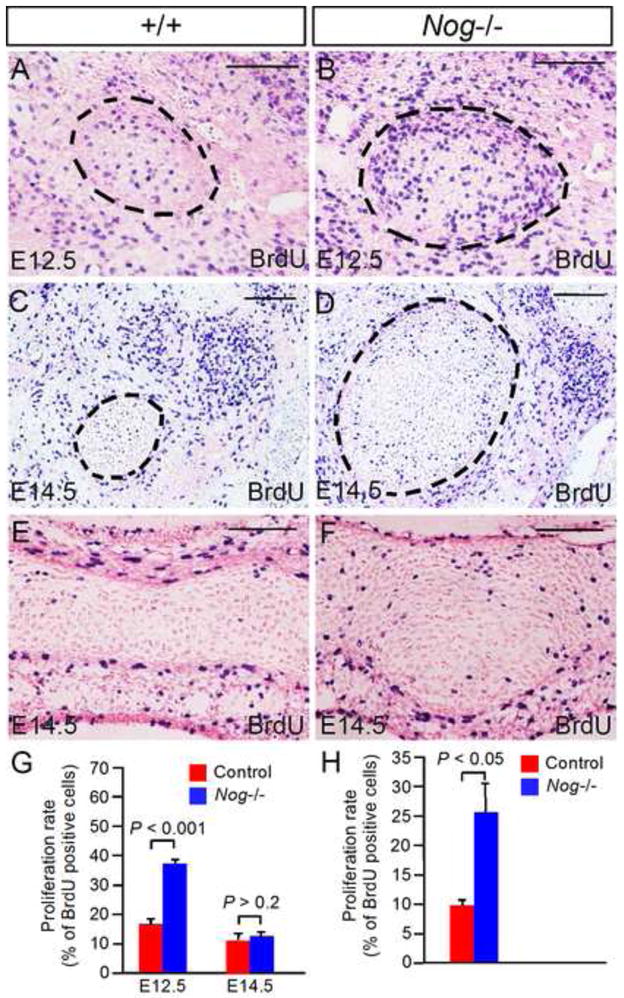
(A–D) Coronal sections show BrdU labeled cells in control and Nog−/− Meckel’s cartilage at E12.5 and E14.5. (E, F) Coronal sections show BrdU labeled cells in presphehoid of E14.5 control (E) and mutant F). (G) Statistical analysis shows significantly increased cell proliferation rate in Nog−/− Meckel’s cartilage at E12.5 (P < 0.001) but not at E14.5 (P > 0.2) as compared to controls. (H) Statistical analysis shows significantly increased cell proliferation rate (P < 0.05) in the presphenoid in E14.5 Nog−/− embryo as compared to that in control. Standard errors are indicated. Scale bar = 100-μm.
Activation of BMPRIa-mediated signaling in Meckel’s cartilage resembles effect of Noggin-deficiency
Because Bmp2 and Bmp7 are expressed in developing Meckel’s cartilage only at early stage (E11.5 and E12.5), but are no longer expressed there from E13.5 on, despite continuous expression of these two Bmps as well as Bmp4 in the surrounding mesenchyme and developing mandubular bones at late stage, we wondered if the withdrawal of BMP signaling in developing Meckel’s cartilage represents a critical step for the arrest of further chondrogenic differentiation and subsequent degeneration. To address this question, we generated conditional transgenic mice that expressed a constitutively active form of either BMPRIa (caBmprIa) or BMPRIb (caBmprIb) in chondrocyte lineage. The inducible transgenic allele Col2a1-CreERT2, upon administration of tamoxifen, gives rise to high specificity and strong efficiency of Cre recombination activity in chondrocytes including Meckel’s cartilage (Chen et al., 2007; Fig. 8A). We found that the expression of caBmprIb, activated by administration of tamoxifen at E12.5, did not produce a phenotype in Meckel’s cartilage (Fig. 8B; N = 5), despite previous evidence for the functional efficiency of this transgenic allele in rescuing tooth defects in mice lacking BmprIa and in causing severe ichthyosis-like skin phenotype in mice expressing this transgenic allele in the skin (Yu et al., 2010; Li et al., 2011). However, when caBmprIa was activated in chondrocytes after tamoxifen administration, enlarged Meckel’s cartilage was found in Col2a1-CreERT2;pMes-caBmprIa mice (Fig. 8C, 8D; N = 5). The enlarged Meckel’s cartilage also underwent endochondral differentiation, as assessed by the expression of Ihh and Col X, mimicking the phenotype observed in Nog−/− mice (Fig. 8E, 8F; N = 2).
Figure 8. Activation of BMPRIa-mediated BMP signaling in chondrocyte-lineage resembles Nog−/− Meckel’s cartilage phenotype.
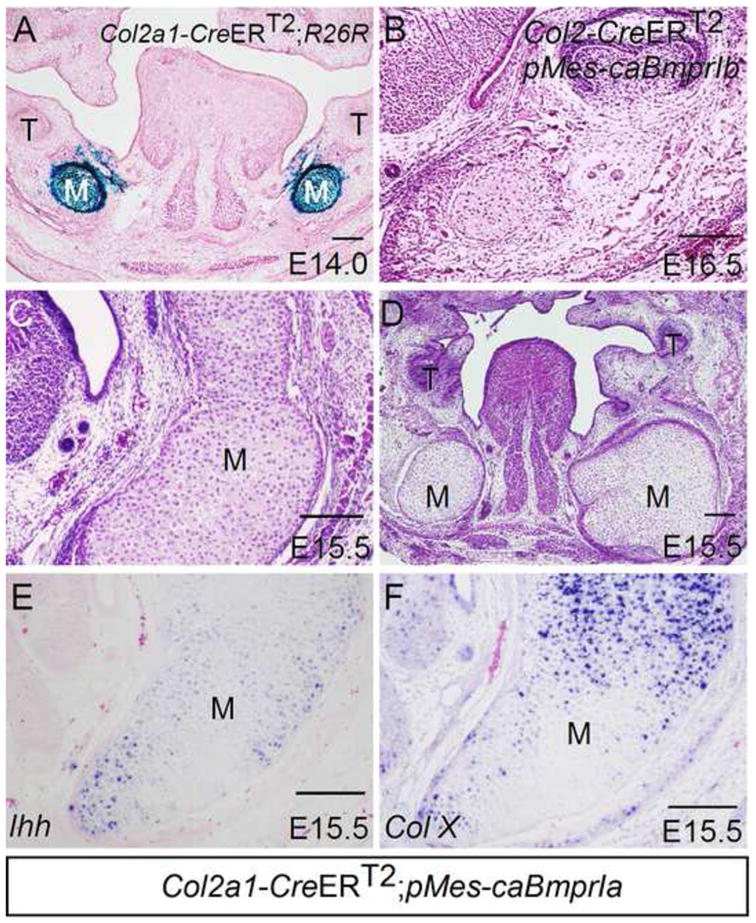
(A) X-gal staining shows Cre activity in chondrocytes of Meckel’s cartilage of an E14.0 Col2a1-CreERT2;R26R embryo that received an administration of tamoxifen at E12.5. (B) A coronal section of an E16.5 Col2a1-CreERT2;pMes-caBmprIb embryo shows unaffected Meckel’s cartilage. (C, D) Coronal sections E15.5 Col2a1-CreERT2;pMes-caBmprIa embryos show significantly enlarged Meckel’s cartilage. (E, F) In situ hybridization assay shows Ihh expression (E) and Col X expression (F) in enlarged Meckel’s cartilage of E15.5 Col2a1-CreERT2;pMes-caBmprIa embryo. M, Meckel’s cartilage; T, tooth. Scale bar = 100-μm.
Discussion
As a transient embryonic structure, Meckel’s cartilage disappears through different ultimate fates of the chondrocytes depending on their position (Bhaskar et al., 1953; Frommer and Margolies, 1971; Richman and Diewert, 1988; Harada and Ishizeki, 1998). It has been well established that the majority of Meckel’s cartilage, the middle portion, undergoes degeneration. Despite that the chondrocytes in this portion become hypertrophic, they do not express chondrogenic differentiation markers, such as Col X and Ihh (Chung et al., 1995; this study), suggesting an arrest of chondrogenic differentiation prior to degeneration. Previous studies have implicated multiple factors and cellular processes in degeneration and resorption of Meckel’s cartilage, including several matrix metalloproteinases (Ishizeki and Nawa, 2000; Sakakura et al., 2007a, 2007b), macrophages (Harada and Ishezeki, 1998; Sakakura et al., 2005; Tsuzurahara et al., 2011), and possible autophagy and apoptosis (Trichilis and Wroblewski, 1997; Yang et al., 2012). However, the underlying mechanism that is responsible for the arrest of chondrogenic differentiation and subsequent degeneration of Meckel’s cartilage remains unknown.
In this study, we investigated the role of BMP signaling in Meckel’s cartilage development using Nog−/− mice as a gain-of BMP function model. We focused our studies on the middle portion of Meckel’s cartilage because of its distinct ultimate fate. We found that in the absence of Noggin, which leads to elevated BMP signaling and cell proliferation rate, the fate of chondrocytes in this portion is changed. Meckel’s cartilage becomes enlarged and undergoes further chondrogenic differentiation and endochondral ossification. This phenotype is resembled by expression of a constitutively active form of BmprIa in chondrocyte lineage, indicating that suppression of BMP activity is essential for normal Meckel’s cartilage development and the ultimate fate of the chondrocytes. Interestingly, the caudal end and the symphysis of Meckel’s cartilage, both undergo endochondral differentiation eventually in wild type animals, also became enlarged in Noggin mutants. However, they exhibited similar chondrogenic differentiation status in the mutants at the end of gestation as compared to the controls, suggesting that loss of Noggin may not alter the fate of chondrocytes in these two compartments.
BMP signaling has been shown to play critical roles in multiple steps of chondrogenesis and endochondral ossification. In developing Meckel’s cartilage, Bmp2 and Bmp7 are expressed in the mesenchymal condensation and chondrocytes of Meckel’s cartilage at early stage, consistent with the role of BMP signaling in promoting differentiation of mesenchymal cells into chondrocytes and stimulating chondrocyte proliferation (Denker et al., 1999; Zehentner et al., 1999; Yoon et al., 2005). However, the expression of these two genes is abruptly inhibited in Meckel’s cartilage at E13.5, and never comes back. In accord with this inhibition of Bmp gene expression is the dramatically reduced BMP signaling activity in Meckel’s cartilage after this stage. The residual BMP signaling activity present in Meckel’s cartilage from E13.5 on could be the consequence of the expression of Bmp2, Bmp4, and Bmp7 in the surrounding mesenchyme and the developing mandibular bone. Since BMP signaling also induces/stimulates chondrocyte hypertrophy and is required for endochondral bone formation (Valcourt et al., 2002; Kobayashi et al., 2005; Retting et al., 2009), the suppression of Bmp expression appears to responsible for the arrest of chondrogenic differentiation and subsequent endochondral ossification. Interestingly, although cellular hypertrophy is seen in the chondrocytes of Meckel’s cartilage, these hypertrophic chondrocytes never express chondrogenic differentiation markers including Ihh and Col X, suggesting an abnormal hypertrophic process. It was shown previously that Col X expression is closely associated with the hypertrophic chondrocyte differentiation that undergoes endochondral ossification (Solursh et al., 1986). Thus the absence of Col X expression in Meckel’s cartilage could be responsible, at least partially, for the lack of endochondral ossification.
BMP signaling is known to be transduced by a type II receptor together with two originally identified type I receptors (BMPRIa and BMPRIb) as well as activin receptor type IA (ActRIa). These type I BMP receptors appear to have distinct but overlapping function during embryogenesis. In the developing chick limb, expression of caBmprIa but not caBmprIb promotes chondrogenic differentiation (Zou et al., 1997). Similarly in mice, expression of caBmprIa in chondrocytes accelerates chondrogenic differentiation in the axial and appendage skeletal elements (Kobayashi et al., 2005). Consistent with these previous findings, in our current study, we show that expression of caBmprIa but not caBmprIb in chondrocyte lineage induces formation of significantly enlarged Meckel’s cartilage and enhances chondrogenic differentiation, indicating that BMPRIa and BMPRIb have distinct function in regulating chondrogenesis of CNC-derived cells in mice. Interesting, tissue-specific inactivation of BmprIa in the CNC lineage did not affect the size of Meckel’s cartilage (Li et al., 2011), suggesting potential functional redundancy between BmprIa and other type I BMP receptors such as BmprIb whose knockout did not give rise to any obvious craniofacial defect (Baur et al., 2000; Yi et al., 2000). On the other hand, in the developing chicken face, mis-expression of either caBmprIa or caBmprIb similarly produced enlarged cranial cartilages including Meckel’s cartilage (Ashique et al., 2002), suggesting a species specific function of BMPRIB-mediated signaling in the regulation of chondrogenesis during craniofacial development. Nevertheless, these observations support the idea that despite different origins of germ layer and different species, BMPRIa-mediated signaling has a conserved role in promoting chondrogenesis.
In conclusion, in this study, we provide evidence that precisely regulated Bmp gene expression and BMP signaling activity is essential for normal development of the middle portion of Meckel’s cartilage and its ultimate chondrocyte fate. Withdrawal of BMP signaling leads to arrest of chondrogenic differentiation and subsequent degeneration of this portion of Meckel’s cartilage during embryogenesis. Elevation BMP signaling activity in the absence of Noggin or maintenance of BMP signaling activity by expression of caBmprIa enhances chondrogenic differentiation, leading to endochondral ossification of the middle portion of Meckel’s cartilage.
Research Highlights.
Bmp genes and Noggin are expressed in developing Meckel’s cartilage.
Noggin mutants exhibit enlarged craniofacial cartilages and Mekcel’s cartilage.
Nog−/− Meckel’s cartilage undergoes chondrogenic differentiation.
Elevated BMP signaling in phenocopies the fate of Nog−/− Meckel’s cartilage.
BMP signaling withdraw is required for normal Meckel’s cartilage development.
Acknowledgments
Y.W. was supported by a fellowship from the Fourth Military Medical University College of Stomatology, P.R. China. Y.Z. was supported by a fellowship from the Department of Health, Fujian Province, P.R. China. This work was supported by NIH grant R01DE17792 and R01DE14044 to Y.P.C.
Footnotes
The authors declare that no conflict of interest exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ashique AM, Fu K, Richman JM. Signaling via type IA and type IB bone morphogenetic protein receptors (BMPR) regulates intrmembranous bone formation, chondrogenesis and feather formation in the chicken embryo. Int J Dev Biol. 2002;46:243–253. [PubMed] [Google Scholar]
- Baur ST, Mai JJ, Dymecki SM. Combinatorial signaling through BMP receptor IB and GDF5: shaping the distal mouse limb and the genetics of distal limb diversity. Development. 2000;127:605–619. doi: 10.1242/dev.127.3.605. [DOI] [PubMed] [Google Scholar]
- Bernick S, Patek PQ. Postnatal development of the rat mandible. J Dent Res. 1969;48:1258–1263. doi: 10.1177/00220345690480062901. [DOI] [PubMed] [Google Scholar]
- Bhaskar SN, Weinmann JP, Schour I. Role of Meckel’s cartilage in the development and growth of the rat mandible. J Dent Res. 1953;48:398–410. doi: 10.1177/00220345530320031401. [DOI] [PubMed] [Google Scholar]
- Bi W, Deng JM, Behringer RR, de Crombrugghe B. Sox9 is required for cartilage formation. Nat Genet. 1999;22:85–89. doi: 10.1038/8792. [DOI] [PubMed] [Google Scholar]
- Brunet LJ, McMahon JA, McMahon AP, Harland RM. Noggin, cartilage morphogenesis, and joint formation in the mammalian skelen. Science. 1998;280:1455–1457. doi: 10.1126/science.280.5368.1455. [DOI] [PubMed] [Google Scholar]
- Chai Y, Mah A, Crohin C, Groff S, Bringas P, Jr, Le T, Santos V, Slavkin HC. Specific transforming growth factor-beta subtypes regulate embryonic mouse Meckel’s cartilage and tooth development. Dev Biol. 1994;162:85–103. doi: 10.1006/dbio.1994.1069. [DOI] [PubMed] [Google Scholar]
- Chen D, Zhao M, Mundy GR. Bone Morphogenetic Proteins. Growth Factors. 2004;22:233–241. doi: 10.1080/08977190412331279890. [DOI] [PubMed] [Google Scholar]
- Chen M, Lichtler AC, Sheu TJ, Xie C, Zhang X, O’Keefe RJ, Chen D. Generation of a transgenic mouse model with chondrocyte-specific and tamoxifen-inducible expression of Cre recombinase. Genesis. 2007;45:44–50. doi: 10.1002/dvg.20261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KS, Park HH, Ting K, Takita H, Apte SS, Kunoki Y, Nishimura I. Modulated expression of type X collagen in the Meckel’s cartilage with different developmental fates. Dev Biol. 1995;170:387–396. doi: 10.1006/dbio.1995.1224. [DOI] [PubMed] [Google Scholar]
- Denker AE, Haas AR, Nicoll SB, Tuan RS. Chondrogenic differentiation of murine C3H10T1/2 multipotential mesenchmal cells: I. Stimulation by bone morphogenetic protein-2 in high-density micromass culture. Differentiation. 1999;64:67–76. doi: 10.1046/j.1432-0436.1999.6420067.x. [DOI] [PubMed] [Google Scholar]
- Dixon AD. Prenatal development of the facial skeleton. In: Dixon AD, Hoyete DAN, Rönning O, editors. Fundamentals of craniofacial growth. CRC Press LLC; 1997. pp. 59–97. [Google Scholar]
- Frommer J, Margolies MR. Contribution of meckel’s cartilage to ossification of mandible in mice. J Dent Res. 1971;50:1260–1267. doi: 10.1177/00220345710500052801. [DOI] [PubMed] [Google Scholar]
- Groppe J, Greenwald J, Wiater E, Rodriguez-Leon J, Economides AN, Kwiatkowski W, Affolter M, Vale WW, Belmonte JC, Choe S. Structural basis of BMP signalling inhibition by the cystine knot protein Noggin. Nature. 2002;420:636–642. doi: 10.1038/nature01245. [DOI] [PubMed] [Google Scholar]
- Harada Y, Ishizeki K. Evidence for transformation of chondrocytes and site-specific resorption during the degradation of Meckel’s cartilage. Anat Embryol. 1998;197:439–450. doi: 10.1007/s004290050155. [DOI] [PubMed] [Google Scholar]
- He F, Xiong W, Wang Y, Matsui M, Yu X, Chai Y, Klingensmith J, Chen YP. Modulation of BMP signaling by Noggin is required for the maintenance of palatal epithelial integrity during palatogenesis. Dev Biol. 2010;347:109–121. doi: 10.1016/j.ydbio.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Wang Y, He F, Li L, Zheng Y, Zhang Y, Chen YP. Noggin is required for early development of murine upper incisor. J Dent Res. 2012;91:394–400. doi: 10.1177/0022034511435939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizeki K, Nawa T. Further evidence for secretion of matrix metalloproteinase-1 by Meckel’s chondrocytes during degradation of the extracellular matrix. Tissues Cell. 2000;32:207–215. doi: 10.1054/tice.2000.0106. [DOI] [PubMed] [Google Scholar]
- Ishizeki K, Takahashi N, Nawa T. Formation of the sphenomandibular ligament by Meckel’s cartilage in the mouse: possible involvement of epidermal growth factor as revealed by studies in vivo and in vitro. Cell Tissue Res. 2001;304:67–80. doi: 10.1007/s004410100354. [DOI] [PubMed] [Google Scholar]
- Ito Y, Bringas P, Jr, Mogharei A, Zhao J, Deng C, Chai Y. Receptor-regulated and inhibitory Smads are critical in regulating transforming growth factorβ-mediated Meckel’s cartilage development. Dev Dyn. 2002;224:69–78. doi: 10.1002/dvdy.10088. [DOI] [PubMed] [Google Scholar]
- Ito Y, Yeo JY, Chytil A, Han J, Bringas P, Jr, Nakajima A, Shuler CF, Moses HL, Chai Y. Conditional inactivation of Tgfbr2 in cranial neural crest causes cleft palate and calvarial defects. Development. 2003;130:5269–5280. doi: 10.1242/dev.00708. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Lyons KM, McMahom AP, Kronenberg HM. BMP signaling stimulates cellular differentiation at multiple steps during cartilage development. Proc Natl Acad Sci USA. 2005;102:18023–18027. doi: 10.1073/pnas.0503617102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lana-Elola E, Tylzanowski P, Takatalo M, Alakurtti K, Veistinen L, Mitsiadis TA, Graf D, Rice R, Luyten FP, Rice DP. Noggin null allele mice exhibit a microform of holoprosencephaly. Hum Mol Genet. 2011;20:4005–4015. doi: 10.1093/hmg/ddr329. [DOI] [PubMed] [Google Scholar]
- Li L, Lin M, Wang Y, Cserjesi P, Chen Z, Chen YP. BmprIa is required in mesenchymal tissue and has limited redundant function with BmprIb in tooth and palate development. Dev Biol. 2011;349:451–461. doi: 10.1016/j.ydbio.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J. TGFβ signaling in context. Nature Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon JA, Takada S, Zimmerman LB, Fan CM, Harland RM, McMahon AP. Noggin-mediated antagonism of BMP signaling is required for growth and patterning of the neural tube and somite. Genes Dev. 1998;12:1438–1452. doi: 10.1101/gad.12.10.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minina E, Wenzel HM, Kreschel C, Karp S, Gaffield W, McMahon AP, Vortkamp A. BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development. 2001;128:4523–4534. doi: 10.1242/dev.128.22.4523. [DOI] [PubMed] [Google Scholar]
- Mori-Akiyama Y, Akiyama H, Rowitch DH, de Crombrugghe B. Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc Natl Acad Sci USA. 2003;100:9360–9365. doi: 10.1073/pnas.1631288100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka K, Shum L, Takahashi I, Takahashi K, Ikura T, Dashner R, Nuckolls GH, Slavkin HC. Convergence of the BMP and EGF signaling pathways on Smad1 in the regulation of chondrogenesis. Int J Dev Biol. 1999;43:795–807. [PubMed] [Google Scholar]
- Retting KN, Song B, Yoon BS, Lyons KM. BMP canonical Smad signaling through Smad1 and Smad5 and Smad5 is required for endochondral bone formation. Development. 2009;136:1093–1104. doi: 10.1242/dev.029926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman JM, Diewert VM. The fate of Meckel’s cartilage chondrocytes in ocular culture. Dev Biol. 1988;129:48–60. doi: 10.1016/0012-1606(88)90160-1. [DOI] [PubMed] [Google Scholar]
- Sakakura Y, Tsuruga E, Irie K, Hosokawa Y, Nakamura H, Yajima T. Immunolocalization of receptor activator of nuclear factor-κB ligand (RANKL) and osteoprotegerin (OPG) in Meckel’s cartilage compared with developing endochondral bones in mice. J Anat. 2005;207:325–337. doi: 10.1111/j.1469-7580.2005.00466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakura Y, Hosokawa Y, Tsuruga E, Irie K, Yajima T. In situ localization of gelatinolytic activity during development and resorption of Meckel’s cartilage in mice. Eur J Oral Sci. 2007a;115:212–223. doi: 10.1111/j.1600-0722.2007.00447.x. [DOI] [PubMed] [Google Scholar]
- Sakakura Y, Hosokawa Y, Tsuruga E, Irie K, Nakamura M, Yajima T. contributions of matrix metalloproteinases toward Meckel’s cartilage resorption in mice: immunohistochemical studies, including comparisons with developing endochondral bones. Cell Tissue Res. 2007b;328:137–151. doi: 10.1007/s00441-006-0329-7. [DOI] [PubMed] [Google Scholar]
- Savostin-Asling I, Asling CW. Resorption of calcified cartilage as seen in Meckel’s cartilage of rats. Anat Rec. 1973;176:345–360. doi: 10.1002/ar.1091760310. [DOI] [PubMed] [Google Scholar]
- Shimo T, Kanyama M, Wu C, Sugito H, Billings PC, Abrams WR, Rosenbloom J, Iwamoto M, Pacifici M, Koyama E. Expression and roles of connective tissue growth factor in Meckel’s cartilage development. Dev Dyn. 2004;231:136–147. doi: 10.1002/dvdy.20109. [DOI] [PubMed] [Google Scholar]
- Solursh M, Jensen KL, Reiter RS, Schmid TM, Linsenmayer TF. Environmental regulation of type X collagen production by cultures of limb mesenchyme, mesectoderm, and sterna chondrocytes. Dev Biol. 1986;117:90–101. doi: 10.1016/0012-1606(86)90351-9. [DOI] [PubMed] [Google Scholar]
- Soriana P. Generalized LacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- St Amand TR, Zhang Y, Semina EV, Zhao X, Hu YP, Nguyen L, Murray JC, Chen YP. Antagonistic signals between BMP4 and FGF8 define the expression of Pitx1 and Pitx2 in mouse tooth-forming anlage. Dev Biol. 2000;217:323–332. doi: 10.1006/dbio.1999.9547. [DOI] [PubMed] [Google Scholar]
- Stottmann RW, Anderson RM, Klingensmith J. The BMP antagonist Chordin and Noggin have essential but redundant roles in mouse mandibular outgrowth. Dev Biol. 2001;240:457–473. doi: 10.1006/dbio.2001.0479. [DOI] [PubMed] [Google Scholar]
- Terao F, Takahashi I, Mitani H, Haruyama N, Sasamo Y, Suzuki O, Takano-Yamamoto T. Fibroblast growth factor 10 regulates cartilage formation during early mandibular morphogenesis in rats. Dev Biol. 2011;350:337–347. doi: 10.1016/j.ydbio.2010.11.029. [DOI] [PubMed] [Google Scholar]
- Trichilis A, Wroblewski J. Expression of p53 and hsp70 in relation to apoptosis during Meckel’s cartilage development in the mouse. Anat Embryol. 1997;196:107–113. doi: 10.1007/s004290050083. [DOI] [PubMed] [Google Scholar]
- Tsuzurahara F, Seota S, Kawawa T, Baba K, Nakamura M. The role of macrophages in the disappearance of Meckel’s cartilage during mandibular development in mice. Acta Histochem. 2011;113:194–200. doi: 10.1016/j.acthis.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Valcourt U, Gouttenoire J, Moustakas A, Herbage D, Mallein-Gerin F. Functions of transforming growth factor-beta family type I receptors and Smad proteins in the hypertrophic maturation and osteoblastic differentiation of chondrocytes. J Biol Chem. 2002;277:228–241. doi: 10.1074/jbc.M202086200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu C, Rohr J, Liu H, He F, Yu J, Sun C, Li L, Gu S, Chen YP. Tissue interaction is required for glenoid fossa development during temporomandibular joint formation. Dev Dyn. 2011;240:2466–2473. doi: 10.1002/dvdy.22748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, He F, Morikawa Y, Yu X, Zhang Z, Lan Y, Jiang R, Cserjesi P, Chen YP. Hand2 is required in the epithelium for palatogenesis in mice. Dev Biol. 2009;330:131–141. doi: 10.1016/j.ydbio.2009.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang RT, Zhang C, Liu Y, Zhou HH, Li ZB. Autophagy prior to chondrocyte cell death during the degeneration of Meckel’s cartilage. Anat Rec. 2012;295:734–741. doi: 10.1002/ar.22433. [DOI] [PubMed] [Google Scholar]
- Yi SE, Daluiski A, Pederson R, Rosen V, Lyons KM. The type I BMP receptor BMPRIB is required for chondrogenesis in the mouse limb. Development. 2000;127:621–630. doi: 10.1242/dev.127.3.621. [DOI] [PubMed] [Google Scholar]
- Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM. BmprIa and BmprIb have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci USA. 2005;102:5062–5067. doi: 10.1073/pnas.0500031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Espinoza-Lewis RA, Sun C, Li L, He F, Xiong W, Yang J, Wang A, Chen YP. Overexpression of constitutively active BMP-receptor-IB in mouse skin causes an ichthyosis-vulgaris-like disease. Cell Tissue Res. 2010;342:401–410. doi: 10.1007/s00441-010-1077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehentner BK, Dony C, Burtscher H. The transcription factor Sox9 is involved in BMP-2 signaling. J Bone Miner Res. 1999;14:1734–1741. doi: 10.1359/jbmr.1999.14.10.1734. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Wlodarczyk BJ, Niederreither K, Venugopalan S, Florez S, Finnell RH, Amendt BA. Fuz regulates craniofacial development through tissue specific responses to signaling factor. PLoS One. 2011;6(9):e24608. doi: 10.1371/journal.pone.0024608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yu X, Zhang Y, Geronimo B, Lovlie A, Fromm SH, Chen YP. Targeted misexpression of constitutively active BMP receptor-IB causes bifurcation and duplication and posterior transformation of digit in mouse limb. Dev Biol. 2000;220:154–167. doi: 10.1006/dbio.2000.9637. [DOI] [PubMed] [Google Scholar]
- Zimmerman LB, De Jesus-Escobar JM, Harland RM. The Spemann organizer signal noggin binds and inactivates bone morphogenetic protein 4. Cell. 1996;86:599–606. doi: 10.1016/s0092-8674(00)80133-6. [DOI] [PubMed] [Google Scholar]
- Zou H, Wieser R, Massagué J, Niswander L. Distinct roles of type I bone morphogenetic protein receptors in the formation and differentiation of cartilage. Genes Dev. 1997;11:2191–2203. doi: 10.1101/gad.11.17.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]