

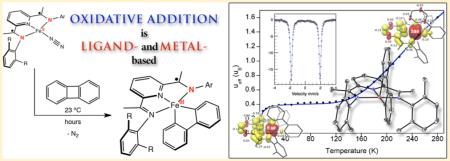
Abstract
Addition of biphenylene to the bis(imino)pyridine iron dinitrogen complexes, (iPrPDI)Fe(N2)2 and [(MePDI)Fe(N2)]2(μ2-N2) (RPDI = 2,6-(2,6-R2—C6H3— N=CMe)2C5H3N; R = Me, iPr), resulted in oxidative addition of a C—C bond at ambient temperature to yield the corresponding iron biphenyl compounds, (RPDI)Fe-(biphenyl). The molecular structures of the resulting bis-(imino)pyridine iron metallacycles were established by X-ray diffraction and revealed idealized square pyramidal geometries. The electronic structures of the compounds were studied by Mössbauer spectroscopy, NMR spectroscopy, magnetochemistry, and X-ray absorption and X-ray emission spectroscopies. The experimental data, in combination with broken-symmetry density functional theory calculations, established spin crossover (low to intermediate spin) ferric compounds antiferromagnetically coupled to bis(imino)pyridine radical anions. Thus, the overall oxidation reaction involves cooperative electron loss from both the iron center and the redox-active bis(imino)pyridine ligand.
INTRODUCTION
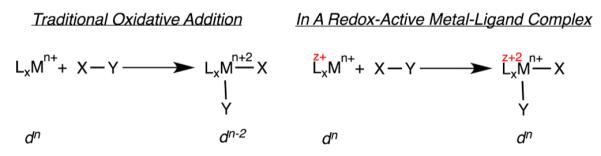
Oxidative addition is a fundamental transformation in organometallic chemistry that is often a key bond activation step responsible for introducing substrates into the metal coordination sphere during catalytic turnover.1 As exemplified by square planar, d8 metal compounds such as Vaska’s complex, (Ph3P)2Ir(CO)Cl, the traditional version of the reaction involves a formal two-electron oxidation at the metal (e.g., d8 to d6; Scheme 1).2
Scheme 1.
Oxidative Addition Reactions
Metal complexes with redox-active ligands, those where the ligands participate directly in the electronic structures of the molecules,3,4 have received renewed attention due to their interesting electronic structures, role in metalloenzymes,5,6 ability to promote unusual group transfer reactivity,7,8 and importance in base metal catalysis.9,10 Enabling redox chemistry at the supporting ligands potentially changes the paradigm for oxidative addition; electron loss could occur at the supporting ligands rather than the metal (Scheme 1). Heyduk and co-workers have provided an illustrative example of this concept with the formal addition of chlorine to a bis(amido)phenolate zirconium(IV) compound to furnish the corresponding dichloride and convincingly demonstrated that electron loss occurs at each of the chelating ligands.11
The potential economic and environmental advantages of iron have renewed interest in developing base metal catalysts as alternatives to precious metals.12–18 Aryl-substituted bis-(imino)pyridine iron dinitrogen compounds, (RPDI)Fe(N2)2 and [(RPDI)Fe(N2)]2(μ2-N2) (ArPDI = 2,6-(2,6-R2—C6H3— N=CMe)2 C5H3N; R = iPr, Et, Me), have emerged as an effective and versatile class of catalyst precursors for olefin and alkyne hydrogenation,19–23 intra-24,25 and intermolecular cyclization26 of unsaturated olefins as well as the regioselective, anti-Markovnikov hydrosilylation of olefins with tertiary silanes.27–29 Despite the high activity of these compounds in olefin hydrogenation and hydrosilylation, many of the mechanistic details surrounding catalytic turnover have yet to be elucidated. The now well-established redox activity of the bis(imino)pyridine chelate30,31 renders such studies more challenging as fundamental transformations such as oxidative addition and reductive elimination likely involve ligand-based redox events.32
Attempts to study the oxidative addition of catalytically relevant substrates such as dihydrogen or PhSiH3 to one of the most well-studied bis(imino)pyridine iron precatalysts, (iPrPDI)Fe(N2)2, furnished the iron σ-complexes, (iPrPDI)Fe-(η2-H2) and (iPrPDI)Fe(η2-PhSiH3)2, offering little direct information about the oxidative addition process.19 Repeating the reaction with D2 yielded (iPrPDI)Fe(η2-D2) with concomitant deuterium incorporation into the isopropyl methyl positions of the 2,6-aryl substituents, suggesting that a net oxidative addition–reductive elimination of C–H bonds was operative (Scheme 2).
Scheme 2.

Treatment of (iPrPDI)Fe(N2)2 with D2 and PhSiH3
Formal oxidative addition of carbon–heteroatom bonds has been observed with bis(imino)pyridine iron and cobalt dinitrogen complexes. Carbon–oxygen bond cleavage in diallyl ether and various esters has been identified as a catalyst deactivation pathway during iron-catalyzed [2π + 2π] cyclo-addition23 and olefin hydrogenation,33 respectively. In stoichiometric chemistry, addition of allyl ether to (iPrPDI)Fe(N2)2 produced an equimolar mixture of the iron allyl and iron alkoxide complexes, (iPrPDI)Fe(η3-CH2CHCH2) and (iPrPDI)-Fe(OCH2CH=CH2), respectively, demonstrating that oxidative chemistry can occur over two iron complexes (Scheme 3). Extension of this chemistry to alkyl and vinyl halides allowed synthesis of various bis(imino)pyridine iron alkyl complexes, including those with β-hydrogens.34 Elucidation of the electronic structures of both (iPrPDI1–)FeIIR (R = alkyl) and (iPrPDI1–)FeIIX (X = Cl, Br) classes of compounds established ferrous compounds with monoreduced, bis(imino)pyridine radical anions.
Scheme 3.

C–X Oxidative Addition to (iPrPDI)FeN2 and (MePDI)CoN2
Studies into the electronic structures of (iPrPDI)Fe(N2)2 and (iPrPDI)FeN2, two compounds in equilibrium in solution, have established redox non-innocent and redox-active bis(imino)pyridine chelates, respectively.35,36 The five-coordinate complex, (iPrPDI)Fe(N2)2, is a highly covalent molecule similar to (iPrPDI)Fe(CO)2, where the oxidation state of the iron is best described as a resonance hybrid between Fe(0) and Fe(II) with the bis(imino)pyridine ligand acting as a classical π-acceptor. This electronic structure description is also applicable to the dimeric bis(imino)pyridine iron dinitrogen compounds, [(RPDI)Fe(N2)]2(μ2-N2), for which there is no evidence for dissociation of N2 or formation of monomeric compounds in solution or the solid state.22 In this bonding situation, the bis(imino)pyridine is termed “redox non-innocent” in accord with Jørgensen’s original definition of a case where the covalency of the metal–ligand interaction creates ambiguity over the metal oxidation state.37
For the four-coordinate compound, (iPrPDI)FeN2, the iron is best described as an intermediate spin iron(II) center antiferromagnetically coupled to a triplet bis(imino)pyridine diradical dianion. Unlike (iPrPDI)Fe(N2)2, the oxidation state of the iron in (iPrPDI)FeN2 is unambiguously intermediate spin ferrous. Accordingly, we use the term “redox active” to distinguish this electronic structure from the description of (iPrPDI)Fe(N2)2, as the oxidation state of the iron in the four-coordinate compound is unambiguous.33 As the four-coordinate compound, (iPrPDI2–)FeIIN2, predominates in solution, the formal oxidative addition of alkyl halides may be viewed as a ligand based process. The triplet diradical bis(imino)pyridine in (iPrPDI2–)FeIIN2 undergoes a one-electron oxidation to yield (iPrPDI1–)FeIIR and (iPrPDI1–)FeIIX while the iron maintains the ferrous oxidation state.
In related chemistry with cobalt, Zhu and Budzelaar have reported the oxidative addition of aryl halides with (MePDI)-CoN2 to yield (MePDI)CoAr (Ar = substituted aryl) and (MePDI)CoX (X = Cl, Br, I)38 and have presented evidence for radical pathways.39 The electronic structure of the (RPDI)CoN2 family of compounds has been established as low-spin, d8 cobalt(I) derivatives with bis(imino)pyridine radical anions, e.g. (MePDI1–)CoIN2, with the SOMO of the complex essentially chelate-based.40 Because both (MePDI1–)CoIIAr and (MePDI1–)-CoIIX are known Co(II) compounds antiferromagnetically coupled to bis(imino)pyridine radical anions,41 the formal oxidative addition process is solely metal based. This behavior is opposite the oxidative addition process in iron chemistry as electron loss occurs solely at the cobalt center and demonstrates the flexibility of the redox-active bis(imino)pyridine chelate (to participate or not) to smoothly adjust to the electronic requirements of a metal complex and a specific redox process.
Although these studies are informative for understanding electron transfer processes in reduced iron and cobalt complexes with redox-active ligands, little is known about oxidative addition reactions relevant to catalytic hydrogenation, hydrosilylation, and hydrogenative cyclization reactions that occur at a single iron center. To circumvent complications from formation of σ-complexes, products with strong metal–carbon bonds were targeted as a method to study oxidative addition
 |
(1) |
reactions with (iPrPDI)Fe(N2)2 and related precatalysts. As amply demonstrated by Jones and co-workers,42 biphenylene is an attractive substrate for this purpose due to the presence of a relatively weak C–C bond (BDE = 65.4 kcal/mol) coupled to the formation of two strong metal–phenyl bonds. Accordingly, well-defined C–C oxidative events have been reported for iron,43 platinum,44 rhodium,45 and nickel.46 Here we describe the oxidative addition of the C–C bond of biphenylene to bis(imino)pyridine iron dinitrogen compounds to yield the corresponding iron metallacycles. These studies highlight the flexibility of the redox-active bis(imino)pyridine chelate to enable catalysis by mitigation of otherwise likely high energy Fe(IV) intermediates.
RESULTS AND DISCUSSION
Synthesis and Structure of Bis(imino)pyridine Iron Biphenyl Compounds
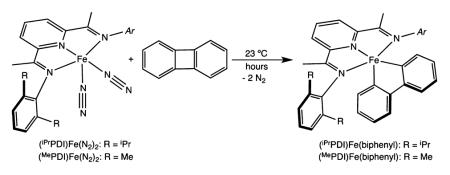
Addition of 1 equiv of biphenylene (per iron center) to a saturated pentane solution containing either (iPrPDI)Fe(N2)2 or [(MePDI)Fe(N2)]2(μ2-N2) at ambient temperature resulted in C–C bond cleavage and yielded the corresponding bis(imino)pyridine iron biphenyl compounds, (RPDI)Fe(biphenyl), as green powders (eq 1). Monomeric iron dinitrogen complexes are depicted in eq 1 for clarity, and all bis(imino)pyridine iron complexes are drawn in their traditional (formal oxidation state notation) form until the electronic structure determinations are presented.
The solid state structures of both (iPrPDI)Fe(biphenyl) and (MePDI)Fe(biphenyl) were determined by X-ray diffraction and are presented in Figure 1. Selected bond distances and angles are reported in Table 1. In both compounds, the overall molecular geometry is best described as idealized square pyramidal with the three nitrogen atoms of the bis(imino)pyridine and one of the carbon atoms of the biphenyldiyl ligand defining the basal plane. The other carbon of the metallacycle defines the apical position. The iron–carbon bond distances of Fe(1)–C(34) and Fe(1)–C(45) of 1.965(3) and 1.943(3) Å, respectively, in (iPrPDI)Fe(biphenyl) are shorter than the iron–carbon bond lengths of 2.062(3) and 2.054(3) Å, respectively, previously reported for (iPrPDI)Fe(CH2SiMe3)2, a molecule with an S = 2 ground state.27,47 In both (iPrPDI)Fe(biphenyl) and (MePDI)Fe(biphenyl), the bond distances of the bis-(imino)pyridine chelate are diagnostic of redox activity and key for indirectly assigning the metal oxidation state.29,39 In both complexes, the iron–nitrogen and iron–carbon bond lengths are suffciently contracted to eliminate a high spin state, and the elongation of the Nimine–Cimine and contraction of Cimine–Cipso distances are consistent with one-electron reduction and hence, a ferric oxidation state assignment.
Figure 1.

Solid state structures for (iPrPDI)Fe(biphenyl) (left) and (MePDI)Fe(biphenyl) (right) at 30% ellipsoids. Only one representative molecule (of two) from the asymmetric unit of the crystal containing (MePDI)Fe(biphenyl) is shown. All solvent molecules and hydrogen atoms are omitted for clarity. Data for (iPrPDI)Fe(biphenyl) collected at 173 K; data for (MePDI)Fe(biphenyl) collected at 100 K.
Table 1.
Selected Bond Distances (Å) and Angles (deg) for (iPrPDI)Fe(biphenyl) and (MePDI)Fe(biphenyl)
| (iPrPDI)Fe(biphenyl) | (MePDI)Fe(biphenyl) | |
|---|---|---|
| Fe(1)–N(1) | 1.959(2) | 1.9402(18) |
| Fe(1)–N(2) | 1.852(2) | 1.8569(16) |
| Fe(1)–N(3) | 1.988(2) | 1.9503(18) |
| Fe(1)–Cbasal | 1.965(3) | 1.9656(19) |
| Fe(1)–Capical | 1.943(3) | 1.9436(19) |
| N(1)–C(2) | 1.315(4) | 1.318(2) |
| N(3)–C(8) | 1.317(4) | 1.320(2) |
| C(2)–C(3) | 1.438(4) | 1.452(3) |
| C(7)–C(8) | 1.433(4) | 1.447(3) |
| N(1)–Fe(1)–N(2) | 79.41(9) | 79.80(7) |
| N(1)–Fe(1)–N(3) | 154.78(9) | 157.38(7) |
| N(1)–Fe(1)–Cbasal | 99.76(10) | 103.06(7) |
| N(1)–Fe(1)–Capical | 99.70(11) | 93.75(7) |
| N(2)–Fe(1)–N(3) | 79.46(10) | 80.11(7) |
| N(2)–Fe(1)–Cbasal | 175.09(12) | 175.93(8) |
| N(2)–Fe(1)–Capical | 92.47(12) | 92.47(7) |
| N(3)–Fe(1)–Cbasal | 104.21(10) | 97.57(7) |
| N(3)–Fe(1)–Capical | 94.81(11) | 97.39(7) |
NMR Spectroscopic Studies
The benzene-d6 1H NMR spectra of (iPrPDI)Fe(biphenyl) and (MePDI)Fe(biphenyl) at 23 °C exhibit a number of resonances consistent with paramagnetic Cs and C2v symmetric iron complexes, respectively. The resonances for each spectrum are shifted over an 86 (R = iPr) and 33 ppm (R = Me) range. The significantly broader resonances of (MePDI)Fe(biphenyl) compared to (iPrPDI)Fe(biphenyl), in combination with the higher C2v symmetry, suggest that the less sterically congested bis(imino)pyridine iron complex allows for rapid rocking of the biphenyl group through the iron-chelate plane at 23 °C. Similar behavior has been reported for five-coordinate compounds with neutral ligands such as (iPrPDI)Fe(N2)2 and (iPrPDI)Fe(CO)2 where C2v symmetry is observed by NMR spectroscopy although distorted square pyramidal structures are observed in the solid state.19 Cooling a toluene-d8 solution of (MePDI)Fe(biphenyl) to 0 °C resulted in a sharpening and increase in the number of the observed resonances. Under these conditions, (MePDI)Fe-(biphenyl) exhibited the number of resonances consistent with a molecule of Cs symmetry over a range of 85 ppm, demonstrating the static limit, analogous to the 23 °C spectrum of (iPrPDI)Fe(biphenyl). Variable temperature 1H NMR experiments for both compounds are reported in the Supporting Information (Figures S1 and S2).
Kinetic Stability of (RPDI)Fe(biphenyl) Compounds
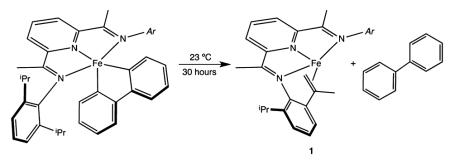
The kinetic stability of the (RPDI)Fe(biphenyl) compounds was investigated as it impacts the ability to isolate and handle pure material used for subsequent spectroscopic studies. Benzene-d6 solutions of (MePDI)Fe(biphenyl) proved to be indefinitely stable at 23 °C under an inert atmosphere. By contrast, allowing a benzene-d6 solution of (iPrPDI)Fe-(biphenyl) to stand for 30 h at 23 °C resulted in liberation of a stoichiometric quantity of biphenyl along with formation of the previously reported bis(imino)pyridine iron intramolecular olefin complex arising from dehydrogenation of the isopropyl aryl substituents (eq 2).48
 |
(2) |
The transfer dehydrogenation reaction, previously observed in the context of bis(imino)pyridine iron-catalyzed enyne and diyne cyclizations,24 was also confirmed by deuterium labeling experiments. Allowing (iPrPDI*)Fe(biphenyl) (* indicates deuteration of the isopropyl methyl groups) to stand in benzene-d6 yielded 2-d1-biphenyl along with isotopologues of 1. Analysis of the liberated biphenyl by 1H and 2H NMR spectroscopy established exclusive deuterium incorporation into the 2-position of the arene. Complete conversion to products required 96 h at 23 °C, consistent with a normal primary kinetic isotope effect on transfer dehydrogenation, similar to observations in related enyne chemistry.24 Because the iron product, 1, is NMR silent and has a triplet ground state, its formation, which is kinetically competitive during preparation of (iPrPDI)Fe(biphenyl), must be taken into account when conducting spectroscopic or magnetic measurements on the starting iron compound.
Magnetochemistry
The observation of paramagnetic compounds by NMR spectroscopy prompted more detailed investigations into the magnetic behavior of (iPrPDI)Fe-(biphenyl) and (MePDI)Fe(biphenyl). Ambient temperature (23 °C) measurements using a magnetic susceptibility balance yielded effective magnetic moments of 1.5 and 2.0 μB for (iPrPDI)Fe(biphenyl) and (MePDI)Fe(biphenyl), consistent with S = 1 molecules. Similarly, effective moments of 1.1 ((iPrPDI)Fe(biphenyl)) and 1.7 μB ((MePDI)Fe(biphenyl)) were determined by the method of Evans in benzene-d6 solution at 23 °C.
Variable temperature measurements were also conducted on both compounds using SQUID magnetometry. The temperature dependence of the magnetic moment for (MePDI)Fe-(biphenyl) is presented in Figure 2. This compound was selected for initial study due to its kinetic stability. At temperatures below 120 K, the magnetic moment plateaued at 0.4 μB. The low temperature data were modeled for a diamagnetic iron compound with 200 × 10−6 cm3/mol of temperature independent paramagnetism containing 0.5% of an unknown S = 5/2 paramagnetic impurity. At temperatures above 120 K, a sharp rise in the magnetic moment is observed, reaching a value of 1.6 μB at 300 K. The overall temperature dependence of the magnetic moment is consistent with a spin crossover (SCO) from an S = 0 to S = 1 ground state and was modeled using the Sorai and Seki domain model, where the high spin fraction is determined by x = 1/[1 + exp{(nΔH/R)(1/T – 1/Tc)}].49,50 Although the S = 1 state is not fully resolved, the high transition temperature of 352 K (transition width/enthalpy, nΔH = 805 cm−1) resulting from the model supports an incomplete population of the higher spin state at 300 K. The low temperature increase in magnetic moment (below 20 K) was modeled with a Weiss temperature (θ) = −5 K in the paramagnetic impurity and likely results from the influence of zero field splitting and magnetization saturation. Because the high spin state is experimentally not fully resolved, zero field splitting (D) = 0 and g = 2 were constrained as they cannot be properly determined. However, a change in the magnitude of either of these parameters would not alter the conclusions regarding the initial and final spin states: (MePDI)Fe(biphenyl) is an S = 0 to S = 1 spin crossover compound. Similar SCO behavior has been observed with bis(imino)pyridine iron imido (S = 0 to S = 1)51 and bis(chelate) bis(imino)pyridine cobalt (S = 1/2 to S = 3/2) compounds.52 The variable temperature magnetic data observed for (iPrPDI)Fe(biphenyl) displays the same general trends as for (MePDI)Fe(biphenyl) (see Supporting Information) but the instability of the compound (vide supra) made collection of reliable magnetic data more challenging. In an attempt to circumvent these issues and to evaluate the influence of impurities, all magnetic samples were analyzed by Mössbauer spectroscopy prior to SQUID, MSB, or Evans measurements. Representative data are reported in the Supporting Information.
Figure 2.
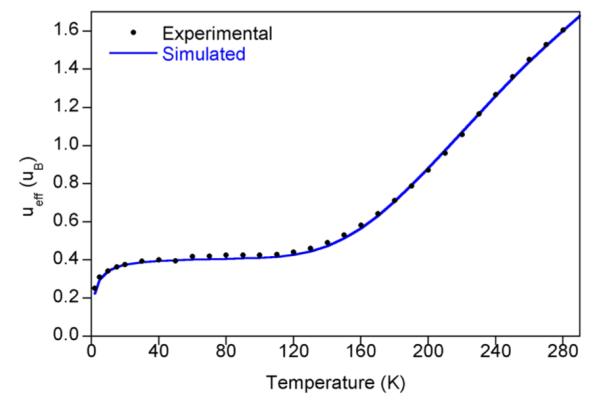
Variable temperature SQUID magnetization data for (MePDI)Fe(biphenyl) from 4 to 300 K. Data (black dots) are corrected for underlying diamagnetism, and the simulation (blue line) depicts a phase transition from an S = 0 to S = 1 state (as fit using the Sorai and Seki domain model)49,50 with a transition temperature of 352 K and enthalpy, nΔH = 805 cm−1. The model also contains 0.5% of a paramagnetic impurity with θ = −5 K.
Mössbauer Spectroscopic Studies
The electronic structures of (iPrPDI)Fe(biphenyl) and (MePDI)Fe(biphenyl) were also studied using zero- and applied-field 57Fe Mössbauer spectroscopy. Experimentally determined isomer shifts (δ) and quadrupole splittings (ΔEQ) are reported in Table 2; representative spectra are presented in Figure 3. Also included in Table 2 are the values of δ and ΔEQ for several other bis(imino)pyridine iron compounds for comparison. The diamagnetic iron dinitrogen complexes, (iPrPDI)Fe(N2) and (iPrPDI)Fe(DMAP),31 were chosen as intermediate spin ferrous (SFe = 1) compounds with two-electron reduced bis(imino)pyridine chelates (SPDI = 1). The iron imido, (iPrPDI)Fe=N(Dipp) (Dipp = 2,6-iPr2–C6H3),32 and dialkyl, (iPrPDI)Fe-(CH2SiMe3)2,28 complexes were chosen as molecules with Fe(III) oxidation states and one-electron reduced chelates. It should be noted that the imido was characterized as an intermediate spin Fe(III) compound, (SFe = 3/2), while the dialkyl was determined to be high spin (SFe = 5/2).
Table 2.
Zero-Field 57Fe Mössbauer Spectroscopic Parameters for Bis(imino)pyridine Iron Compounds Relevant to This Study (Data Collected at 80 K)
Figure 3.
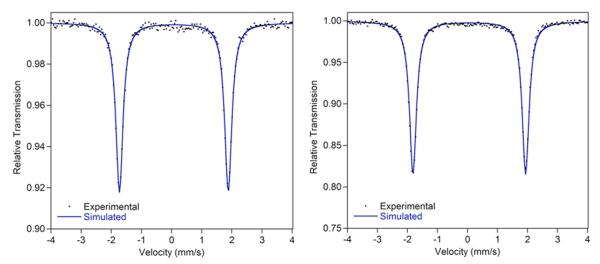
Zero-field 57Fe Mössbauer spectra of (iPrPDI)Fe(biphenyl) (left) and (MePDI)Fe(biphenyl) (right). Data collected at 80 K.
The isomer shifts of the (RPDI)Fe(biphenyl) compounds, 0.07 (R = iPr) and 0.05 mm/s (R = Me), are lower than those of the known intermediate spin ferrous derivatives, (iPrPDI)-Fe(N2) and (iPrPDI)Fe(DMAP), and the intermediate and high spin ferric species, (iPrPDI)Fe=N(Dipp) and (iPrPDI)Fe-(CH2SiMe3)2, respectively. The values of δ are comparable to that measured for (iPrPDI)Fe(CO)2, a highly covalent molecule that is best described by (iPrPDI2–)FeII(CO)2 and (iPrPDI0)-Fe0(CO)2 resonance forms and the alkyl-substituted iron imido complex, (iPrPDI)FeN1Ad, an established spin crossover compound.51
57Fe Mössbauer spectroscopy has been applied to detect spin crossover behavior in iron coordination compounds.53 The most well-studied examples are octahedral complexes of iron(II) where the spectral parameters of the low- and high-spin states are different and the time scale of the experiment (10−7 s) allows detection of the two different spin isomers.49,53 In bis(imino)pyridine chemistry, Mössbauer spectroscopy distinguished the low- and high-spin states of (iPrPDI)FeN2Ad and allowed determination of the energetics of the spin transition.51 To probe whether such behavior could be detected in organometallic iron complexes, variable temperature data were collected on both (iPrPDI)Fe(biphenyl) and (MePDI)Fe-(biphenyl). For the former compound, spectra were obtained at 10, 80, 150, 200, and 250 K. The parameters obtained at 250 K (δ = 0.01 mm/s and ΔEQ = 3.51 mm/s) are indistiguishable from the values (δ = 0.07 mm/s and ΔEQ = 3.58 mm/s) obtained at 80 K. Similar behavior was observed with (MePDI)Fe(biphenyl), where parameters of δ = −0.02 mm/s and ΔEQ = 3.43 mm/s were measured at 295 K indistiguishable from the values of δ = 0.05 mm/s and ΔEQ = 3.69 mm/s at 80 K. Representative spectra for both compounds are presented in the Supporting Information.
Although SCO behavior has been detected in Fe(III) compounds previously,55 the differences in isomer shift and quadrupole splitting between the low and high spin forms may be suffciently small to be differentiated by the experiment. As will be described in the Computational Studies section, the predicted parameters for the S = 1 state of (iPrPDI)Fe-(biphenyl) are δ = 0.18 mm/s and ΔEQ = 3.70 mm/s. These values, when considering the accepted errors in computed Mössbauer parameters,56 are indistinguishable from the values predicted (and observed) at 80 K.
To determine the sign of the quadrupole splitting and gain additional insight into the low temperature magnetic ground state, applied field Mössbauer measurements were made from 1 to 7 T at 4.2 and 120 K using (iPrPDI)Fe(biphenyl) as a representative example. The spectra exhibit characteristic magnetic splitting upon the application of an external field (Figure 4) and were modeled using an S = 0 ground state and confirmed the diamagnetism of the molecule at low temperatures. Simulation of the data yielded an isomer shift of 0.07 mm/s, a positive quadrupole splitting, ΔEQ = +3.58(2) mm/s, and an asymmetry parameter for the electric field gradient, η = 0.45(5). The large and positive quadrupole splittings for (RPDI)Fe(biphenyl) (the value for (MePDI)Fe(biphenyl) is likely also positive) indicate a large asymmetric field gradient along the z-axis.53
Figure 4.

Applied field 57Fe Mössbauer spectra of (iPrPDI)Fe-(biphenyl) recorded at 1 T, 4.2 K (bottom); 4 T, 4.2 K; 7 T, 4.2 K; 7 T, 120 K (top).
X-ray Absorption and Emission Studies
To gain additional insight into the oxidation and spin state of the iron, (iPrPDI)Fe(biphenyl) was also studied by X-ray Absorption Spectroscopy (XAS). The pre-edge region of an XAS spectrum (~7110–7116 eV) is a well-known indicator of oxidation state, with shifts of ~1 eV occurring for each one-electron oxidation event at the iron center,57,58 and this technique has been previously used to aid in the understanding of the electronic structure of bis(imino)pyridine compounds.36,51 Here, at low (10 K) and high (298 K) temperatures, XAS is also applied to provide a second experimental probe of the SCO behavior which was demonstrated by the SQUID data but was invisible to the zero-field Mössbauer experiment. As XAS is a molecular technique, it potentially eliminates the detection of bulk magnetic effects and is less sensitive to impurities than traditional magnetic measurements. Previous studies of light,59 magnetic field,60 and temperature61–63 induced SCO events in iron compounds have demonstrated the sensitivity of XAS for detecting small electronic structural differences in SCO compounds. It is generally observed that, upon increasing spin state, the rising edge shifts to lower energy due to longer metal–ligand bonds, which make the iron center appear “more reduced.” In the pre-edge region, a decrease in pre-edge intensity is observed for the higher spin state, consistent with longer metal–ligand bonds for the higher spin state, which results in a decrease in covalently mediated metal 3d–4p mixing.62–64
XAS data for (iPrPDI)Fe(biphenyl) were obtained at 10, 35–43, 57–66, and 298 K (Figure 5). The spectra up to 66 K all overlay, and the data are presented together with the Fe K-edge spectrum of (iPrPDI0)FeIICl2, an established five-coordinate, high spin ferrous compound.30 At 10 K, the 1s to 3d pre-edge feature appears at 7111.8 eV for (iPrPDI)FeIICl2 while (iPrPDI)Fe(biphenyl) exhibits two pre-edge features with similar intensities at 7111.5 and 7112.5 eV (Figure 5b), consistent with a ferric oxidation state. The pre-edge of (iPrPDI)Fe(biphenyl) is 0.7–1 eV lower in energy than known Fe(IV) compounds,57,65 suggesting that oxidative addition of biphenylene to (iPrPDI2–)FeIIN2 was not exclusively iron-based with retention of the dianionic chelate. Instead, the rising edge (at ~7121.5 eV) of (iPrPDI)Fe(biphenyl) is also consistent with a ferric rather than ferrous oxidation state and confirms participation of the bis(imino)pyridine chelate in the overall electronic structure and in the course of the oxidative addition reaction.
Figure 5.
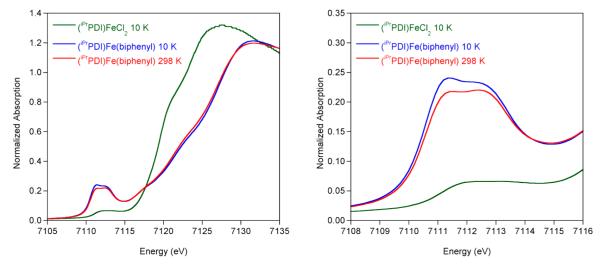
Comparison of the normalized Fe K-edge XAS spectra for (iPrPDI)FeCl2 and (iPrPDI)Fe(biphenyl). The data for the latter were collected at 10 (blue) and 298 (red) K. The data for (iPrPDI)FeCl2 were taken from ref 36. See Supporting Information for figures detailing the full set of experiments.
Variable temperature X-ray emission spectroscopy (XES) studies were also performed to study the spin crossover behavior of (iPrPDI)Fe(biphenyl). X-ray emission spectroscopy detects emission from the relaxation of a 3p electron into a 1s hole created by excitation of the core electron and thus allows different electronic states to be accessed as compared to XAS. XES is of particular interest for SCO compounds because it is a molecular technique that is sensitive to the metal spin state. Additionally, XES of SCO compounds has not been widely explored,59a,62,66 and further studies are necessary to establish frameworks such that this method could be used for unknown systems, or systems for which not enough material can be isolated for traditional magnetic measurements. (iPrPDI)Fe-(biphenyl) offers a unique system for applying XES, in terms of both exploring the electronic structure of the compound and understanding what effect spin state changes in SCO compounds have upon spectral features in XES.
The main line (7030–7080 eV) of the spectrum, which includes the Kβ’ (~7040–7050 eV) and Kβ1,3 (~7055–7065 eV) features, contains the most information about the metal spin state (Figure 6, left).62,67 In low-spin compounds, a Kβ’ feature is absent but is present in high-spin derivatives (~7040–7050 eV).62,67 Similarly, the Kβ1,3 transition (7055–7065 eV) shifts 1–2 eV higher in energy for high-spin compounds and is dominated by 3p–3d exchange interactions with smaller contributions from 3p spin–orbit coupling.62,67 The energy splitting between the Kβ’ and Kβ1,3 is a result of the exchange interaction between 3p and 3d electrons and is therefore sensitive to the d-orbital occupancy and metal oxidation state.68–70 The Kβ2,5 and Kβ” feature, or valence to core region (V2C, 7080–7120 eV), results from a ligand np or ns to metal 1s transition and is predominantly sensitive to ligand identity. The V2C has also been shown to have a lower intensity for high-spin complexes as compared to low-spin complexes, and these trends have been confirmed in bis-(imino)pyridine iron compounds.36
Figure 6.

Variable temperature XES spectra of (iPrPDI)Fe(biphenyl) highlighting the main line (left) and V2C (right) at 100 K (blue) and 298 K (red).
As shown in Figure 6 (left), the spectrum of (iPrPDI)Fe-(biphenyl) at 100 K exhibits essentially no Kβ’ feature and is consistent with a low spin iron center at this temperature. This is in agreement with established parameters for diamagnetic bis(imino)pyridine iron compounds.34 Increasing the temperature to 298 K produced no significant change in the Kβ’ region. The Kβ1,3 shifts by ~0.4 eV to higher energy upon increasing temperature (from 7058.2 eV at 10 K to 7058.6 eV at 298 K), consistent with the presence of some higher spin state component at higher temperatures. Typical energies for the Kβ1,3 for low spin Fe(III) compounds are from 7057.8 to 7059.1 eV, and those for high spin Fe(III) compounds fall between 7060.0 and 7060.4 eV.67 The Kβ1,3 energies at both 10 and 298 K fall within the energy regime for low spin Fe(III) compounds and are clearly outside the range of high spin Fe(III) compounds. For bis(imino)pyridine Fe(II) compounds the Kβ1,3 energy difference for low spin versus intermediate spin compounds (not SCO) is 0.6–0.8 eV, suggesting approximately 50% conversion from low spin to intermediate spin (iPrPDI)Fe(biphenyl) at 298 K.
The V2C shows a slight shift (0.3 eV) to higher energy at 298 K but displays no significant changes in area. In a simple picture, one would expect a decrease in the V2C XES area upon increasing spin state due to longer metal–ligand bond lengths. Previous studies67 have shown that high-spin ferric complexes have approximately twice the intensity of low spin ferric complexes. However, in this case a smaller change is predicted, as only an intermediate spin ferric state is accessed. The observed shift and similar areas for the low spin and intermediate spin V2C XES spectra are further supported by calculations (vide infra). Hence the XES data are consistent with a diamagnetic ground state for (iPrPDI)Fe(biphenyl) which crosses to a higher spin state at higher temperatures (but is not fully accessed at 298 K). The data presented here indicate that the Kβ1,3 is sensitive to spin state changes of 0.4–0.6 μB.
Computational Studies
The electronic structures of the high and low spin forms of (iPrPDI)Fe(biphenyl) were investigated with full molecule DFT calculations. Geometry optimizations were initiated from the experimental solid-state structures, and calculations were performed with the ORCA program with the B3LYP functional as with other bis(imino)pyridine metal complexes.36,61 For computational expediency, XAS and XES calculations were performed using BP86. This functional has also shown better agreement with experimental spectra (as compared to B3LYP) and improves computational effciency.71,77 Unrestricted Kohn–Sham (UKS) calculations were performed for singlet and triplet ground states as well as the corresponding broken symmetry possibilities, BS(1,1) and BS(3,1). In the broken symmetry notation BS(m, n) describes a state in which there are m unpaired spin-up electrons and n unpaired spin-down electrons on separate fragments.72–74 Both the singlet and triplet calculations converged to the respective BS(1,1) and BS(3,1) broken symmetry solutions. A Restricted Kohn–Sham (RKS) solution was also evaluated.
The BS(3,1) solution was found to be the lowest in energy, whereas the BS(1,1) solution was 7.9 kcal higher in energy, and the RKS solution was the highest in energy by 12.5 kcal (Table 3). Although these results are opposite the experimental data, where the S = 0 ground state is preferred, DFT methods are known to erroneously favor higher spin states.75 It has been suggested that computed energy differences of HS – LS = −6 to 0 kcal or less are indicative of spin-crossover compounds.75 While our data are slightly outside this range, we believe the energy differences are most consistent with a spin-crossover compound where the DFT favors the higher spin state.
Table 3.
Experimental and Computed 57Fe Mössbauer Spectroscopic Parameters and Relative Computed Ground State Energies for (iPrPDI)Fe(biphenyl)
| exptl 80 K |
exptl 250 K |
BS(3,1) | BS(1,1) | RKS | |
|---|---|---|---|---|---|
| relative calcd energy (kcal) |
na | na | 0.00 | +7.91 | +12.47 |
| δ (mm/s) | 0.07 | 0.01 | 0.18 | 0.08 | 0.14 |
| ΔEQ (mm/s) | +3.58 | 3.51 | +3.70 | +2.64 | −2.98 |
| η | 0.45 | — | 0.42 | 0.26 | 0.88 |
The BS(1,1) geometry optimization reproduced experimental crystallographic parameters, although the metal–ligand bond distances are slightly elongated, as is common when using the B3LYP functional (Table 4).76 A qualitative molecular orbital (MO) diagram and spin density plot are presented in Figure 7. The BS(1,1) solution establishes a low spin iron(III) center that is antiferromagnetically coupled to a bis(imino)pyridine radical giving rise to a diamagnetic ground state. The computed Mössbauer parameters of δ = 0.08 mm/s, ΔEQ = +2.64 mm/s, and η = 0.26 are in good agreement with the 80 K experimental values of 0.08, +3.58, and 0.45, respectively, thereby validating the computational results. For bis(imino)pyridine iron compounds, computational quadrupole splittings often do not agree completely with experiment and are especially challenging for SCO compounds (errors of 1.00 mm/s are considered reasonable).55
Table 4.
Experimental and Computed Bond Lengths (Å) and Angles (deg) for (iPrPDI)Fe(biphenyl)
| exptl | BS(3,1) | BS(1,1) | RKS | |
|---|---|---|---|---|
| Fe(1)–N(1) | 1.959(2) | 2.188 | 2.057 | 2.048 |
| Fe(1)–N(2) | 1.852(2) | 1.916 | 1.889 | 1.879 |
| Fe(1)–N(3) | 1.988(2) | 2.179 | 2.050 | 2.047 |
| Fe(1)–C(34) | 1.965(3) | 1.986 | 1.987 | 1.978 |
| Fe(1)–C(45) | 1.943(3) | 2.032 | 1.951 | 1.961 |
| N(1)–C(2) | 1.315(4) | 1.311 | 1.316 | 1.307 |
| N(3)–C(8) | 1.317(4) | 1.313 | 1.318 | 1.307 |
| N(2)–C(3) | 1.360(4) | 1.369 | 1.363 | 1.355 |
| N(2)–C(7) | 1.362(4) | 1.369 | 1.364 | 1.355 |
| C(2)–C(3) | 1.438(4) | 1.457 | 1.457 | 1.465 |
| C(7)–C(8) | 1.433(4) | 1.456 | 1.455 | 1.465 |
| N(2)–Fe(1)–C(34) | 175.09(12) | 178.6 | 179.3 | 176.7 |
| N(2)–Fe(1)–C(45) | 92.47(12) | 98.9 | 96.4 | 92.0 |
Figure 7.

Qualitative molecular orbital diagram (left) and spin density plot (right) obtained from the BS(1,1) solution for (iPrPDI)Fe(biphenyl). Spin density plot obtained from a Löwdin population analysis of the BS(1,1) solution for (iPrPDI)Fe(biphenyl) (red = positive spin density, yellow = negative spin density). Total electron densities are Fe = +0.85, PDI = −0.78, biphenyl = −0.07.
The BS(3,1) solution also reproduces the experimentally determined crystallographic parameters within computational error, but with a slightly larger deviation from experiment than the BS(1,1) solution (Table 4). Figure 8 depicts a qualitative MO diagram and spin density plot for the BS(3,1) solution described by an intermediate spin iron(III) center antiferromagnetically coupled to a bis(imino)pyridine radical (S = 0.68) giving rise to the overall S = 1 spin state. Analogous to the BS(1,1) solution, the dyz iron orbital mediates antiferromagnetic coupling to the bis(imino)pyridine ligand. Upon increasing the spin state, an electron is promoted from the dxz orbital into the previously unoccupied dz2 orbital. The computed Mössbauer parameters of δ = 0.18 mm/s, ΔEQ = +3.70 mm/s, η = 0.42 are in excellent agreement with the experimental values of δ = 0.01 mm/s, ΔEQ = 3.51 mm/s determined at 250 K.
Figure 8.

Qualitative molecular orbital diagram (left) and spin density plot (right) obtained from the BS(3,1) solution for (iPrPDI)Fe(biphenyl). Spin density plot obtained from a Löwdin population analysis of the BS(3,1) solution for (iPrPDI)Fe(biphenyl) (red = positive spin density, yellow = negative spin density). Total electron densities are Fe = +2.65, PDI = −0.57, biphenyl = −0.08.
The computational results for (MePDI)Fe(biphenyl) were analogous to those obtained for (iPrPDI)Fe(biphenyl). The BS(3,1) solution was once again computed to be lowest in energy. The diamagnetic BS(1,1) solution was +7.2 kcal higher in energy, and the RKS solution was highest in energy by +13.1 kcal (see Supporting Information for bond distances and angles, MO diagrams, and spin density plots). The low spin calculation similarly converged to the BS(1,1) solution, and the intermediate spin calculation converged to the BS(3,1) solution. The computed Mössbauer parameters displayed similar correlations to experimental parameters, as in (iPrPDI)-Fe(biphenyl).
The XAS pre-edge was calculated using time-dependent DFT (TDDFT) with the BP86 functional on the geometry-optimized coordinates.36 This functional offers improved accuracy and speed over B3LYP and has proven successful for XAS calculations for coordination compounds, metalloenzymes, and bis(imino)pyridine compounds.36,58,77,78 Although experimentally observed transitions occur between states, simplified DFT models based on MO theory have been found to correlate well to experimentally observed features. The BP86 solution was compared to the B3LYP solution to ensure that the same electronic structure description was obtained. For the broken symmetry solutions, BP86 produced a more covalent solution, but the overall electronic structure description remained the same. The calculated XAS spectra for (iPrPDI)Fe(biphenyl) are presented in Figure 9. The BS(1,1) calculation contains two pre-edge features at 7111.1 and 7112.4 eV, which are in agreement with the experimentally observed pre-edge features at 7111.5 and 7112.5 eV. In a simplified molecular orbital model, the first feature is due to a transition to the dyz orbital, and the second feature is due to a transition to the dz2 orbital. Both features have significant bis(imino)pyridine contributions. The BS(3,1) calculation only displays one pre-edge feature at 7112.6 eV which has contributions from several transitions with dxz, dyz, and dz2 contributions. An increase in iron spin state from BS(1,1) to BS(3,1) is in agreement with the experimental data in which the intensity of the 7111.5 eV pre-edge feature decreases relative to the feature at 7112.5 eV.
Figure 9.

Experimental (top) and calculated (bottom) XAS spectra of (iPrPDI)Fe(biphenyl) using TD-DFT. A shift of 181.25 eV and broadening of 1.5 eV have been applied to the computed spectra.
XES calculations were performed using a simple one-electron approximation, as has been previously described and applied to a variety of systems.36 The calculated BS(1,1) and BS(3,1) XES spectra show similar intensities and features (see Supporting Information) which derive from various bis(imino)pyridine orbital combinations, although the BS(3,1) solution is shifted to slightly higher energy by 0.6 eV. The experimentally observed shift of 0.3 eV (between 100 and 298 K) suggests that approximately 50% conversion to the high spin state (intermediate spin Fe(III)) has been reached and supports the lack of a decrease in intensity upon accessing the higher spin state (see Supporting Information). The computed electronic structure descriptions for three different spectroscopic methods (Mössbauer, XAS, XES) are in agreement with experimentally observed spectra, lending support for the electronic structure description of a (iPrPDI−1)-Fe(low spin III)(biphenyl) S = 0 state at low temperatures which undergoes a spin transition to a (i P rPDI−1)-Fe(intermediate spin III)(biphenyl) S = 1 state at more elevated temperatures.
The elucidation of the electronic structure of the (RPDI)-Fe(biphenyl) compounds by spectroscopic, magnetochemical, structural, and computational studies allows for an accounting of the electron flow in the C–C oxidative addition from the corresponding iron dinitrogen precursors. As highlighted in Scheme 4, the oxidative addition to (iPrPDI)Fe(N2) is therefore comprised of both metal and ligand redox events. Both the iron and bis(imino)pyridine undergo concomitant one-electron oxidations resulting in overall two-electron cleavage of the C–C bond. Similar metal–ligand cooperativity in two-electron oxidation events are well-established in metalloenzyme chemistry, viz. cytochrome P-450.6 We note that intermediates, such as arene π-complexes,42 may also form on the reaction coordinate for oxidative addition and therefore influence the pathway of electron flow. However, we have not observed such intermediates and accordingly not deduced their electronic structures. Thus, the electron flow associated with oxidative addition in this work concerns only the transformation from reactants to products.
Scheme 4.
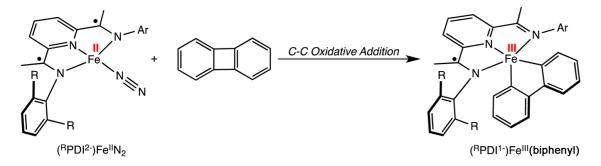
Electron Flow in C–C Activation by a Redox-Active Bis(imino)pyridine Iron Dinitrogen Compound
CONCLUDING REMARKS
Carbon–carbon oxidative addition of biphenylene has been observed at ambient temperature with bis(imino)pyridine iron dinitrogen compounds. The redox activity of the bis(imino)pyridine chelate raises the question as to whether the formal electron transfer events are metal- or ligand-based. Characterization of the resulting (RPDI)Fe(biphenyl) compounds by X-ray diffraction; zero-field and applied field 57Fe Mössbauer, XAS, and XES spectroscopies; and variable temperature SQUID magnetometry established spin crossover compounds arising from ferric centers antiferromagnetically coupled to ligand radical anions. At low temperature, diamagnetic ground states are observed and slightly higher energy S = 1 states are populated at increased temperatures. Accounting for the electron flow in the net two-electron C–C cleavage reaction involves cooperative one-electron oxidation at both the metal and bis(imino)pyridine ligand and avoids formation of presumably high energy iron(IV) compounds. Gaining a more detailed understanding of the nature of oxidative addition reactions with redox-active iron complexes also provides important insight into a fundamental transformation that likely constitutes a key substrate activation step in catalytic cycles and provides an important starting point for understanding the mechanism of turnover.
EXPERIMENTAL SECTION
Preparation of (iPrPDI)Fe(biphenyl)
Biphenylene (0.061 g, 0.40 mmol) was added to a 20 mL pentane solution containing 0.232 g of (iPrPDI)Fe(N2)2 (0.39 mmol) in pentane. The resulting slurry was stirred for 14 h after which time a green precipitate formed. The solid (0.110 g, 0.16 mmol) was collected by filtration. The supernatant was reduced to approximately half the original volume, stored at −35 °C, and yielded an additional 0.059 g (0.086 mmol) of a green powder. The combined mass of the isolated green solid was 0.169 g (62% yield) and was identified as (iPrPDI)Fe(biphenyl). Anal. Calcd for C45H51FeN3: C, 78.36; H, 7.45; N, 6.09. Found: C, 77.92; H, 7.28; N, 6.00. Magnetic susceptibility (Evans): μeff = 1.1 μB (benzene-d6, 23 °C). Magnetic susceptibility balance (22 °C): μeff = 1.5 μB. 1H NMR (benzene-d6) δ = 46.94 (40 Hz, 1H, p-pyr), 33.86 (43 Hz, 1H), 26.45 (24 Hz, 6H), 9.56 (20 Hz, 1H), 8.50 (22 Hz, 2H), 5.74 (22 Hz, 1H), 5.23 (19 Hz, 2H), 4.71 (110 Hz, 1H), 4.57 (23 Hz, 2H), 2.79 (18 Hz, 6H, CH(CH3)2), 2.16 (21 Hz, 6H, CH(CH3)2), 1.62 (6 Hz, 6H, CH(CH3)2), 0.57 (23 Hz, 6H, CH(CH3)2), −1.30 (24 Hz, 1H), −3.91 (30 Hz, 3H), −14.03 (38 Hz, 3H, C(CH3),), −15.21 (71 Hz, 1H), −21.79 (40 Hz, 1H), −39.02 (93 Hz, 1H).
Preparation of (MePDI)Fe(biphenyl)
A 20 mL scintillation vial was charged with biphenylene (0.096 g, 0.64 mmol), [(MePDI)Fe-(N2)]2(μ2-N2) (0.292 g, 0.312 mmol), and approximately 10 mL of pentane. The mixture was stirred for 18 h after which time a green precipitate formed. The mixture was cooled to −35 °C, and the solid was collected by filtration. The product was recrystallized from a mixture of toluene and diethyl ether at −35 °C to yield 0.199 g (55% yield) of a green solid identified as (MePDI)Fe(biphenyl). Anal. Calcd for C45H51FeN3: C, 76.95; H, 6.11; N, 7.28. Found: C, 76.71; H, 5.72; N, 7.05. Magnetic susceptibility (Evans): μeff = 1.7 μB (benzene-d6, 23 °C). Magnetic susceptibility balance (22 °C) μeff = 2.0 μB. 1H NMR (benzene-d6, 22 °C) δ = 51.94 (36 Hz, 1H), 37.98 (55 Hz, 6H), 5.32 (2,500 Hz), 3.72 (14 Hz, 6H), −19.11 (37 Hz, 2H).
Supplementary Material
ACKNOWLEDGMENTS
P.J.C. and K.W. thank the U.S. National Science Foundation and Deutsche Forschungsgemeinschaft for a Cooperative Activities in Chemistry between U.S. and German Investigators grant. S.D. acknowledges the Department of Chemistry and Chemical Biology at Cornell University, the Max Planck Society, the Sloan Foundation, and an ACS Petroleum Research Fund Grant (50270-DN13) for generous financial support. J.M.D. also acknowledges support from the National Institute of General Medical Sciences (Award Number T32GM008500). S.C.E.S. thanks the NSF for a graduate research fellowship (DGE-0646086) for support. S.P.S. thanks the Natural Sciences and Engineering Research Council of Canada for a predoctoral fellowship (PGS-D). This work is based, in part, upon research conducted at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the National Science Foundation and the National Institutes of Health/National Institute of General Medical Sciences under NSF Award DMR-0936384. Portions of the research were carried out at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of the DOE, BES. The SSRL SMB Program is supported by DOE, BER and NIH, NCRR, BMTP. We also thank Daigoro Hirai and Robert J. Cava for help in collecting additional SQUID measurements.
Footnotes
Supporting Information Additional experimental procedures, including general considerations, spectroscopic and computational details. Crystallographic data for (iPrPDI)Fe(biphenyl) and (MePDI)Fe(biphenyl) in cif format. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Hartwig J. Organotransition Metal Chemistry: From Bonding to Catalysis. University Science Books; Sausalito, CA: 2009. [Google Scholar]
- (2).Vaska L. Acc. Chem. Res. 1968;1:335. [Google Scholar]
- (3).Chirik P. J. Inorg. Chem. 2011;50:9737. doi: 10.1021/ic201881k. [DOI] [PubMed] [Google Scholar]
- (4) (a).Jørgensen CK. Coord. Chem. Rev. 1966;1:164. [Google Scholar]; (b) Pierpont CG. Coord. Chem. Rev. 2001;216:99. [Google Scholar]; (c) Evangelio E, Ruiz-Molina D. Eur. J. Inorg. Chem. 2005:2957. [Google Scholar]; (d) Ray K, Petrenko T, Wieghardt K, Neese F. Dalton Trans. 2007:1552. doi: 10.1039/b700096k. [DOI] [PubMed] [Google Scholar]; (e) De Bruin B, Hetterscheid DGH, Koekkoek AJJ, Grutzmacher H. Prog. Inorg. Chem. 2007;55:247–354. [Google Scholar]
- (5) (a).Stubbe J, Van der Donk WA. Chem. Rev. 1998;98:706. doi: 10.1021/cr980059c. [DOI] [PubMed] [Google Scholar]; (b) Jazdewski BA, Tolman WB. Coord. Chem. Rev. 2000;200–202:633. [Google Scholar]; (c) Lewis EA, Tolman WB. Chem. Rev. 2004;104:1047. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]; (d) Kaim W, Schwederski B. Coord. Chem. Rev. 2010;254:1580. [Google Scholar]
- (6).Rittle J, Green MT. Science. 2010;330:933. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- (7) (a).Heyduk AF, Zarkesh RA, Nguyen AI. Inorg. Chem. 2011;50:9849. doi: 10.1021/ic200911b. [DOI] [PubMed] [Google Scholar]; (b) Nguyen N, Zarkesh RA, Lacy DC, Thorson MK, Heyduk AF. Chem. Sci. 2011;2:166. [Google Scholar]; (c) Zarkesh RA, Ziller JW, Heyduk AF. Angew. Chem., Int. Ed. 2008;47:4715. doi: 10.1002/anie.200800812. [DOI] [PubMed] [Google Scholar]
- (8).King ER, Hennessy ET, Betley TA. J. Am. Chem. Soc. 2011;133:4917. doi: 10.1021/ja110066j. [DOI] [PubMed] [Google Scholar]
- (9).Chirik PJ, Wieghardt K. Science. 2010;327:794. doi: 10.1126/science.1183281. [DOI] [PubMed] [Google Scholar]
- (10).Blanchard S, Derat E, Desage-El Murr M, Fensterbank, Malacria M, Mouries-Mansuy V. Eur. J. Inorg. Chem. 2012:376. [Google Scholar]
- (11).Blackmore KJ, Ziller JW, Heyduk AF. Inorg. Chem. 2005;44:5559. doi: 10.1021/ic050997c. [DOI] [PubMed] [Google Scholar]
- (12).Bianchini C, Giambastiani G, Guerrero I, Mantovani G, Meli A, Segarra AM. Coord. Chem. Rev. 2006;250:1391. [Google Scholar]
- (13).Gaillard S, Renaud J-L. ChemSusChem. 2008;1:505. doi: 10.1002/cssc.200800071. [DOI] [PubMed] [Google Scholar]
- (14).Enthaler S, Junge K, Beller M. Angew. Chem., Int. Ed. 2008;47:3317. doi: 10.1002/anie.200800012. [DOI] [PubMed] [Google Scholar]
- (15).Bauer EB. Curr. Org. Chem. 2008;47:1341. [Google Scholar]
- (16).Bolm C, Legros J, Paith JL, Zani L. Chem. Rev. 2004;104:6217. doi: 10.1021/cr040664h. [DOI] [PubMed] [Google Scholar]
- (17).Chirik PJ. Chapter 4. In: Bullock RM, editor. Catalysis Without Precious Metals. Wiley-VCH; Weinheim: 2010. [Google Scholar]
- (18).Daida EJ, Peters JC. Inorg. Chem. 2004;43:7474. doi: 10.1021/ic0488583. [DOI] [PubMed] [Google Scholar]
- (19).Bart SC, Lobkovsky E, Chirik PJ. J. Am. Chem. Soc. 2004;126:13794. doi: 10.1021/ja046753t. [DOI] [PubMed] [Google Scholar]
- (20).Archer AM, Bouwkamp MW, Cortez M-P, Lobkovsky E, Chirik PJ. Organometallics. 2006;25:4269. [Google Scholar]
- (21).Trovitch RJ, Lobkovsky E, Bill E, Chirik PJ. Organometallics. 2008;27:1470. [Google Scholar]
- (22).Russell SK, Darmon JM, Lobkovsky E, Chirik PJ. Inorg. Chem. 2010;49:2782. doi: 10.1021/ic902162z. [DOI] [PubMed] [Google Scholar]
- (23).Yu RP, Darmon JM, Hoyt, Margulieux GW, Turner ZR, Chirik PJ. ACS Catal. 2012;2:1760. doi: 10.1021/cs300358m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bouwkamp MW, Bowman AC, Lobkovsky E, Chirik PJ. J. Am. Chem. Soc. 2006;128:13340. doi: 10.1021/ja064711u. [DOI] [PubMed] [Google Scholar]
- (25).Sylvester KT, Chirik PJ. J. Am. Chem. Soc. 2009;131:8772. doi: 10.1021/ja902478p. [DOI] [PubMed] [Google Scholar]
- (26).Russell SK, Lobkovsky E, Chirik PJ. J. Am. Chem. Soc. 2011;133:8858. doi: 10.1021/ja202992p. [DOI] [PubMed] [Google Scholar]
- (27).Tondreau AM, Atienza CCHA, Weller KJ, Nye SA, Lewis KM, Delis JGP, Chirik PJ. Science. 2012;335:567. doi: 10.1126/science.1214451. [DOI] [PubMed] [Google Scholar]
- (28).Tondreau A, Atienza CCH, Darmon JM, Milsmann C, Hoyt HM, Weller KJ, Nye SA, Lewis KN, Boyer J, Delis JGP, Lobkovsky E, Chirik PJ. Organometallics. 2012;31:4886. [Google Scholar]
- (29).Atienza CCH, Tondreau AM, Weller KJ, Lewis KM, Cruse R, Nye SA, Boyer JL, Delis JP, Chirik PJ. ACS Catal. 2012;2:2169. [Google Scholar]
- (30).Knijnenburg Q, Gambarotta S, Budzelaar PHM. Dalton Trans. 2006:5442. doi: 10.1039/b612251e. [DOI] [PubMed] [Google Scholar]
- (31).Bart SC, Chlopek K, Bill E, Bouwkamp MW, Lobkovsky E, Neese F, Wieghardt K, Chirik PJ. J. Am. Chem. Soc. 2006;128:13901. doi: 10.1021/ja064557b. [DOI] [PubMed] [Google Scholar]
- (32).Bart SC, Lobkovsky E, Bill E, Chirik PJ. J. Am. Chem. Soc. 2006;128:5302. doi: 10.1021/ja057165y. [DOI] [PubMed] [Google Scholar]
- (33).Trovitch RJ, Lobkovsky E, Bouwkamp MW, Chirik PJ. Organometallics. 2008;27:6264. [Google Scholar]
- (34).Trovitch RJ, Lobkovsky E, Chirik PJ. J. Am. Chem. Soc. 2008;130:11631. doi: 10.1021/ja803296f. [DOI] [PubMed] [Google Scholar]
- (35).Chirik PJ. Inorg. Chem. 2011;50:9737. doi: 10.1021/ic201881k. [DOI] [PubMed] [Google Scholar]
- (36).Stieber SCE, Milsmann C, Hoyt JM, Turner ZR, Finkelstein KD, Wieghardt K, DeBeer S, Chirik PJ. Inorg. Chem. 2012;51:3770. doi: 10.1021/ic202750n. [DOI] [PubMed] [Google Scholar]
- (37).Jørgensen CK. Coord. Chem. Rev. 1966;1:164. [Google Scholar]
- (38).Zhu D, Budzelaar PHM. Organometallics. 2010;29:5759. [Google Scholar]
- (39).Zhu D, Korobkov I, Budzelaar PHM. Organometallics. 2012;31:3958. [Google Scholar]
- (40).Bowman AC, Milsmann C, Atienza CCH, Lobkovsky E, Wieghardt K, Chirik PJ. J. Am. Chem. Soc. 2010;132:1676. doi: 10.1021/ja908955t. [DOI] [PubMed] [Google Scholar]
- (41).Knijnenburg Q, Hetterscheid D, Kooistra TM, Budzelaar PHM. Eur. J. Inorg. Chem. 2004:1204. [Google Scholar]
- (42).Perthuisot C, Edelbach BL, Zubris DL, Simhai N, Iverson CN, Müller C, Satoh T, Jones WD. J. Mol. Catal. A. 2002;189:157. [Google Scholar]
- (43).Yeh W-Y, Hsu SCN, Peng S-M, Lee G-H. Organometallics. 1998;17:2477. [Google Scholar]
- (44).Edelbach BL, Lachicotte RJ, Jones WD. J. Am. Chem. Soc. 1998;120:2843. [Google Scholar]
- (45).Iverson CN, Jones WD. Organometallics. 2001;20:5745. [Google Scholar]
- (46).Edelbach BL, Lachicotte RJ, Jones WD. Organometallics. 1999;18:4660. [Google Scholar]
- (47).Bouwkamp MW, Bart SC, Hawrelak EJ, Trovitch RJ, Lobkovsky E, Chirik PJ. Chem. Commun. 2005:3406. doi: 10.1039/b504063a. [DOI] [PubMed] [Google Scholar]
- (48).Bart SC, Bowman AC, Lobkovsky E, Chirik PJ. J. Am. Chem. Soc. 2007;129:7212. doi: 10.1021/ja070056u. [DOI] [PubMed] [Google Scholar]
- (49).Kahn O. Molecular Magnetism. VCH; New York: 1993. [Google Scholar]
- (50).Sorai M, Seki S. J. Phys. Chem. Solids. 1974;35:555. [Google Scholar]
- (51).Bowman AC, Milsmann C, Bill E, Turner ZR, Lobkovsky E, DeBeer S, Wieghardt K, Chirik PJ. J. Am. Chem. Soc. 2011;133:17353. doi: 10.1021/ja205736m. [DOI] [PubMed] [Google Scholar]
- (52).Bowman AC, Milsmann C, Bill E, Lobkovsky E, Weyhermüller T, Wieghardt K, Chirik PJ. Inorg. Chem. 2010;49:6110. doi: 10.1021/ic100717w. [DOI] [PubMed] [Google Scholar]
- (53).Gütlich P, Bill E, Trautwein AX. Mössbauer Spectroscopy and Transition Metal Chemistry, Fundamentals and Applications. Springer; 2010. [Google Scholar]
- (54).Dézsi I, Molnar B, Tarnozci T, Tompa K. J. Inorg. Nucl. Chem. 1967;29:2486. [Google Scholar]
- (55).Merrithew PB, Rasmussen PG. Inorg. Chem. 1972;11:325. [Google Scholar]
- (56).Römelt R, Ye S, Neese F. Inorg. Chem. 2009;48:784. doi: 10.1021/ic801535v. [DOI] [PubMed] [Google Scholar]
- (57).Westre TE, Kennepohl P, DeWitt JG, Hedman B, Hodgson KO, Solomon EI. J. Am. Chem. Soc. 1997;119:6297. [Google Scholar]
- (58).Chandrasekaran P, Stieber SCE, Collins TJ, Que L, Neese F, DeBeer S. Dalton Trans. 2011;40:11070. doi: 10.1039/c1dt11331c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59) (a).Osawa H, Iwazumi T, Shoji H, Hirai E, Nakamura T, Ogawa Y, Ishikawa T, Koshihara S, Isozumi Y, Nanao S. Phase Trans. 2002;75:919. [Google Scholar]; (b) Oyanagi H, Tayagaki T, Tanaka K. Phys. Scr. 2005;T115:107. [Google Scholar]; (c) Khalil M, Marcus MA, Smeigh AL, McCusker JK, Chong HHW, Schoenlein RW. J. Phys. Chem. A. 2006;110:38. doi: 10.1021/jp055002q. [DOI] [PubMed] [Google Scholar]; (d) Milne C, Pham V-T, Gawelda W, Nahhas AE, Van der Veen RM, Johnson SL, Beaud P, Ingold G, Borca C, Grolimund D, Abela R, Chergui M, Bressler C. Acta Phys. Pol., A. 2010;117:391. [Google Scholar]
- (60).Her JL, Matsuda YH, Nakano M, Niwa Y, Inada Y. J. Appl. Phys. 2012;111:053921. [Google Scholar]
- (61) (a).Oyanagi H, Iizuka T, Matsushita T, Saigo S, Makino R, Ishimura Y, Ishiguro T. J. Phys. Soc. Jpn. 1987;56:3381. [Google Scholar]; (b) Winkler H, Sawaryn A, Trautwein AX, Yousif AS, Hermes C, Toftlund H, Herber RH. Hyperfine Interact. 1988;42:921. [Google Scholar]; (c) Young NA. Dalton Trans. 1996:1275. [Google Scholar]; (d) Lin Y, Hsu I, Hsieh H, Lee J, Chen J, Lee J, Wang Y. J. Chin. Chem. Soc.-Taip. 2006;53:1571. [Google Scholar]
- (62).Vankó G, Neisius T, Molnár G, Renz F, Kárpáti S, Shukla A, de Groot FMF. J. Phys. Chem. B. 2006;110:11647. doi: 10.1021/jp0615961. [DOI] [PubMed] [Google Scholar]
- (63).Jackson Rudd D, Goldsmith CR, Cole AP, Stack TDP, Hodgson K, Hedman B. Inorg. Chem. 2005;44:1221. doi: 10.1021/ic048765l. [DOI] [PubMed] [Google Scholar]
- (64).DeBeer George S, Brant P, Solomon EI. J. Am. Chem. Soc. 2005;127:667. doi: 10.1021/ja044827v. [DOI] [PubMed] [Google Scholar]
- (65) (a).Jensen MP, Costas M, Ho RYN, Kaizer J, Mairata i Payeras A, Münck E, Que L, Jr., Rohde J-U, Stubna A. J. Am. Chem. Soc. 2005;127:10512. doi: 10.1021/ja0438765. [DOI] [PubMed] [Google Scholar]; (b) Jackson TA, Rohde J-U, Sook Seo M, Sastri CV, DeHont R, Stubna A, Ohta T, Kitagawa T, Münck E, Nam W, Que L., Jr. J. Am. Chem. Soc. 2008;130:12394. doi: 10.1021/ja8022576. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) England J, Martinho M, Farquhar ER, Frisch JR, Bominaar EL, Münck E, Que L., Jr. Angew. Chem. 2009;121:3676. doi: 10.1002/anie.200900863. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) McDonald AR, Bukowski MR, Farquhar ER, Jackson TA, Koehntop KD, Sook Seo M, DeHont RF, Stubna A, Halfen JA, Münck E, Nam W, Que L., Jr. J. Am. Chem. Soc. 2010;132:17118. doi: 10.1021/ja1045428. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Bigi JP, Harman WH, Lassalle-Kaiser B, Robles DM, Stich TA, Yano J, Britt RD, Chang CJ. J. Am. Chem. Soc. 2012;134:1536. doi: 10.1021/ja207048h. [DOI] [PubMed] [Google Scholar]
- (66).Lin J, Struzhkin VV, Jacobsen SD, Hu MY, Chow P, Kung J, Liu H, Mao H, Hemley RJ. Nature. 2005;436:377. doi: 10.1038/nature03825. [DOI] [PubMed] [Google Scholar]
- (67).Lee N, Petrenko T, Bergmann U, Neese F, DeBeer S. J. Am. Chem. Soc. 2010;132:9715. doi: 10.1021/ja101281e. [DOI] [PubMed] [Google Scholar]
- (68) (a).de Groot F. Chem. Rev. 2001;101:1779. doi: 10.1021/cr9900681. [DOI] [PubMed] [Google Scholar]; (b) Glatzel P, Bergmann U. Coord. Chem. Rev. 2005;249:65. [Google Scholar]
- (69).Taguchi T, Gupta R, Lassalle-Kaiser B, Boyce DW, Yachandra VK, Tolman WB, Yano J, Hendrich MP, Borovik AS. J. Am. Chem. Soc. 2012;134:1996. doi: 10.1021/ja210957u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70) (a).Tsutsumi K, Nakamori H, Ichikawa K. Phys. Rev. B. 1976;13:929. [Google Scholar]; (b) Thole BT, Cowan RD, Sawatzky GA, Fink J, Fuggle JC. Phys. Rev. B. 1985;31:6865. doi: 10.1103/physrevb.31.6856. [DOI] [PubMed] [Google Scholar]
- (71).Römelt R, Ye S, Neese F. Inorg. Chem. 2009;48:784. doi: 10.1021/ic801535v. [DOI] [PubMed] [Google Scholar]
- (72).Ginsberg AP. J. Am. Chem. Soc. 1980;102:111. [Google Scholar]
- (73).Noodleman L, Peng CY, Case DA, Mouesca JM. Coord. Chem. Rev. 1995;144:199. [Google Scholar]
- (74).Kirchner B, Wennmohs F, Ye S, Neese F. Curr. Opin. Chem. Biol. 2007;11:134. doi: 10.1016/j.cbpa.2007.02.026. [DOI] [PubMed] [Google Scholar]
- (75).Ye S, Neese F. Inorg. Chem. 2010;49:772. doi: 10.1021/ic902365a. [DOI] [PubMed] [Google Scholar]
- (76).Neese F. J. Biol. Inorg. Chem. 2006;11:702. doi: 10.1007/s00775-006-0138-1. [DOI] [PubMed] [Google Scholar]
- (77).DeBeer George S, Petrenko T, Neese F. J. Phys. Chem. A. 2008;112:12936. doi: 10.1021/jp803174m. [DOI] [PubMed] [Google Scholar]
- (78).DeBeer George S, Neese F. Inorg. Chem. 2010;49:1849. doi: 10.1021/ic902202s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.