

Abstract
Ataxia with Oculomotor Apraxia type 2 (AOA2) is one of the most frequent types of autosomal degenerative cerebellar ataxia. The first objective of this work was to identify specific cerebellar atrophy using MRI in patients with AOA2. Since increased iron deposits have been reported in degenerative diseases, our second objective was to report iron deposits signals in the dentate nuclei in AOA2. Five patients with AOA2 and 5 age-matched controls were subjects in a 3T MRI experiment that included a 3D turbo field echo T1-weighted sequence. The normalized volumes of twenty-eight cerebellar lobules and the percentage of atrophy (relative to controls) of the 4 main cerebellar regions (flocculo-nodular, vermis, anterior and posterior) were measured. The dentate nucleus signals using 3D fast field echo sequence for susceptibility-weighted images (SWI) were reported, as a measure of iron content. We found that all patients had a significant atrophy of all cerebellar lobules as compared to controls. The percentage of atrophy was the highest for the vermis, consistent with patients' oculomotor presentation, and for the anterior lobe, consistent with kinetic limb ataxia. We also describe an absence of hypointensity of the iron signal on SWI in the dentate nucleus of all patients compared to control subjects. This study suggests that patients with Ataxia with Oculomotor Apraxia type 2 present MRI patterns consistent with their clinical presentation. The absence of SWI hypointensity in dentate nucleus is a new radiological sign which was identified in all patients. The specificity of this absence of signal must be further determined in AOA2.
Keywords: Autosomal recessive cerebellar ataxias, Degeneration, Saccades, Iron signal
Highlights
-
•
Oculomotor apraxia in AOA2 points to a dysfunction of cerebellar oculomotor vermis.
-
•
AOA2 is associated with a predominant cerebellar atrophy in vermis and anterior lobe.
-
•
A decrease in iron signal of dentate nucleus is unexpectedly observed in all patients.
1. Introduction
Autosomal recessive cerebellar ataxias (ARCAs) belong to the wide group of disorders known as inherited ataxias (Anheim et al., 2012). This group encompasses a large number of diseases, the most frequent ones being Friedreich ataxia (FRDA, estimated prevalence 2–4/100,000) and ataxia telangiectasia (AT, 1–2.5/100,000). In the past 15 years, new forms of ARCAs have been identified, and among them were ataxia with oculomotor apraxia type 1 (AOA1) (Moreira et al., 2001), and type 2 (AOA2) (Anheim et al., 2010; Bomont et al., 2000).
AOA2 was first described in Japanese (Watanabe et al., 1998) and Pakistani (Nemeth et al., 2000) families and according to geographic origin, may be the second most frequent ARCA after FRDA (Campuzano et al., 1996; Durr et al., 1996; Le Ber et al., 2004; Nicolaou et al., 2008). Clinical features include onset between 10 and 22 years of age, progressive cerebellar ataxia, oculomotor apraxia (OMA), strabismus, chorea and/or dystonia, axonal sensorimotor neuropathy, elevated serum alpha-fetoprotein (AFP) levels, and cerebellar atrophy as shown by MRI (Anheim et al., 2009; Bomont et al., 2000; Nemeth et al., 2000; Watanabe et al., 1998). Although oculomotor apraxia appears to be a mandatory symptom as the name of the disease implies, it is described in only 50% of patients in AOA2. Furthermore, oculomotor apraxia is not a pathognomonic sign since it is described in other ARCAs, such as AT and ataxia–telangiectasia-like disorder (ATLD) (Le Ber et al., 2006). Oculomotor apraxia is best defined by the failure of voluntary gaze shifts with preservation of random eye movements (Cogan, 1952) and normal saccadic velocity (and main sequence) (Zee et al., 1977). In congenital forms (including hereditary diseases), oculomotor apraxia is usually associated with a characteristic head thrust (Cogan, 1952). In both AOA1 (Le Ber et al., 2003) and AOA2 (Panouilleres et al., in press), clinical descriptions and ocular motor recording mainly indicate stair-case hypometric horizontal saccades, which contribute to gaze failure. Both saccadic hypometria and stair-case saccades could be attributed to a dysfunction of the superior colliculus (Robinson, 1972; Schiller and Stryker, 1972) or the basal ganglia (Sauleau et al., 2007, 2008; Shaikh et al., 2011); although an impairment of cerebellar vermis (Solomon et al., 2008), involved in the control of saccade metrics, is predominantly suggested in those patients with predominant cerebellar symptoms.
Anatomically, the cerebellum is divided into three lobes: the flocculonodular lobe, the anterior lobe (lobules I to V) and the posterior lobe (lobules VI to IX). Functionally, the cerebellum is divided into three parts that do not strictly correspond to the anatomical subdivisions: the vestibulocerebellum, the spinocerebellum and the pontocerebellum. The vestibulocerebellum [flocculus, paraflocculus, nodulus and uvula (lobule IX)] is connected to the fastigial and vestibular nuclei and is important for steady gaze holding, smooth pursuit, the vestibulo-ocular reflex and postural control mechanisms (Kandel et al., 2000). The spinocerebellum [superior vermis (lobules I to V), adjacent medial half of the anterior lobe, inferior vermis (lobules VI to VIII)] is divided into the vermal zone, connected to the fastigial and vestibular nuclei, and the paravermal zone, connected to the interpositus nuclei (Kandel et al., 2000). Most of the vermal zone is involved in postural control, while the so-called oculomotor vermis (lobules VI, VII) is mainly involved in the control of saccades, but also contributes to smooth pursuit and vergence (Manto et al., 2012). The paravermal zone is mainly involved in the metrics and sensory feedback of limb motor control (Kandel et al., 2000). Finally, the pontocerebellum (lateral cerebellar cortex of lobules VI to VIII) is connected to the dentate nucleus and is involved in the planning of complex motor actions (Kandel et al., 2000). It may also be added that hemispheric lobule VIIa (crus I and crus II) participates in both saccadic and smooth pursuit control (Nitschke et al., 2004; Panouilleres et al., 2012; Ron and Robinson, 1973). Given this anatomo-functional organization of the cerebellum, one aim of this study was to search for region-specific atrophy which correlates with clinical dysfunction. Indeed, in patients with AOA2, the particular saccadic patterns seen, point mainly to the oculomotor vermis.
Apart from the anatomical degeneration, some neurodegenerative disorders have been shown to be associated with abnormal iron deposits that can be observed using susceptibility weighted imaging (SWI) on MRI (Gasparotti et al., 2011). On SWI MRI, a high concentration of paramagnetic iron is known to have a shortening effect on the longitudinal relaxation time and, thus, iron deposits appear as hypointensities (i.e., dark) (Deoni and Catani, 2007; Dimitrova et al., 2002). Iron plays an important role in normal brain metabolism (Connor et al., 2001; Koeppen, 1995). It is known, for example, to be a cofactor for enzymes involved in neurotransmitter synthesis, and a component of cytochromes essential for energy production. However, iron is also known to react with oxygen resulting in the production of neurotoxic free radicals. Iron accumulation has thus been associated with normal aging and neurodegenerative diseases. In particular, the iron concentration in the dentate nucleus increases with age in healthy human subjects (Hallgren and Sourander, 1958; Maschke et al., 2004). Excessive iron accumulation is considered to be a pathogenetic factor in various neurodegenerative diseases such as Parkinson's disease, Hallervorden–Spatz syndrome, Alzheimer's disease, Huntington's disease as well as different types of hereditary cerebellar ataxia like Friedreich's ataxia and aceruloplasminemia (Berg et al., 2001; Chiueh, 2001; Miyajima et al., 2001; Smith and Perry, 1995; Waldvogel et al., 1999). To our knowledge, there is no data on susceptibility weighted images of the dentate nucleus in ARCAs except for FRDA. Our hypothesis is that, in a similar manner to FRDA, the iron concentration is increased in the dentate nucleus of AOA2 patients.
The aim of this work was to identify specific MRI abnormalities in patients with AOA2. We first investigated the regional specificity of cerebellar degeneration using T1-weighted MRI sequences. We then measured iron deposits in the dentate nuclei using susceptibility-weighted MRI.
2. Subjects and methods
2.1. Subjects
Approval was received from the National French ethical committee on human experimentation, in agreement with French law (March 4, 2002) and the Declaration of Helsinki (n° 2002-303). Written informed consent was obtained from all subjects participating in the study. Five patients (3 females), including two pairs of siblings, and five (3 females) controls were included. The median age was 37 in the patient group [range: 26–42] and 34 in the control group [range: 26–44].
Clinical data of the 5 patients are summarized in Table 1. The median age at onset was 17 [range: 12–20]. The median disease duration at examination was 17 [range: 10–30]. The initial symptom was gait ataxia in 80% of patients, which was present in 100% of patients at examination. Patients also presented with major kinetic limb ataxia with decomposition and dysmetria of knee–tibia movement and finger-to-nose tests associated with intention tremor. Clinical signs of peripheral neuropathy (i.e. sensory loss, reflex depression or abolition) were constant, moderate to severe. Three patients were confined to a wheelchair at examination. Two had difficulty swallowing, one had head tremor, one had chorea of the left hand and two had sphincter disturbance. Finally, symptoms in one patient were increased after pregnancy.
Table 1.
Phenotypic characteristics of the 5 patients with AOA2.
| Family | Patient | Nucleotide change (exon) | Age at onset | Disease duration | Initial symptom | Oculomotor phenotype | Tendon reflexes | Deep sensory loss | Functional score | ICARS total (/100) | MMSE (/30) | FAB (/18) | EMG | AFP level | Height (cm) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I | I-1 | 5413C > T (10) | 18 | 10 | Gait ataxia, writing ataxia, dysarthria | Gaze-evoked nystagmus, saccadic smooth pursuit, absent fixation inhibition of VOR and staircase hypometric saccades (oculomotor apraxia) *For family I: additional positional induced central vestibular nystagmus and vestibulo-ocular hyper-reflexia |
Abolished | Present | 5 | 69 | 29 (draw) | 18 | Axonal sensory–motor neuropathy | 60 | 168 |
| I | I-2 | 12 | 14 | Dysarthria | Abolished | Present | 4 | 50 | 29 (write) | 17 (luria) | Axonal sensory–motor neuropathy predominant in sensory | 45.9 | 180 | ||
| II | II-1 | 7267G > A (23) | 20 | 17 | Gait ataxia | Abolished | Present | 6 | 69 | 24/28 (write, calculation) | 13 | Axono-myelinic neuropathy | 22.5 | 171 | |
| II | II-2 | 17 | 24 | Gait ataxia | Abolished | Present | 6 | 68 | 24 (calculation) | 18 | ND | 15 | 158 | ||
| III | III-3 | 6292C > T (15) | 12 | 30 | Gait ataxia | Periodic alternating nystagmus | Abolished | Present | 6 | 56 | 29 (reminder) | 17 (fluency) | ND | 79 | 176 |
Functional score of motor disability was assessed by a seven-stage functional scale: 0 = normal; 1 = mild modifications at examination; 2 = mild functional disability, able to walk and run; 3 = able to walk without help up to 500 m, unable to run; 4 = needs unilateral help to walk; 5 = needs bilateral help to walk; 6 = wheelchair-bound; 7 = bedridden. ICARS total, Mini mental state examination (MMSE), Frontal assessment battery (FAB), electromyogram (EMG), Alpha-Fetoprotein (AFP) levels. ND: no data.
Four patients (I-1, I-2, II-1, II-2) presented with a similar oculomotor phenotype including gaze-evoked nystagmus, saccadic smooth pursuit, an absent fixation inhibition of the vestibulo-ocular reflex and lastly staircase hypometric horizontal and vertical saccades. The two sibling patients (I-1, I-2) presented with an additional positional induced central vestibular nystagmus (in either direction), and vestibulo-ocular hyper-reflexia. The last patient (III) showed mainly isolated periodic alternating nystagmus.
To quantify the patients' symptoms, we used the one-hundred-point semi-quantitative International Cooperative Ataxia Rating Scale (ICARS) (Trouillas et al., 1997). This test translates the classical symptomatology of ataxia into semi-quantitative scores and 4 compartments (I, postural and stance disturbances; II, limb movements disturbances; III, speech disorders; IV, oculomotor disorders); the higher the scores on the scale, the more severe the ataxia. The median score was 62 [range: 39–69].
Cognitive impairment was mild on the Mini Mental State Examination (MMSE) (median score 27.3 [range: 24–29], limited on the most part by writing difficulties). The patient with a score of 24 had not attended school and had not learned arithmetic. Patients were mildly impaired on the Frontal Assessment Battery (FAB; median score 16.8 [range: 13–18]) (Dubois et al., 2000).
2.2. Methods
2.2.1. Image acquisition
MR imaging acquisitions for patients and control subjects were all performed on the same 3T MR system (Achieva 3T, Philips Medical system, Best, The Netherlands). The MRI protocol included a non-contrast 3D turbo field echo T1-weighted sequence (TR/TE = 6.59 / 2.95; Matrix = 268 × 164) yielding 200 sagittal 0.9-mm thick sections, for cerebellar volume measurements. A 3D fast field echo sequence was also used to acquire 143 axial 1.4-mm thick sections of susceptibility-weighted images (TR/TE = 20.50 / 28.93; Matrix = 316 × 314), for iron deposit evaluation.
2.2.2. Post-processing and image analysis
The cerebellum was isolated from the T1-weighted volumes using a knowledge-based image segmentation procedure applying a spatially unbiased atlas template of the cerebellum and brainstem (SUIT, http://www.icn.ucl.ac.uk/motorcontrol/imaging/suit.htm). We first isolated the cerebellum using John Ashburner's segmentation algorithm (Ashburner and Friston, 2005). We registered the individual cerebellum into the SUIT atlas template. Next we resliced the images using the deformation map in order to resample them into the new atlas space, and then back into the individual subject's space. Twenty-eight lobular volumes (V) were finally extracted using the Schmahmann atlas (Schmahmann et al., 1999). The volumes were then normalized to the participant's size by dividing volumes by the participant's height (in mm) (Cotton et al., 2005) and expressed in mm3/mm. For further analysis, we pooled the volumes into four cerebellar regions corresponding mainly to the functional parts of the cerebellum: 1) vermis (vermal spinocerebellum: vermis of lobule VI to VIII); 2) anterior lobe (lateral part of lobule IV and V, corresponding mainly to the paravermal spinocerebellum, located anteriorly to the postero-superior sulcus); 3) posterior lobe (lateral part of lobule VI to VIII); and 4) flocculo-nodular lobe (lateral and vermal parts of lobule IX and X). For each of these regions, and for each patient (Vp), an atrophy index was calculated with respect to the median volume measured in controls (VCmedian), as follows:
Volumetric measurements of cerebral area focussing on the frontal and parietal eye fields were also performed using Freesurfer image analysis (see Supplementary data 1).
2.2.3. Statistics
Analyses were performed using R software (Team, 2011). Given the size of the samples, non-parametric tests were used, as implemented within the Coin package (Hothorn et al., 2006, 2008). When comparing cerebellar volume for each of the 28 lobules between controls and patients, bilateral exact 2-sample permutation tests were used. When comparing patients' atrophy index across the 4 regions, the Friedman test for multiple dependent samples was used. Finally, when pairwise comparisons were performed, adjusted p-values were obtained from a single-step max-T procedure (Westfall and Stanley Young, 1993).
The susceptibility-weighted images were visually compared in the patient and the control groups.
3. Results and discussion
3.1. Cerebellar volume measurements
This study confirmed the presence of cerebellar atrophy in patients with AOA2 by showing that patients had a cerebellum volume of 53 ± 15% (median value) less than controls. Fig. 1 presents representative examples of T1 images for a control subject and for a patient. In the sagittal view, the cerebellar atrophy of the patient is emphasized by the empty space surrounding the cerebellar vermis. In the frontal view, the deeper sulci on the patient's MRI relative to the control are easily noticeable. In the patient group, the median (standard deviation) normalized cerebellar volume was of only 36 ± 9 mm3/mm whereas it was of 70 ± 3 mm3/mm in the control group (Fig. 2).
Fig. 1.
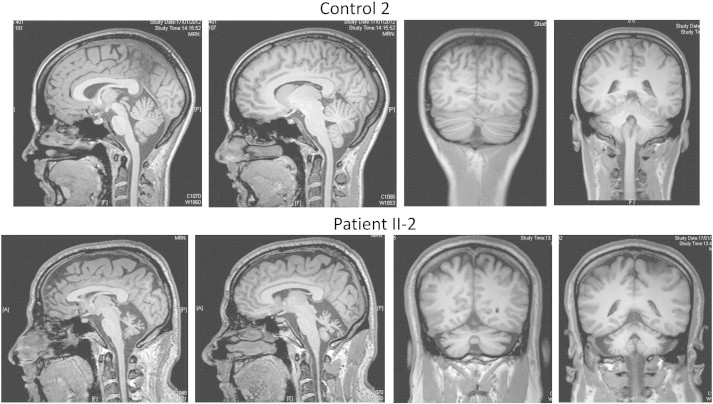
An example of non-contrast 3D turbo field echo T1-weighted sequence of brain MRI in one control and one patient, in sagittal (left) and coronal (right) views.
Fig. 2.
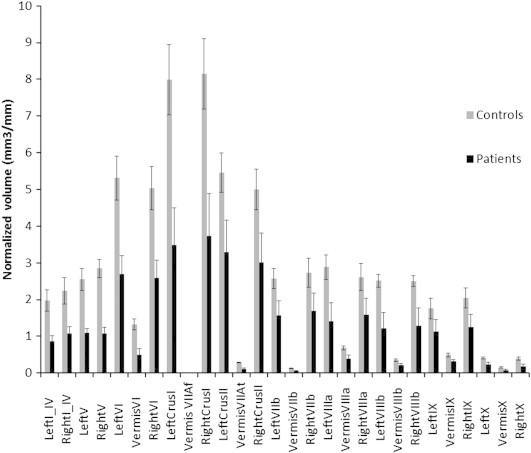
Median cerebellar volumes (in mm3/mm) measured in the 28 lobules, in controls (gray bars) and in patients (dark bars). Error bars = standard deviation. The volume of all lobules was significantly smaller in patients versus controls (all p ≤ 0.012), illustrating a general cerebellar atrophy.
Each of the twenty-eight cerebellar regions was significantly smaller in patients than in controls (bilateral exact 2-sample permutation tests, all p ≤ 0.012).
Moreover, we revealed a region-specific pattern of atrophy, which was more severe in the vermis and the anterior lobe of the cerebellum. As described in Methods section, these twenty-eight volumes were pooled into four different cerebellar regions: the vermis, the anterior lobe, the posterior lobe and the flocculonodular lobe. For each region, an atrophy index was computed for each patient with respect to the control group. Statistical tests showed that this atrophy was different across regions (exact Friedman test: χ2(3) = 14.04, p = 0.0001). Indeed, pairwise comparisons showed that the anterior lobe (59 ± 6%) and the vermis (57 ± 15%) were more atrophied than the flocculo-nodular lobe (42 ± 16%) (p = 0.003 and 0.039, respectively) (Fig. 3). The posterior lobe (46 ± 14%) did not show any significant difference in atrophy compared to other regions.
Fig. 3.
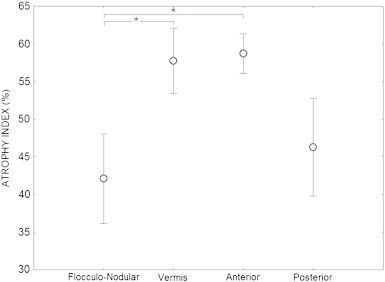
Mean atrophy index in the 4 cerebellar regions in patients. Error bars = standard deviation. * = p < 0.05.
In ARCAs, studies mainly highlight a predominant vermian atrophy of the cerebellum. In FRDA, a progressive mild cerebellar atrophy and mild vermian atrophy are found in advanced stages of the disease (Ormerod et al., 1994). A moderate cerebellar atrophy similar to that encountered in FRDA was found in Ataxia with Vitamin E Deficiency (AVED) (Anheim et al., 2010). MRI of autosomal recessive spastic ataxia of Charlevoix–Saguenay showed a predominantly vermian cerebellar atrophy as well as linear hypointensities on T2 and T2 fluid-attenuated inversion recovery-weighted images in the pons (Martin et al., 2007). In AOA2, previous imaging studies revealed extensive vermian atrophy (Anheim et al., 2009; Bernard et al., 2008; Le Ber et al., 2004; Nicolaou et al., 2008; Schols et al., 2008), which is observed shortly after the onset of the disease and remains stable at more advanced stages (Anheim et al., 2009). Our study confirms the predominance of vermian atrophy in AOA2. Although not specific with regards to the anatomical data in ARCAs, the predominant involvement of the vermis in AOA2 is consistent with the saccadic abnormalities. Indeed, as for AOA1, “oculomotor apraxia” seems to correspond mainly to hypometric saccades (stair cases). Hypometry of saccades suggests a dysfunction of lobules VI and VII of the cerebellar vermis (Barash et al., 1999; Noda, 1991; Takagi et al., 1998), which calibrate saccade amplitude (Sato and Noda, 1992). Our study further underlines an atrophy of the anterior part of the cerebellum. This last result is consistent with the findings of a postmortem examination in one 79 year-old AOA2 patient showing that the cerebellar atrophy was most evident at the level of the vermis and the anterior lobe (Criscuolo et al., 2006). The predominant involvement of the anterior cerebellum can be associated with the kinetic ataxia of limb movements observed in all of our patients.
Finally, we found a discrete and non-significant atrophy of frontal gyri including the frontal eye fields (Supplementary data). This discrete frontal atrophy could also explain in part the oculomotor disturbances of oculomotor apraxia.
3.2. Iron deposition on susceptibility-weighted images
We did not find the expected iron deposits in the dentate nucleus in patients with AOA2 (Fig. 4). In contrast, because the normal appearance of the dentate nucleus is a highly convoluted structure in deep cerebellar white matter with a typical dark signal related to brain iron deposits, it was easily detectable in all control subjects (arrows). The other iron cerebral structures such as nucleus rubrum, nucleus lenticularis, and cerebellar peduncles were all identifiable in the susceptibility sequence in all patients and controls (data not shown).
Fig. 4.
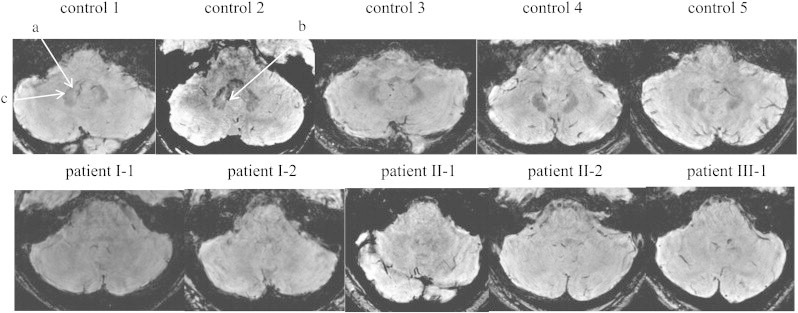
Representative axial slices of 3D fast field echo sequences of susceptibility-weighted images in controls (top raw) and in patients (bottom raw). a: Dentate nucleus; b: the hilus; c: toothed appearance after which the dentate nucleus is named.
To our knowledge, this is the first report in a degenerative disease of a disappearance of the iron-induced signal normally observed in the dentate nucleus. Our hypothesis was to find increased iron-induced signal in AOA2, such as that found in FRDA (Koeppen et al., 2007; Waldvogel et al., 1999). This finding is however consistent with a previous anatomo-pathological study in AOA2 that showed a reduced number of dentate nuclei neurons, which could be linked to the decrease of iron concentration (Criscuolo et al., 2006). The dentate nucleus is part of the functional ponto-cerebellum, which is involved in the coordination and timing of fine and skilled voluntary movements. Efferent fibers from the dentate nucleus pass through the superior cerebellar peduncles and cross over the midline at the pontomesencephalic junction to synapse in the red nucleus and the ventrolateral thalamus, which then projects to the motor cortex. It is responsible for the planning, initiation and control of volitional movements (Ito, 1993; Leiner et al., 1986; Mathiak et al., 2002). In addition to the degeneration of the cerebellar anterior lobe, the lack of iron signal of dentate nuclei in our AOA2 patients could be associated with the kinetic aspect of cerebellar syndrome. There is some discordance between our findings of predominant atrophy in the anterior lobe as compared to the posterior lobe and the lack of iron signal of dentate nuclei. However, given the weak signal of interpositus nuclei in normal subjects, we could not verify the integrity of its signal using susceptibility weighted images in patients. It is therefore possible that the abnormal signal applies to all deep cerebellar nuclei. Furthermore, even if less atrophied than the anterior lobe and the vermis, the non-vermal areas of the posterior lobe (especially Crus I) which are connected to the dentate nucleus, still showed large atrophy (46%) as compared to control subjects.
4. Conclusion
We identified and quantified cerebellar atrophy in five patients presenting with ataxia with oculomotor apraxia type 2. In addition to the general atrophy of the entire cerebellum, a significantly larger atrophy was found for the vermis, consistent with their oculomotor deficits, as well as an atrophy in the anterior lobe of the cerebellum, also concordant with their kinetic limb ataxia. For the first time, an absence of SWI hypointensity was observed in the dentate nucleus for all patients. It remains to be determined whether this poor iron concentration is specific to AOA2.
Acknowledgments
We thank Dr Sappey-Marinier, CERMEP-Imagerie du vivant and CREATIS, INSERM U1044/CNRS UMR 5220, Lyon, F-69000, France for technical support.
This work was supported by the Hospices Civils de Lyon (2002-303). We thank Dr Aarlenne Khan for her help in grammatical improvement. This work was performed within the framework of the LABEX CORTEX (ANR-11-LABX-0042) of Université de Lyon, within the program "Investissements d'Avenir" (ANR-11-IDEX-0007) operated by the French National Research Agency (ANR).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supplementary data
Supplementary material.
References
- Anheim M., Monga B., Fleury M., Charles P., Barbot C., Salih M., Delaunoy J.P., Fritsch M., Arning L., Synofzik M., Schols L., Sequeiros J., Goizet C., Marelli C., Le Ber I., Koht J., Gazulla J., De Bleecker J., Mukhtar M., Drouot N., Ali-Pacha L., Benhassine T., Chbicheb M., M'Zahem A., Hamri A., Chabrol B., Pouget J., Murphy R., Watanabe M., Coutinho P., Tazir M., Durr A., Brice A., Tranchant C., Koenig M. Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain. 2009;132:2688–2698. doi: 10.1093/brain/awp211. [DOI] [PubMed] [Google Scholar]
- Anheim M., Tranchant C., Koenig M. The autosomal recessive cerebellar ataxias. The New England Journal of Medicine. 2012;366:636–646. doi: 10.1056/NEJMra1006610. [DOI] [PubMed] [Google Scholar]
- Anheim M., Fleury M., Monga B., Laugel V., Chaigne D., Rodier G., Ginglinger E., Boulay C., Courtois S., Drouot N., Fritsch M., Delaunoy J.P., Stoppa-Lyonnet D., Tranchant C., Koenig M. Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: implications for clinical management. Neurogenetics. 2010;11:1–12. doi: 10.1007/s10048-009-0196-y. [DOI] [PubMed] [Google Scholar]
- Ashburner J., Friston K.J. Unified segmentation. NeuroImage. 2005;26:839–851. doi: 10.1016/j.neuroimage.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Barash S., Melikyan A., Sivakov A., Zhang M., Glickstein M., Thier P. Saccadic dysmetria and adaptation after lesions of the cerebellar cortex. Journal of Neuroscience. 1999;19:10931–10939. doi: 10.1523/JNEUROSCI.19-24-10931.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg D., Gerlach M., Youdim M.B., Double K.L., Zecca L., Riederer P., Becker G. Brain iron pathways and their relevance to Parkinson's disease. Journal of Neurochemistry. 2001;79:225–236. doi: 10.1046/j.1471-4159.2001.00608.x. [DOI] [PubMed] [Google Scholar]
- Bernard V., Stricker S., Kreuz F., Minnerop M., Gillessen-Kaesbach G., Zuhlke C. Ataxia with oculomotor apraxia type 2: novel mutations in six patients with juvenile age of onset and elevated serum alpha-fetoprotein. Neuropediatrics. 2008;39:347–350. doi: 10.1055/s-0029-1214424. [DOI] [PubMed] [Google Scholar]
- Bomont P., Watanabe M., Gershoni-Barush R., Shizuka M., Tanaka M., Sugano J., Guiraud-Chaumeil C., Koenig M. Homozygosity mapping of spinocerebellar ataxia with cerebellar atrophy and peripheral neuropathy to 9q33–34, and with hearing impairment and optic atrophy to 6p21–23. European Journal of Human Genetics. 2000;8:986–990. doi: 10.1038/sj.ejhg.5200586. [DOI] [PubMed] [Google Scholar]
- Campuzano V., Montermini L., Molto M.D., Pianese L., Cossee M., Cavalcanti F., Monros E., Rodius F., Duclos F., Monticelli A., Zara F., Canizares J., Koutnikova H., Bidichandani S.I., Gellera C., Brice A., Trouillas P., De Michele G., Filla A., De Frutos R., Palau F., Patel P.I., Di Donato S., Mandel J.L., Cocozza S., Koenig M., Pandolfo M. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- Chiueh C.C. Iron overload, oxidative stress, and axonal dystrophy in brain disorders. Pediatric Neurology. 2001;25:138–147. doi: 10.1016/s0887-8994(01)00266-1. [DOI] [PubMed] [Google Scholar]
- Cogan D.G. A type of congenital ocular motor apraxia presenting jerky head movements. Transactions of the American Academy of Ophthalmology and Otolaryngology. 1952;56:853–862. [PubMed] [Google Scholar]
- Connor J.R., Menzies S.L., Burdo J.R., Boyer P.J. Iron and iron management proteins in neurobiology. Pediatric Neurology. 2001;25:118–129. doi: 10.1016/s0887-8994(01)00303-4. [DOI] [PubMed] [Google Scholar]
- Cotton F., Euvrard T., Durand-Dubief F., Pachai C., Cucherat M., Ramirez Rozzi F., Bonmartin A., Guihard-Costa A.M., Tran Minh V.A., Vallee B., Froment J.C. Correlation between cranial vault size and brain size over time: preliminary MRI evaluation. Journal of Neuroradiology. 2005;32:131–137. doi: 10.1016/s0150-9861(05)83128-1. [DOI] [PubMed] [Google Scholar]
- Criscuolo C., Chessa L., Di Giandomenico S., Mancini P., Sacca F., Grieco G.S., Piane M., Barbieri F., De Michele G., Banfi S., Pierelli F., Rizzuto N., Santorelli F.M., Gallosti L., Filla A., Casali C. Ataxia with oculomotor apraxia type 2: a clinical, pathologic, and genetic study. Neurology. 2006;66:1207–1210. doi: 10.1212/01.wnl.0000208402.10512.4a. [DOI] [PubMed] [Google Scholar]
- Deoni S.C., Catani M. Visualization of the deep cerebellar nuclei using quantitative T1 and rho magnetic resonance imaging at 3 Tesla. NeuroImage. 2007;37:1260–1266. doi: 10.1016/j.neuroimage.2007.06.036. [DOI] [PubMed] [Google Scholar]
- Dimitrova A., Weber J., Redies C., Kindsvater K., Maschke M., Kolb F.P., Forsting M., Diener H.C., Timmann D. MRI atlas of the human cerebellar nuclei. NeuroImage. 2002;17:240–255. doi: 10.1006/nimg.2002.1124. [DOI] [PubMed] [Google Scholar]
- Dubois B., Slachevsky A., Litvan I., Pillon B. The FAB: a Frontal Assessment Battery at bedside. Neurology. 2000;55:1621–1626. doi: 10.1212/wnl.55.11.1621. [DOI] [PubMed] [Google Scholar]
- Durr A., Cossee M., Agid Y., Campuzano V., Mignard C., Penet C., Mandel J.L., Brice A., Koenig M. Clinical and genetic abnormalities in patients with Friedreich's ataxia. The New England Journal of Medicine. 1996;335:1169–1175. doi: 10.1056/NEJM199610173351601. [DOI] [PubMed] [Google Scholar]
- Gasparotti R., Pinelli L., Liserre R. New MR sequences in daily practice: susceptibility weighted imaging. A pictorial essay. Insights Imaging. 2011;2:335–347. doi: 10.1007/s13244-011-0086-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallgren B., Sourander P. The effect of age on the non-haemin iron in the human brain. Journal of Neurochemistry. 1958;3:41–51. doi: 10.1111/j.1471-4159.1958.tb12607.x. [DOI] [PubMed] [Google Scholar]
- Hothorn T., Hornik K., Van De Wiel M.A., Zeileis A. A lego system for conditional inference. The American Statistician. 2006;60:257–263. [Google Scholar]
- Hothorn T., Hornik K., Van De Wiel M.A., Zeileis A. Implementing a class of permutation tests: the coin package. Journal of Statistical Software. 2008;28:1–23. [Google Scholar]
- Ito M. Movement and thought: identical control mechanisms by the cerebellum. Trends in Neurosciences. 1993;16:448–450. doi: 10.1016/0166-2236(93)90073-u. (discussion 453-444) [DOI] [PubMed] [Google Scholar]
- Kandel E.R., Schwartz J.H., Jessel T.M. 4th ed. McGraw-Hill; 2000. Principles of Neural Science. [Google Scholar]
- Koeppen A.H. The history of iron in the brain. Journal of Neurological Sciences. 1995;134:1–9. doi: 10.1016/0022-510x(95)00202-d. (Suppl.) [DOI] [PubMed] [Google Scholar]
- Koeppen A.H., Michael S.C., Knutson M.D., Haile D.J., Qian J., Levi S., Santambrogio P., Garrick M.D., Lamarche J.B. The dentate nucleus in Friedreich's ataxia: the role of iron-responsive proteins. Acta Neuropathologica. 2007;114:163–173. doi: 10.1007/s00401-007-0220-y. [DOI] [PubMed] [Google Scholar]
- Le Ber I., Moreira M.C., Rivaud-Pechoux S., Chamayou C., Ochsner F., Kuntzer T., Tardieu M., Said G., Habert M.O., Demarquay G., Tannier C., Beis J.M., Brice A., Koenig M., Durr A. Cerebellar ataxia with oculomotor apraxia type 1: clinical and genetic studies. Brain. 2003;126:2761–2772. doi: 10.1093/brain/awg283. [DOI] [PubMed] [Google Scholar]
- Le Ber I., Bouslam N., Rivaud-Pechoux S., Guimaraes J., Benomar A., Chamayou C., Goizet C., Moreira M.C., Klur S., Yahyaoui M., Agid Y., Koenig M., Stevanin G., Brice A., Durr A. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients. Brain. 2004;127:759–767. doi: 10.1093/brain/awh080. [DOI] [PubMed] [Google Scholar]
- Le Ber I., Rivaud-Pechoux S., Brice A., Durr A. Autosomal recessive cerebellar ataxias with oculomotor apraxia. Revue Neurologique (Paris) 2006;162:177–184. doi: 10.1016/s0035-3787(06)74997-9. [DOI] [PubMed] [Google Scholar]
- Leiner H.C., Leiner A.L., Dow R.S. Does the cerebellum contribute to mental skills? Behavioral Neuroscience. 1986;100:443–454. doi: 10.1037//0735-7044.100.4.443. [DOI] [PubMed] [Google Scholar]
- Manto M., Bower J.M., Conforto A.B., Delgado-Garcia J.M., da Guarda S.N., Gerwig M., Habas C., Hagura N., Ivry R.B., Marien P., Molinari M., Naito E., Nowak D.A., Oulad Ben Taib N., Pelisson D., Tesche C.D., Tilikete C., Timmann D. Consensus paper: roles of the cerebellum in motor control—the diversity of ideas on cerebellar involvement in movement. Cerebellum. 2012;11:457–487. doi: 10.1007/s12311-011-0331-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M.H., Bouchard J.P., Sylvain M., St-Onge O., Truchon S. Autosomal recessive spastic ataxia of Charlevoix–Saguenay: a report of MR imaging in 5 patients. AJNR. American Journal of Neuroradiology. 2007;28:1606–1608. doi: 10.3174/ajnr.A0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maschke M., Weber J., Dimitrova A., Bonnet U., Bohrenkamper J., Sturm S., Kindsvater K., Muller B.W., Gastpar M., Diener H.C., Forsting M., Timmann D. Age-related changes of the dentate nuclei in normal adults as revealed by 3D fast low angle shot (FLASH) echo sequence magnetic resonance imaging. Journal of Neurology. 2004;251:740–746. doi: 10.1007/s00415-004-0420-5. [DOI] [PubMed] [Google Scholar]
- Mathiak K., Hertrich I., Grodd W., Ackermann H. Cerebellum and speech perception: a functional magnetic resonance imaging study. Journal of Cognitive Neuroscience. 2002;14:902–912. doi: 10.1162/089892902760191126. [DOI] [PubMed] [Google Scholar]
- Miyajima H., Adachi J., Kohno S., Takahashi Y., Ueno Y., Naito T. Increased oxysterols associated with iron accumulation in the brains and visceral organs of acaeruloplasminaemia patients. QJM. 2001;94:417–422. doi: 10.1093/qjmed/94.8.417. [DOI] [PubMed] [Google Scholar]
- Moreira M.C., Barbot C., Tachi N., Kozuka N., Mendonca P., Barros J., Coutinho P., Sequeiros J., Koenig M. Homozygosity mapping of Portuguese and Japanese forms of ataxia–oculomotor apraxia to 9p13, and evidence for genetic heterogeneity. American Journal of Human Genetics. 2001;68:501–508. doi: 10.1086/318191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth A.H., Bochukova E., Dunne E., Huson S.M., Elston J., Hannan M.A., Jackson M., Chapman C.J., Taylor A.M. Autosomal recessive cerebellar ataxia with oculomotor apraxia (ataxia–telangiectasia-like syndrome) is linked to chromosome 9q34. American Journal of Human Genetics. 2000;67:1320–1326. doi: 10.1016/s0002-9297(07)62962-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou P., Georghiou A., Votsi C., Middleton L.T., Zamba-Papanicolaou E., Christodoulou K. A novel c.5308_5311delGAGA mutation in Senataxin in a Cypriot family with an autosomal recessive cerebellar ataxia. BMC Medical Genetics. 2008;9:28. doi: 10.1186/1471-2350-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitschke M.F., Binkofski F., Buccino G., Posse S., Erdmann C., Kompf D., Seitz R.J., Heide W. Activation of cerebellar hemispheres in spatial memorization of saccadic eye movements: an fMRI study. Human Brain Mapping. 2004;22:155–164. doi: 10.1002/hbm.20025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda H. Cerebellar control of saccadic eye movements: its neural mechanisms and pathways. The Japanese Journal of Physiology. 1991;41:351–368. doi: 10.2170/jjphysiol.41.351. [DOI] [PubMed] [Google Scholar]
- Ormerod I.E., Harding A.E., Miller D.H., Johnson G., MacManus D., du Boulay E.P., Kendall B.E., Moseley I.F., McDonald W.I. Magnetic resonance imaging in degenerative ataxic disorders. Journal of Neurology, Neurosurgery, and Psychiatry. 1994;57:51–57. doi: 10.1136/jnnp.57.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panouilleres M., Neggers S.F., Gutteling T.P., Salemme R., van der Stigchel S., van der Geest J.N., Frens M.A., Pelisson D. Transcranial magnetic stimulation and motor plasticity in human lateral cerebellum: dual effect on saccadic adaptation. Human Brain Mapping. 2012;33:1512–1525. doi: 10.1002/hbm.21301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panouilleres M., Frismand S., Sillan O., Urquizar C., Vighetto A., Pelisson D., Tilikete C. Saccades and eye-head coordination in ataxia with oculomotor apraxia type 2. Cerebellum. 2013 doi: 10.1007/s12311-013-0463-1. (in press) [DOI] [PubMed] [Google Scholar]
- Robinson D.A. Eye movements evoked by collicular stimulation in the alert monkey. Vision Research. 1972;12:1795–1808. doi: 10.1016/0042-6989(72)90070-3. [DOI] [PubMed] [Google Scholar]
- Ron S., Robinson D.A. Eye movements evoked by cerebellar stimulation in the alert monkey. Journal of Neurophysiology. 1973;36:1004–1022. doi: 10.1152/jn.1973.36.6.1004. [DOI] [PubMed] [Google Scholar]
- Sato H., Noda H. Saccadic dysmetria induced by transient functional decortication of the cerebellar vermis [corrected] Experimental Brain Research. 1992;88:455–458. doi: 10.1007/BF02259122. [DOI] [PubMed] [Google Scholar]
- Sauleau P., Pollak P., Krack P., Pelisson D., Vighetto A., Benabid A.L., Tilikete C. Contraversive eye deviation during stimulation of the subthalamic region. Movement Disorders. 2007;22:1810–1813. doi: 10.1002/mds.21650. [DOI] [PubMed] [Google Scholar]
- Sauleau P., Pollak P., Krack P., Courjon J.H., Vighetto A., Benabid A.L., Pelisson D., Tilikete C. Subthalamic stimulation improves orienting gaze movements in Parkinson's disease. Clinical Neurophysiology. 2008;119:1857–1863. doi: 10.1016/j.clinph.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Schiller P.H., Stryker M. Single-unit recording and stimulation in superior colliculus of the alert rhesus monkey. Journal of Neurophysiology. 1972;35:915–924. doi: 10.1152/jn.1972.35.6.915. [DOI] [PubMed] [Google Scholar]
- Schmahmann J.D., Doyon J., McDonald D., Holmes C., Lavoie K., Hurwitz A.S., Kabani N., Toga A., Evans A., Petrides M. Three-dimensional MRI atlas of the human cerebellum in proportional stereotaxic space. NeuroImage. 1999;10:233–260. doi: 10.1006/nimg.1999.0459. [DOI] [PubMed] [Google Scholar]
- Schols L., Arning L., Schule R., Epplen J.T., Timmann D. “Pseudodominant inheritance” of ataxia with ocular apraxia type 2 (AOA2) Journal of Neurology. 2008;255:495–501. doi: 10.1007/s00415-008-0707-z. [DOI] [PubMed] [Google Scholar]
- Shaikh A.G., Xu-Wilson M., Grill S., Zee D.S. ‘Staircase’ square-wave jerks in early Parkinson's disease. British Journal of Ophthalmology. 2011;95:705–709. doi: 10.1136/bjo.2010.179630. [DOI] [PubMed] [Google Scholar]
- Smith M.A., Perry G. Free radical damage, iron, and Alzheimer's disease. Journal of Neurological Sciences. 1995;134:92–94. doi: 10.1016/0022-510x(95)00213-l. (Suppl.) [DOI] [PubMed] [Google Scholar]
- Solomon D., Ramat S., Leigh R.J., Zee D. A quick look at slow saccades after cardiac surgery: where is the lesion? Progress in Brain Research. 2008;171:587–590. doi: 10.1016/S0079-6123(08)00685-7. [DOI] [PubMed] [Google Scholar]
- Takagi M., Zee D.S., Tamargo R.J. Effects of lesions of the oculomotor vermis on eye movements in primate: saccades. Journal of Neurophysiology. 1998;80:1911–1931. doi: 10.1152/jn.1998.80.4.1911. [DOI] [PubMed] [Google Scholar]
- Team R.D.C. R: A Language and Environment for Statistical Computing. In: Team R.D.C., editor. R Foundation for Statistical Computing; Vienna, Austria: 2011. [Google Scholar]
- Trouillas P., Takayanagi T., Hallett M., Currier R.D., Subramony S.H., Wessel K., Bryer A., Diener H.C., Massaquoi S., Gomez C.M., Coutinho P., Ben Hamida M., Campanella G., Filla A., Schut L., Timann D., Honnorat J., Nighoghossian N., Manyam B. International cooperative ataxia rating scale for pharmacological assessment of the cerebellar syndrome. The Ataxia Neuropharmacology Committee of the World Federation of Neurology. Journal of Neurological Sciences. 1997;145:205–211. doi: 10.1016/s0022-510x(96)00231-6. [DOI] [PubMed] [Google Scholar]
- Waldvogel D., van Gelderen P., Hallett M. Increased iron in the dentate nucleus of patients with Friedrich's ataxia. Annals of Neurology. 1999;46:123–125. doi: 10.1002/1531-8249(199907)46:1<123::aid-ana19>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Watanabe M., Sugai Y., Concannon P., Koenig M., Schmitt M., Sato M., Shizuka M., Mizushima K., Ikeda Y., Tomidokoro Y., Okamoto K., Shoji M. Familial spinocerebellar ataxia with cerebellar atrophy, peripheral neuropathy, and elevated level of serum creatine kinase, gamma-globulin, and alpha-fetoprotein. Annals of Neurology. 1998;44:265–269. doi: 10.1002/ana.410440220. [DOI] [PubMed] [Google Scholar]
- Westfall P.H., Stanley Young S. Wiley; New York: 1993. Resampling-Based Multiple Testing. [Google Scholar]
- Zee D.S., Yee R.D., Singer H.S. Congenital ocular motor apraxia. Brain. 1977;100:581–599. doi: 10.1093/brain/100.3.581. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.