

Abstract
Introduction
Post thrombotic syndrome therapy is primarily palliative, and the associated vein wall inflammatory mechanisms are unclear. Vein wall fibrotic injury following deep venous thrombosis (VT) is associated with elevated matrix metalloproteinases (MMPs). Whether and by what mechanism MMP9 directly contributes to vein wall remodeling after VT is unknown.
Methods
WT and MMP9 -/- mice underwent stasis VT by ligation of the inferior vena cava (IVC) and tissue was harvested at 2, 8, and 21 days. Assessment of thrombus size, and gene, protein and structural vein wall determinations were done.
Results
VT resolution was increased in MMP9-/- mice as compared with controls at 21d only. The primary phenotypic fibrotic vein wall differences occurred at 8d post VT, with significantly less vein wall collagen content as assessed by Picosirius red staining in MMP9 -/- mice as compared with WT. Increased monocytic vein wall influx with less IL-1b and TGFb was found in MMP9 -/- vein walls as compared with WT. Corresponding levels of PAI-1 were increased in MMP9 -/- compared with WT, and no difference in FSP-1 + cells as compared with controls.
Conclusions
In stasis VT, MMP9 modulates midterm vein wall collagen content, with an altered local inflammatory and profibrotic environment, likely directed by monocytes. Thus, MMP9 plays a role in both vein wall responses as well as late thrombus resolution.
Keywords: Venous thrombosis, inflammation, matrix metalloproteinases, monocytes
Introduction
Deep venous thrombosis (DVT) is a significant health care problem in this country, with over 250,000 patients affected yearly, although these figures may be conservative.1 An additional 200,000 patients are affected by the late sequelae of post thrombotic syndrome (PTS), characterized by leg pain, sensations of heaviness, limb edema, discoloration and occasionally ulceration.2 The consequence to the vein after DVT is the conversion of a compliant, thin walled vein with functional valves to a thick walled vessel, often with nonfunctioning valves.3 The physiological result of damaged veins is pooling of blood in the dependent extremities, producing venous hypertension, fluid transudation, edema, and potentially ulceration.
Despite effective anticoagulant therapy treatments for DVT, there are no therapies that specifically target PTS.4 While surgical and interventional therapy may be beneficial for selected patients, treatment of PTS is mainly supportive, consisting of limb compression and wound care. While largely effective, compression does not correct the fundamental changes that occur in the venous system and compliance with such ongoing regimens is often difficult.
Rodent studies have suggested that the mechanism and duration of thrombosis affects the magnitude of post-thrombotic vein wall damage seen, with alteration in collagen structure and venous compliance.5, 6 In addition to the intense inflammatory response seen following DVT, there is a phenotypic alteration in the extracellular matrix, associated with activation of matrix metalloproteinases (MMPs), including MMP2 and 9.6-8 While inhibition broadly may decrease some measures of injury,9, 10 the specific role of MMP9 has not been defined in the vein wall after VT. Moreover, recent studies suggest certain biomarkers may be predictive of PTS well as associated with DVT resolution in humans, including MMPs.4, 11
We hypothesized that matrix remodeling via genetic deletion of MMP9 would lessen the damage seen in the vein wall following stasis venous thrombosis (VT), associated with less inflammation.
Methods
Animal Model
Male mice 6 – 8 weeks old, MMP9 -/- (kindly donated by Dr. Robert Senior) and their wild types (WT) B6129 SvEv were used for the experiments. With all surgical procedures, the mice were anesthetized using inhalational Isoflurane and O2, and continuously monitored. Animal studies were approved by the University of Michigan Committee on Use and Care of Animals.
Experimental VT was created in the mouse using infrarenal inferior vena cava (IVC) ligation as previously described.6, 12-14 Briefly, mice were anesthetized, and underwent midline laparotomy. The IVC was ligated with a 7 - 0 prolene suture immediately below the renal veins. Back (lumbar) branches were ablated with cautery, and all visible side branches were interrupted with 7 - 0 prolene suture. Mice were sacrificed on days 2, 8, and 21d, post ligation. At sacrifice, the thrombosed IVC segment was carefully dissected and removed for formalin fixation and paraffin embedding (for histology/immunohistochemistry) or immediately snap frozen (-70° C) to preserve for tissue processing.
At days 2 and 8, the thrombus was separated from the vein wall for processing; however, at 21d, the thrombus and vein wall form a segment of scar tissue, which cannot be separated without tissue disruption, and those segments were processed together.
SDS-PAGE Gelatin Zymography and Reverse Zymography
Activity of the gelatinases (MMP2 and MMP9, active and latent forms) was determined by gelatin zymography on 10% SDS-polyacrylamide gels, as previously described.5, 13, 15 Activity was visualized as light staining bands on a dark background, and normalized to the total amount of protein present in each sample. Activity of the TIMPs (active and latent forms of TIMP-1, TIMP-2) was determined by reverse zymography on 10% SDS polyacrylamide gels, copolymerized with gelatin and human pro-MMP2, which degrades gelatin and is susceptible to inhibition by all TIMPs, as previously described.6
Histology / Immunohistochemical/ Apoptosis/ Collagen Staining
Tissue samples were formalin fixed, paraffin embedded, and cut into 5 μm sections as described.12, 13 Nonspecific antigen sites were blocked with normal serum to decrease non-specific staining, and sections were incubated with primary antibodies to Mac2 (1:200, Cedarlane Laboratories, Burlington, NC), von Willebrand's Factor (vWF; 1:500, Abcam, Cambridge, MA), Ki-67 (1:100, Abcam, Cambridge, MA) and FSP-1(1:100, eBioscience). A species-specific ABC peroxidase kit for either rabbit or rat (Vector Laboratories Inc., Burlingame, California) was used according to the manufacturer's instructions for the corresponding secondary antibody and subsequent steps. The slides were counterstained with hematoxylin. In a blinded fashion, positive cells in 5 high power fields (1000×) radially around the IVC were counted and totaled.
The presence of apoptotic cell death was assessed in the vein wall by using a commercially available kit to determine the presence of characteristic DNA breaks by the TUNEL method (Trevigen Inc., Gaithersburg, MD).
Picrosirius red staining to quantify collagen content was performed as described.16, 17 These sections were then analyzed in crossed-plane polarized light from a monochromatic source to assess cross linked collagen. Two images for each were obtained using a Zeiss Axio M1 scope and Zeiss AxioVision software (Carl Zeiss Microimaging GmbH, Göttingen, Germany) at 0 and 90 degrees to the plane of polarization, in order to capture the birefringence of fibers extinguished in one direction. The images were analyzed blindly utilizing NIH Image J software. The area corresponding to the vein wall was selected as a region of interest, and then the image underwent threshold segmentation to differentiate collagen from other (mainly cellular and empty space) components of the vein wall. A vein wall collagen score was assigned by the formula [(%birefringent area) × (measured vein wall area)] / (total specimen area).
To account for non-collagen vein wall changes, intimal thickness scoring was assessed from H and E sections as described.18 A consistent mid-section thrombosed IVC segment was used for all histological analysis.14, 15, 19
PAI-1 Western immunoblot
Protein levels of PAI-1 (1/1000 Santa Cruz, sc-8979) were measured via Western blot from vein wall harvested tissue as described.15 For normalization of proteins, the membranes were stripped and probed with anti-actin antibodies conjugated with HRP (Santa Cruz Biotechnology, Santa Cruz, CA). The membranes were developed with the West-Pico ECL kit (Pierce, Rockford, IL). Densitometry was performed using Image J program.15
Antigen Analysis by ELISA
Vein wall tissue was homogenized, subjected to ultrasonic sonication, followed by centrifugation samples centrifuged at 10,000×g for 5 minutes, and the supernatant was collected for analysis. Quantification of the analyzed antigens was normalized to the total protein present in the sample, using a modified Bradford assay (Pierce Inc., Rockford, IL). ELISA for mouse tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), transforming growth factor-beta (TGFβ), and monocyte chemotactic protein-1 (MCP-1) was performed according to manufacturer's instructions (all from R and D, Minneapolis, MN).5, 12
Quantitative (real-time) RT-Polymerase Chain Reaction (PCR)
The levels of expression for genes of interest were determined by isolating total RNA (RNA) via Trizol extraction as previously described.13, 14 Then, the RNA underwent reverse transcription. The cDNA was then subjected to a real-time reverse transcriptase reaction using Taq polymerase (Promega, Madison, WI) in a SmartCycler quantitative PCR system (Cepheid, Sunnyvale, CA). SYBR green intercalating dye (Roche, Indianapolis, IN) was used to monitor levels of cDNA amplification for each gene. β-actin was used as an internal control for reference in each sample. The sequence numbers were: Col1a2 - RefSeq# NM_007743.3; Col3a1- RefSeq# NM_009930.2; tropoelastin - RefSeq # NM_ 0036834.3; beta Actin- RefSeq# NM_007393.3.
Statistical Analysis
All data are presented as mean +/- SE. Comparisons were made using an unpaired Student's t-test or ANOVA with Dennett's multiple comparison tests as appropriate, using GraphPad Prism version 4.0 for Windows (Graphpad Software, San Diego, CA).
Results
MMP9 deletion affects late VT resolution
The weight:length ratio of a resolving thrombus provides a simple but reliable measure of thrombus resolution.6, 12-14 Given that the inflammatory response is crucial for resolving VT, manipulations which affect inflammation might impair thrombus resolution. No significant differences were observed in VT size in MMP9 -/- at 2 or 8d, but at 21d, the VT were 18% smaller in the MMP9 -/- as compared with WT (P < .001, N = 22 - 24) (Figure 1). Thrombus neovascularization is associated with resolution12 and MMPs may affect angiogenesis. There was no difference in 8d thrombus neovascularization as assessed by vWF + channel staining in MMP9-/- as compared with WT (3 ± 2 vs. 6 ± 1 channels/5 hpf; (N = 4-5, P = .18)
Figure 1.
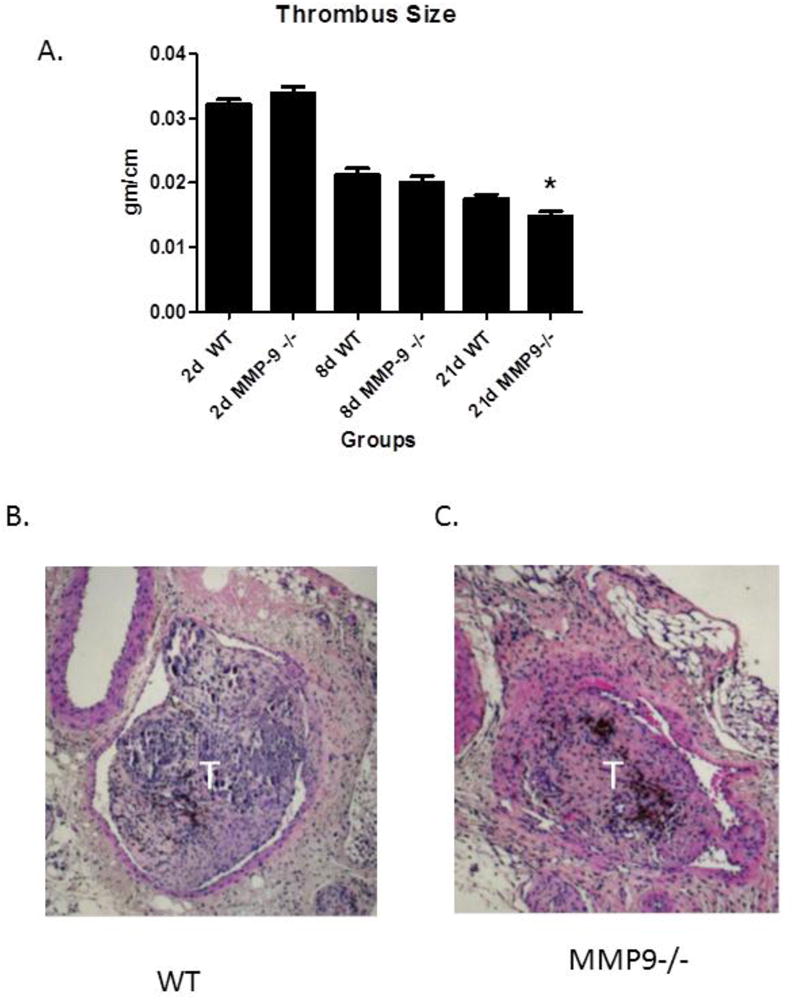
Thrombus size as measured by weight/length ratio in MMP9 -/- (A) and MMP2 -/- mice. (B) 21d H & F section of thrombosed IVC. (C) 21d section of MMP9 -/- vein wall. W = wall; T = thrombus. White bar = 10 μm.
Post-Thrombotic Vein Wall Collagen Metabolism is altered MMP9 -/- mice
The 8 and 21d time points were chosen to evaluate the effect of MMP9 deletion as this time frame is consistent with the PTS phenotype, with a significantly thicker and collagen dense vein wall. Moreover, MMP9 activity is present in the thrombosed vein wall.5, 6 The vein walls of MMP9 -/- mice had approximately 45% less collagen content as compared to WT at 8d (N = 4 − 5; P < .01) (Figure 2a - c), and trended so at 21d. However, intimal thickness was not significantly different between MMP9 -/- and WT (3.6 +/- .4 vs. 3.5 +/- .2 AU; N = 5, P = .87) at 8d. In MMP9 -/- mice, there was a paradoxical ∼2-fold increase in the expression of procollagen I as compared with WT (N = 5, P = .015) at 2 and 8d (Figure 3d). Procollagen III expression was increased 12.5 fold in MMP9 -/- at 8d, as compared with WT (N = 5, P = .009)(Figure 3e). Tropoelastin was upregulated ∼8-fold in MMP9 -/- mice as compared with WT at 8d (N = 4 − 5, P < .001)(Figure 3f).
Figure 2.
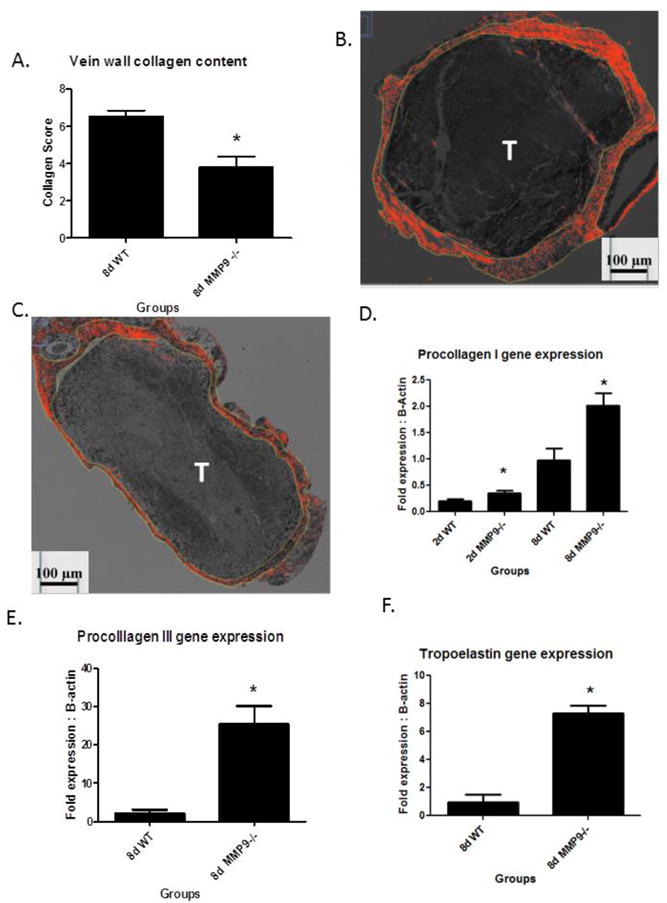
A) Vein wall collagen content was less in MMP9 -/- as compared to WT at 8d; B) WT thrombosed vein shows collagen marked by Sirius Red polarized fibers (outlined). C) Thinner and less collagen is present in MMP9 -/- 8d thrombosed vein. D) Procollagen I gene expression at 2 and 8d was greater in MMP-9 -/- vein wall as compared with WT. Procollagen III (E) and tropoelastin (F) gene expression was elevated in MMP9 -/- as compared to WT at 8d.* = P < .05. T = thrombus.
Figure 3.

(A) Increased MMP2 activity was present in MMP9 -/- mice at 8 and 21d as compared to WT. (B) Increased PAI-1 antigen was observed in 8d MMP9 -/-vein wall as compared with WT. Representative blot shown. *P < .05.
Matrix Metalloproteinase Activity and PAI-1
MMP activity is controlled by both secretion and activation, but also by inhibition by TIMPs, primarily TIMP1 and TIMP2.20 As assessed by reverse zymography, there were no significant differences in the vein wall TIMP1 or 2 activity in MMP9 -/- vein wall as compared with WT (not shown). Interestingly, a reciprocal increase of MMP2 activity was found in the MMP9 -/- mice. By gelatin zymography, MMP2 activity in the MMP9 -/- mice was elevated 2 – 3 fold at 8 and 21d (N = 5; P = .018; P < .01) as compared to WT (Figure 4). PAI-1 is the primary inhibitor of plasmin in the venous system,21 and has pleotropic effects. As PAI-1 is involved with vein wall remodeling,22 we assessed this at 8 and 21d. We found that PA1-1 antigen was significantly elevated in MMP9 -/- vein wall as compared with WT at 8d (N = 3, P = .02) (Figure 4). No difference was found at 21d.
Figure 4.
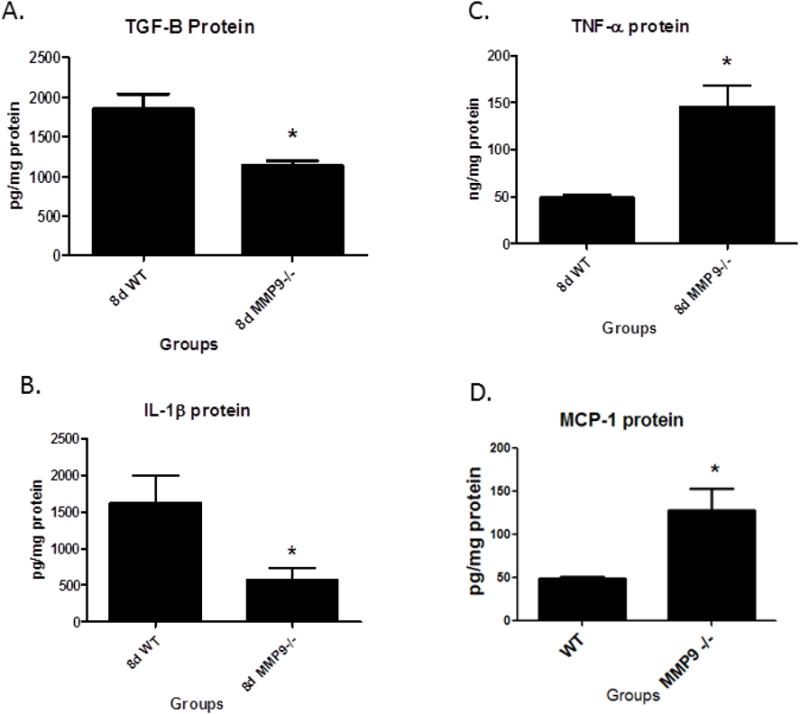
Vein wall TGFβ protein was reduced (A), while TNFα was increased (B) and IL1β was reduced (C) and MCP-1 was increased (D), in MMP9 -/- compared to WT, all at 8d. * = P < .05.
Inflammatory, Fibrotic factors, and Leukocytes
Inflammatory and fibrotic mediators are present in the thrombus and vein wall after VT, including TNFα, IL1β, TGFβ, and MCP-15, 12, 13 (Figure 5). The MMP9 -/- mice had ∼30% less TGFβ protein in the vein wall as compared with WT at 8d (N = 5 − 6; P = .008). There was 2.5 fold greater TNFα (N = 4 − 5; P = .03), but 3 fold less IL-1β protein in the MMP9 -/- vein wall compared to WT (N = 4 − 5; P = .02). Vein wall MCP-1 was elevated 2 fold in MMP9-/- as compared with control (N = 3 - 5, P = .05).
Figure 5.
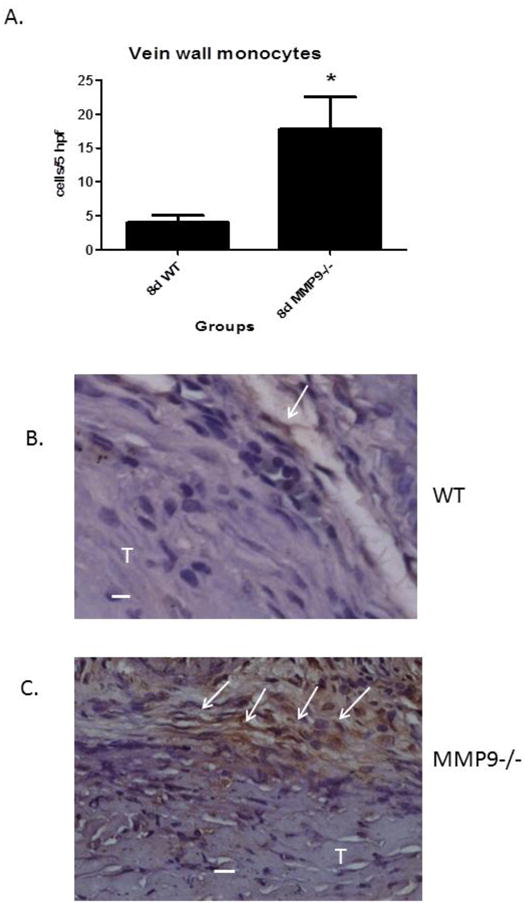
(A) Vein wall monocytes were increased in the MMP9 -/- mice as compared with WT at 8d. (B) Photo micrograph showing few F4/80+ cells in WT while many are present in MMP9 -/-. W = wall; T = thrombus; arrows mark (+) cells; * p < .05. Bar = 10 μm.
Since MMP2 is both produced by leukocytes in the early inflammatory response, and, in part mediates their migration across collagen-rich matrices such as the basement membrane,23 we investigated whether the loss of MMP9 would affect the number of inflammatory cells present in the vein wall. Vein wall monocytes were increased ∼4 fold in the MMP9 -/- mice as compared with WT at 8d (N = 4 − 5; P = .02) (Figure 5 c - f). Medial cellular proliferation and apoptosis contributes to post vascular injury fibrosis.20, 24 To evaluate for the contribution of cellular proliferation, Ki67 antigen staining was evaluated in the medial vein layer.5 No significant differences were observed in Ki67 + cells in MMP9 -/- mice as compared with WT at 8d. No difference in apoptosis (TUNEL + cells) in MMP9 -/- mice as compared with WT was found. We also find no significant differences in myofibroblasts in WT as compared to MMP9-/- vein walls (FSP-1 + cells; 18±2 vs. 18±4; N = 4-5, P = .95).
Discussion
No specific medical therapies are available to target the vascular inflammatory and fibrotic processes that result in PTS.2, 4 Remodeling of the ECM in vessels is mediated in part by the MMPs,20 and activity has been visualized in vivo in arterial injury25 and veins after VT.26 Resolving VT and the vein wall response is associated with MMP9 time dependent activity changes, but the direct role has not been assessed.6, 7 We have recently evaluated the role of MMP2 in vein wall response and VT resolution.15 (Deatrick, KB, etal: JVS, in press) In this study, we demonstrate late VT resolution was accelerated in MMP9 -/- mice, and that the midterm post thrombotic vein wall fibrotic response is dependent on MMP9.
Although not the primary focus of these experiments, VT resolution was accelerated in MMP9 -/- mice at the late time point (21d), evidenced by smaller VT. This may be related to compensatory MMP2 activity at 8 and 21d in the MMP9 -/- mice, where MMP2 activity was elevated as compared with WT. Increased MMP2 has been associated with VT resolution and may be a complementary mechanism for later VT resolution.15 Prior work in our lab has also shown that deletion or inhibition of MMP2 is associated with impaired thrombus resolution at 4d,13, 15 and 8d (Deatrick, KB etal, JVS, in press).Thrombi resolve in part by neovascularization,8 and MMP9 is critical for neovascularization.27 Consistently, deletion of MMP9 did not affect this process as VT size was similar at 2 and 8d. It is also possible other MMP activities may be increased in the MMP9-/- mice, and account for the smaller VT at 21d.13
Numerous studies have highlighted the role of MMPs in the response to vascular injury, including VT resolution.6, 20 Matrix metalloproteinases are zinc containing endoproteinases with multiple targets, including matrix and non-matrix substrates.23 In multiple models of tissue injury, early activation of MMP's occurs prior to the end stage-fibrotic process,20 including our own model.6 In this study, we demonstrate MMP9 deletion is associated with less vein wall collagen at midterm after the stasis thrombosis injury. Consistent with our findings is that MMP9 gene deletion is associated with less constrictive fibrosis in direct and flow mediated arterial injury models.24, 28-30 Although we did not specifically investigate the MMP9 -/- venous vascular smooth muscle cell (vSMC) migration potential, these reports suggest significant migration impairment in vitro.
These data also highlight the disconnect between thrombus size and vein wall injury. That is, vein wall fibrotic injury was less at 8d, yet no difference in VT size was observed. This suggests that blunting specific aspects of inflammation may not impair VT resolution, but may decrease vein wall injury. Recent data in PAI-1 -/- mice with increased plasmin activity, showed an increase in vein wall injury at day 8.22 Conversely, PAI-1 overexpressing mice have larger VT, decreased plasmin, decreased MMP9 activity, and less vein wall fibrotic injury (unpublished data).
In general, the MMPs regulate vessel collagen by degradation as well as cellular influx and function.20, 23 There was a paradoxical increase in pro-collagen and tropoelastin gene expression in the MMP9 -/- vein walls. Explanations include impaired post translational gene regulation, altered collagen turnover with loss of feedback inhibition, or altered growth factor and cytokine production. TGFβ in particular is a well-established mediator of fibrotic injury and is implicated in post translational signaling and endothelial to mesenchymal transformation.31, 32 In the MMP9 -/- mice, decreased TGFβ may have led to impaired gene translation of procollagen I and III. Moreover, MMP9 is important for post collagen compaction,28 and this lack of structural feedback may have stimulated procollagen gene expression. However, it is unlikely that mesenchymal transformation accounted for the difference in collagen content, the number of myofibroblasts did not significantly differ between the MMP-/- and WT mice. Similarly, no difference in the proliferation - apoptotic balance of the medial vein cells was found between the genotypes. Diminished vein wall fibrotic response and major phenotype differences were observed at 8d post VT. In TIMP1 -/- mice, which have significantly increased MMP2 and 9 activities at 8 and 21d, a corollary increase was not found in vein wall fibrotic injury (unpublished data). This suggests that increased MMP2 and 9 activity itself does not worsen parameters of vein wall healing in the stasis VT model, contrasting with solid organ injury models.33 Thus, an indirect effect of MMP9 gene deletion may be responsible for these divergent results. However, the increased MMP2 compensatory activity may have accounted for no decrease in vein wall injury at 21d.
Vein wall monocytes in the vein wall predominate at 8d post-thrombosis and may or may not promote fibrotic tissue repair.5, 12, 13, 34 Interestingly, MMP9 -/- mice had increased vein wall monocyte influx at this time point, possible due to compensatory increase in MMP-2 which is primarily macrophage-derived.6, 35 Another mechanism accounting for increased monocytes may involve MMP chemokine processing,23 as MCP-1 was significantly elevated at 8d in MMP9 -/- mice. Consistent with the current report is that P-selectin inhibition after stasis VT is associated with increased vein wall monocytes, but less vein wall fibrosis.18
Recent investigations suggest that monocyte subtypes confer differing inflammatory or anti-inflammatory responses depending on the environment.34 In vitro, engagement of monocytes to the vitronectin receptor αvβ3 results in transformation to a pro-inflammatory phenotype.36 PAI-1 is a potent inhibitor of vitronectin-mediated cellular adhesion,37 and the increase in PAI-1 in the MMP9-/- vein walls may have inhibited conversion of monocytes to a pro-injurious phenotype. Supporting this observation, we have found PAI-1 gene deleted mice have increased vein wall fibrosis at 8d post thrombosis.22 Current investigations are focused on the role of vitronectin on vein wall remodeling.
Targeting of MMPs has been evaluated in various models of cardiovascular injury and VT with variable responses.10, 20, 38 While the deletion of MMP9 in these experiments correlates with diminished fibrosis at 8d, the significance of this effect is lost by 21 days. Thus, the role of MMP9 in the fibrotic response is temporal to the duration of thrombus contact. Translationally, the timing and duration of MMP9 inhibition needs to guide future studies in treatment of PTS, and a practical means to reduce up-regulation of MMPs may be by limiting the thrombus-vein wall contact time via pharmacomechanical therapies.4 For example, it may be most advantageous to administer a MMP inhibitor after the DVT has lysed, to decrease the unbalanced plasmin effect on MMP activation and vein wall fibrotic injury. These findings also suggest a complex interplay of factors that is likely not simply related to collagen or matrix turnover.20, 23
Acknowledgments
Supported by: HL083918 and HL092129 (PKH), and T32HL092129 (TW)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger V, Rosamond W, Sacco R, Sorlie P, Stafford R, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J. Circulation. 2009. Heart disease and stroke statistics--2010 update. A report from the american heart association. [DOI] [PubMed] [Google Scholar]
- 2.Bergan JJ, Schmid-Schonbein GW, Smith PD, Nicolaides AN, Boisseau MR, Eklof B. Chronic venous disease. N Engl J Med. 2006;355:488–498. doi: 10.1056/NEJMra055289. [DOI] [PubMed] [Google Scholar]
- 3.Johnson BF, Manzo RA, Bergelin RO, Strandness DE., Jr Relationship between changes in the deep venous system and the development of the postthrombotic syndrome after an acute episode of lower limb deep vein thrombosis: A one- to six-year follow-up. J Vasc Surg. 1995;21:307–312. doi: 10.1016/s0741-5214(95)70271-7. discussion 313. [DOI] [PubMed] [Google Scholar]
- 4.Henke PK, Comerota AJ. An update on etiology, prevention, and therapy of postthrombotic syndrome. J Vasc Surg. 2011;53:500–509. doi: 10.1016/j.jvs.2010.08.050. [DOI] [PubMed] [Google Scholar]
- 5.Henke PK, Varma MR, Moaveni DK, Dewyer NA, Moore AJ, Lynch EM, Longo C, Deatrick CB, Kunkel SL, Upchurch GR, Jr, Wakefield TW. Fibrotic injury after experimental deep vein thrombosis is determined by the mechanism of thrombogenesis. Thromb Haemost. 2007;98:1045–1055. [PubMed] [Google Scholar]
- 6.Deatrick KB, Eliason JL, Lynch EM, Moore AJ, Dewyer NA, Varma MR, Pearce CG, Upchurch GR, Wakefield TW, Henke PK. Vein wall remodeling after deep vein thrombosis involves matrix metalloproteinases and late fibrosis in a mouse model. J Vasc Surg. 2005;42:140–148. doi: 10.1016/j.jvs.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 7.Dahi S, Lee JG, Lovett DH, Sarkar R. Differential transcriptional activation of matrix metalloproteinase-2 and membrane type-1 matrix metalloproteinase by experimental deep venous thrombosis and thrombin. J Vasc Surg. 2005;42:539–545. doi: 10.1016/j.jvs.2005.04.051. [DOI] [PubMed] [Google Scholar]
- 8.Henke PK, Varma MR, Deatrick KB, Drewyer NA, Lynch EM, Moore AJ, Dubay DA, Sukheepod P, Pearce CG, Upchurch GR, Jr, Kunkel SL, Franz MG, Wakefield TW. Neutrophils modulate post-thrombotic vein wall remodeling but not thrombus neovascularization. Thromb Haemost. 2006;95:272–281. doi: 10.1160/TH05-02-0099. [DOI] [PubMed] [Google Scholar]
- 9.Dewyer NA, Sood V, Lynch EM, Luke CE, Upchurch GR, Jr, Wakefield TW, Kunkel S, Henke PK. Plasmin inhibition increases mmp-9 activity and decreases vein wall stiffness during venous thrombosis resolution. J Surg Res. 2007;142:357–363. doi: 10.1016/j.jss.2007.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sood V, Luke C, Miller E, Mitsuya M, Upchurch GR, Jr, Wakefield TW, Myers DD, Henke PK. Vein wall remodeling after deep vein thrombosis: Differential effects of low molecular weight heparin and doxycycline. Ann Vasc Surg. 2010;24:233–241. doi: 10.1016/j.avsg.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deatrick KB, Elfline M, Baker N, Luke CE, Blackburn S, Stabler C, Wakefield TW, Henke PK. Postthrombotic vein wall remodeling: Preliminary observations. J Vasc Surg. 53:139–146. doi: 10.1016/j.jvs.2010.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henke PK, Varga A, De S, Deatrick CB, Eliason J, Arenberg DA, Sukheepod P, Thanaporn P, Kunkel SL, Upchurch GR, Wakefield TW. Deep vein thrombosis resolution is modulated by monocyte cxcr2-mediated activity in a mouse model. Arterioscler Thromb Vasc Biol. 2004;24:1130–1137. doi: 10.1161/01.ATV.0000129537.72553.73. [DOI] [PubMed] [Google Scholar]
- 13.Henke PK, Pearce CG, Moaveni DM, Moore AJ, Lynch EM, Longo C, Varma M, Dewyer NA, Deatrick KB, Upchurch GR, Jr, Wakefield TW, Hogaboam C, Kunkel SL. Targeted deletion of ccr2 impairs deep vein thombosis resolution in a mouse model. J Immunol. 2006;177:3388–3397. doi: 10.4049/jimmunol.177.5.3388. [DOI] [PubMed] [Google Scholar]
- 14.Henke PK, Mitsuya M, Luke CE, Elfline MA, Baldwin JF, Deatrick KB, Diaz JA, Sood V, Upchurch GR, Wakefield TW, Hogaboam C, Kunkel SL. Toll-like receptor 9 signaling is critical for early experimental deep vein thrombosis resolution. Arterioscler Thromb Vasc Biol. 31:43–49. doi: 10.1161/ATVBAHA.110.216317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sood V, Luke CE, Deatrick KB, Baldwin J, Miller EM, Elfline M, Upchurch GR, Jr, Wakefield TW, Henke PK. Urokinase plasminogen activator independent early experimental thrombus resolution: Mmp2 as an alternative mechanism. Thromb Haemost. 2010;104:1174–1183. doi: 10.1160/TH10-03-0184. [DOI] [PubMed] [Google Scholar]
- 16.Junqueira LC, Bignolas G, Brentani RR. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem J. 1979;11:447–455. doi: 10.1007/BF01002772. [DOI] [PubMed] [Google Scholar]
- 17.Cuttle L, Nataatmadja M, Fraser JF, Kempf M, Kimble RM, Hayes MT. Collagen in the scarless fetal skin wound: Detection with picrosirius-polarization. Wound Repair Regen. 2005;13:198–204. doi: 10.1111/j.1067-1927.2005.130211.x. [DOI] [PubMed] [Google Scholar]
- 18.Myers DD, Jr, Henke PK, Bedard PW, Wrobleski SK, Kaila N, Shaw G, Meier TR, Hawley AE, Schaub RG, Wakefield TW. Treatment with an oral small molecule inhibitor of p selectin (psi-697) decreases vein wall injury in a rat stenosis model of venous thrombosis. J Vasc Surg. 2006;44:625–632. doi: 10.1016/j.jvs.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 19.Moaveni DK, Lynch EM, Luke C, Sood V, Upchurch GR, Wakefield TW, Henke PK. Vein wall re-endothelialization after deep vein thrombosis is improved with low-molecular-weight heparin. J Vasc Surg. 2008;47:616–624. doi: 10.1016/j.jvs.2007.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: The good, the bad, and the ugly. Circ Res. 2002;90:251–262. [PubMed] [Google Scholar]
- 21.Singh I, Burnand KG, Collins M, Luttun A, Collen D, Boelhouwer B, Smith A. Failure of thrombus to resolve in urokinase-type plasminogen activator gene-knockout mice: Rescue by normal bone marrow-derived cells. Circulation. 2003;107:869–875. doi: 10.1161/01.cir.0000050149.22928.39. [DOI] [PubMed] [Google Scholar]
- 22.Baldwin JF, Sood V, Elfline MA, Luke CE, Dewyer NA, Diaz JA, Myers DD, Wakefield T, Henke PK. The role of urokinase plasminogen activator and plasmin activator inhibitor-1 on vein wall remodeling in experimental deep vein thrombosis. J Vasc Surg. 2012;56:1089–1097. doi: 10.1016/j.jvs.2012.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 24.Cho A, Reidy MA. Matrix metalloproteinase-9 is necessary for the regulation of smooth muscle cell replication and migration after arterial injury. Circ Res. 2002;91:845–851. doi: 10.1161/01.res.0000040420.17366.2e. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, Nie L, Razavian M, Ahmed M, Dobrucki LW, Asadi A, Edwards DS, Azure M, Sinusas AJ, Sadeghi MM. Molecular imaging of activated matrix metalloproteinases in vascular remodeling. Circulation. 2008;118:1953–1960. doi: 10.1161/CIRCULATIONAHA.108.789743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ripplinger CM, Kessinger CW, Li C, Kim JW, McCarthy JR, Weissleder R, Henke PK, Lin CP, Jaffer FA. Inflammation modulates murine venous thrombosis resolution in vivo: Assessment by multimodal fluorescence molecular imaging. Arterioscler Thromb Vasc Biol. 2012;32:2616–2624. doi: 10.1161/ATVBAHA.112.251983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pepper MS. Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler Thromb Vasc Biol. 2001;21:1104–1117. doi: 10.1161/hq0701.093685. [DOI] [PubMed] [Google Scholar]
- 28.Johnson C, Galis ZS. Matrix metalloproteinase-2 and -9 differentially regulate smooth muscle cell migration and cell-mediated collagen organization. Arterioscler Thromb Vasc Biol. 2004;24:54–60. doi: 10.1161/01.ATV.0000100402.69997.C3. [DOI] [PubMed] [Google Scholar]
- 29.Kuzuya M, Kanda S, Sasaki T, Tamaya-Mori N, Cheng XW, Itoh T, Itohara S, Iguchi A. Deficiency of gelatinase a suppresses smooth muscle cell invasion and development of experimental intimal hyperplasia. Circulation. 2003;108:1375–1381. doi: 10.1161/01.CIR.0000086463.15540.3C. [DOI] [PubMed] [Google Scholar]
- 30.Johnson JL, Dwivedi A, Somerville M, George SJ, Newby AC. Matrix metalloproteinase (mmp)-3 activates mmp-9 mediated vascular smooth muscle cell migration and neointima formation in mice. Arterioscler Thromb Vasc Biol. 2011;31:e35–44. doi: 10.1161/ATVBAHA.111.225623. [DOI] [PubMed] [Google Scholar]
- 31.Yan X, Chen YG. Smad7: Not only a regulator, but also a cross-talk mediator of tgf-beta signalling. Biochem J. 2011;434:1–10. doi: 10.1042/BJ20101827. [DOI] [PubMed] [Google Scholar]
- 32.Ghosh AK, Vaughan DE. Pai-1 in tissue fibrosis. J Cell Physiol. 2012;227:493–507. doi: 10.1002/jcp.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vaillant B, Chiaramonte MG, Cheever AW, Soloway PD, Wynn TA. Regulation of hepatic fibrosis and extracellular matrix genes by the th response: New insight into the role of tissue inhibitors of matrix metalloproteinases. J Immunol. 2001;167:7017–7026. doi: 10.4049/jimmunol.167.12.7017. [DOI] [PubMed] [Google Scholar]
- 34.Wynn TA, Barron L. Macrophages: Master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245–257. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nosaka M, Ishida Y, Kimura A, Kondo T. Immunohistochemical detection of mmp-2 and mmp-9 in a stasis-induced deep vein thrombosis model and its application to thrombus age estimation. Int J Legal Med. 2010;124:439–444. doi: 10.1007/s00414-010-0484-y. [DOI] [PubMed] [Google Scholar]
- 36.Antonov AS, Antonova GN, Fujii M, ten Dijke P, Handa V, Catravas JD, Verin AD. Regulation of endothelial barrier function by tgf-beta type i receptor alk5: Potential role of contractile mechanisms and heat shock protein 90. J Cell Physiol. 2012;227:759–771. doi: 10.1002/jcp.22785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Preissner KT, Reuning U. Vitronectin in vascular context: Facets of a multitalented matricellular protein. Semin Thromb Hemost. 2011;37:408–424. doi: 10.1055/s-0031-1276590. [DOI] [PubMed] [Google Scholar]
- 38.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: Structure, function, and biochemistry. Circ Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]