

Abstract
Inhibition of GSK-3β has been well documented to account for the behavioral actions of the mood stabilizer lithium in various animal models of mood disorders. Recent studies have showed that genetic or pharmacological inhibition of GSK-3β resulted in anxiolytic-like and pro-social behavior. In our ongoing efforts to develop GSK-3β inhibitors for the treatment of mood disorders, SAR studies on maleimide-based compounds were undertaken. We present herein for the first time that some of these GSK-3β inhibitors, in particular analogs 1 and 9, were able to stimulate progesterone production in the MA-10 mouse tumor Leydig cell model of steroidogenesis without any significant toxicity. These two compounds were tested in the SmartCube® behavioral assay and showed anxiolytic-like signatures following daily dose administration (50 mg/kg, i.p.) for 13 days. Taken together, these results support the hypothesis that GSK-3β inhibition could influence neuroactive steroid production thereby mediating the modulation of anxiety-like behavior in vivo.
Keywords: kinase inhibitor, GSK-3, maleimides, steroidogenesis, lithium, anxiolytic
INTRODUCTION
The mood stabilizer lithium, commonly available in the carbonate form (Eskalith®), has remained as a primary treatment of choice for manic-depressive illness with demonstrated anti-suicidal and neuroprotective efficacies.1, 2 Although the exact molecular mechanisms underlying the actions of lithium are still not completely understood, it has been reported to directly inhibit a series of biologically relevant enzymes including, but not limited to, glycogen synthase kinase-3 (GSK-3), inositol monophosphatase, phosphoglucomutase, other inositol polyphosphatases, and monophosphoesterases.3 Other proposed mechanisms of lithium action in psychiatric disorders also emerged, such as the ionic hypothesis, modulation of neurotransmitter signaling, arachidonic acid metabolism, and modulation of the hypothalamus-pituitary-adrenal (HPA) axis activity.4, 5 Among these reported possible molecular mechanisms of lithium’s action, the inhibition of GSK-3—either directly or through the destabilization of the Akt/β-arrestin/protein phosphatase 2A complex—has been well documented to account for the behavioral actions of lithium in animal models.6-10 GSK-3 is a multifunctional serine/threonine kinase responsible for the control of a number of central physiological events, such as cell membrane signal-to-gene transcription, metabolic homeostasis, cell development, neurogenesis, and cell survival/apoptosis.11, 12 Two homologs of GSK-3 exist: GSK-3α (51 kDa) and GSK-3β (47 kDa) which share more than 95% amino acid sequence homology in the catalytic site, with the latter being predominantly expressed in the nervous system. GSK-3 is constitutively active in basal non-stimulated cells, thereby catalyzing the phosphorylation of numerous signaling molecules, such as glycogen synthase, β-catenin transcription factor, and tau proteins, leading to their degradation.13, 14 Phosphorylation of the Tyr279 and Tyr 216 on GSK-3α and GSK-3β, respectively, is required for their activation, while Ser21/Ser9 phosphorylation at GSK-3α/β is necessary for their enzymatic inhibition. Significantly, GSK-3 has also been shown to be regulated not only by the mood stabilizers lithium and valproate but also by antidepressants and antipsychotics that modulate the key neurotransmitters signaling in the brain such as dopamine and serotonin.15, 16 Taken together, these findings support the role of GSK-3 inhibition as a key convergence point for at least some neuropsychiatric drugs routinely used for the treatment of mental disorders although the molecular mechanisms by which GSK-3 inhibition elicits mood stabilization remains elusive.17, 18
Recently, selective deletion of forebrain GSK-3β in mice was reported to exert anxiolytic-like and pro-social behavioral effects.19 In line with this study, lithium per se or in combination with other drugs has been demonstrated to suppress anxiety-like symptoms in different behavioral paradigms using mouse models of Fragile X syndrome, Huntington’s disease, and cerebral ischemia.20-24 In the context of anxiety disorders, alteration of the levels of neuroactive steroids such as pregnenolone and progesterone has been implicated in the disease pathophysiology.25 The anxiolytic effects of these neuroactive steroids are attributed to the binding to GABAA receptors which manifests in the potentiation of GABA-induced Cl- currents. A significant body of research in stress physiology has revealed the important roles of progesterone and its metabolite allopregnanolone in the modulation of HPA axis.26 Of particular relevance to the pathophysiology of anxiety disorders, dysregulation of the HPA system has been observed in patients and normalization with lithium therapy suggested that interactions of lithium with the HPA axis may contribute to its therapeutic effects.5 Interestingly, inhibition of GSK-3β (increased Ser9 phosphorylation) and the concomitant β-catenin accumulation has been reported to be an important signaling pathway for the ability of luteal cells to induce progesterone secretion when exposed to luteinizing hormone.27 Our group previously identified maleimide 1 (Figure 1) as a potent and selective, brain-penetrant, GSK-3β inhibitor which attenuates hyperactivity in a mouse model of mania induced by amphetamine and chlordiazepoxide.28 On the basis of the hypothesis that GSK-3 inhibition could induce the production of neurosteroids which in turn modulate the anxiety-like behavior in vivo, we sought to investigate the ability of these ATP-competitive maleimide-based GSK-3β inhibitors to control steroid formation in a steroidogenic cell model.
Figure 1.
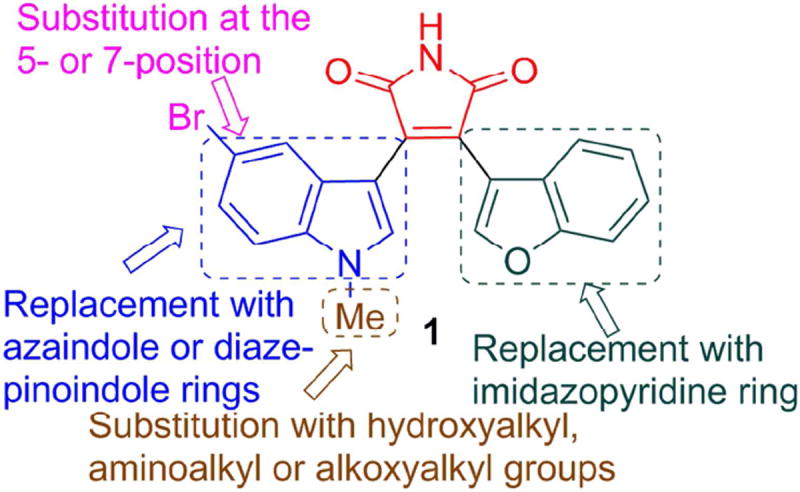
Synthetic modifications to improve the potency and water solubility of 3-(benzofuran-3-yl)-4-(5-bromo-1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione 1.
CHEMISTRY
Based upon a body of SAR information accumulated over the years for the development of benzofuran-3-yl-(indol-3-yl)maleimides as GSK-3β inhibitors, 6-substitution on the indole ring has generally been shown to decrease GSK-3β inhibitory activity. On the other hand, substitution at the 5- or 7-position of the indole ring is generally well tolerated,29 as exemplified by 5-halogen bearing analogs which include the previously reported maleimide 1. A new generation of maleimides 2–31 was synthesized with the aim to improve upon potency and water solubility (Figure 1) following the synthetic routes depicted in Schemes 1-4. Commercially available indoles 32a and 32b were initially alkylated with O-trityl-3-bromopropan-1-ol or O-trityl-2-bromoethan-1-ol in N,N-dimethylformamide (DMF) as a solvent and sodium hydride as a base (Scheme 1). Treatment of the alkylated indoles 33 with ethyl chlorooxoacetate in diethyl ether afforded the glyoxalates 34. Perkin-type condensation30 between the glyoxalates and benzofuran-3-yl-acetamide28 generated the desired maleimides 2–4. Alkylation of 32a and 32b with tert-butyl (2-bromoethyl)carbamate followed by treatment of the intermediary indoles 35a and 35b with ethyl chlorooxoacetate gave the glyoxalates 36a and 36b. Maleimide formation with benzofuran-3-yl-acetamide and subsequent Boc deprotection using trifluoroacetic acid (TFA) in chloroform yielded the target compounds 5 and 6. The N-methylaminopropyl (7, 8) and 2-methoxyethyl analogs (9–12) were synthesized in a similar fashion. Commercially available 7-azaindoles 37a and 37b were subjected to the sequential N-alkylation, glyoxylation and condensation with benzofuran-3-yl-acetamide to give maleimides 13–16 (Scheme 2).31, 32 6-Aza-5-methoxyindole was used as a starting material for target compounds 17 and 18. 2-(Imidazo[1,2-a]pyridin-3-yl)acetamide33 38 was condensed with the appropriate glyoxalates to afford the final maleimides 19 and 20. For compounds 21–23, indole-7-carboxaldehyde 39 was first alkylated with the appropriate alkyl bromides and subjected to sequential Wittig reaction with methylenetriphenylphosphorane and then hydroboration. Alkylation of the intermediary alcohols 40a and 40b followed by treatment with ethyl chlorooxoacetate resulted in the formation of the glyoxalates 41a–c, which upon condensation with benzofuran-3-yl-acetamide gave the desired maleimides 21–23.
Scheme 1. Synthesis of Target Compounds 2–12.a.

a Reagents and conditions: (a) (i) NaH (1.5 equiv.), DMF, rt, 0.5 h; (ii) O-trityl-3-bromopropan-1-ol or O-trityl-2-bromoethan-1-ol (1.2 equiv.), 60 °C, 16 h, 80–99%; (b) ethyl chlorooxoacetate (5 equiv.), Et2O, 0 °C to rt, 16 h, 42–74%; (c) benzofuran-3-yl-acetamide (1.1 equiv.), t-BuOK (2.5 equiv.), THF or DMF, 0 °C to rt, 16 h, 22–51%; (d) (i) NaH (1.5 equiv.), DMF, rt, 0.5 h; (ii) tert-butyl (2-bromoethyl)carbamate (1.2 equiv.), 60 °C, 16 h, 70–90%; (e) trifluoroacetic acid, CHCl3, rt, 2 h; (f) (i) NaH (1.5 equiv.), DMF, rt, 0.5 h; (ii) tert-butyl N-(3-bromopropyl)-N-methylcarbamate (1.2 equiv.), 60 °C, 16 h, 76–99%.
Scheme 4. Synthesis of Target Compound 26.a.

a Reagents and conditions: (a) 2-chloroethylamine hydrochloride (1.1 equiv.), K2CO3, DMF, sealed tube, 110 °C, 16 h, 25%; (b) 36% aq. HCHO (1.2 equiv.), AcOH, H2SO4, 70 °C, 16 h; (c) (Boc)2O (1 equiv.), THF, aq. K2CO3, 0 °C to rt, 5 h, 65% over 2 steps; (d) DDQ (1.2 equiv.), Et2O, PhMe, rt, 3 h, 50%; (e) ethyl chlorooxoacetate (5 equiv.), Et2O, 0 °C to rt, 16 h, 39%; (f) benzofuran-3-yl-acetamide (1.1 equiv.), t-BuOK (2.5 equiv.), THF or DMF, 0 °C to rt, 16 h, 30%; (g) trifluoroacetic acid, CHCl3, rt, 2 h.
Scheme 2. Synthesis of Target Compounds 13–23.a.
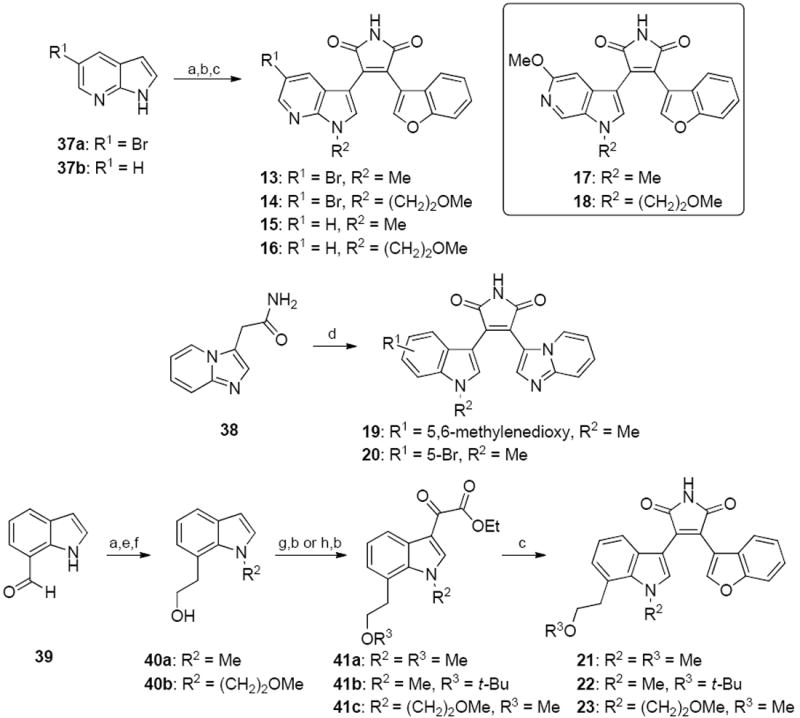
a Reagents and conditions: (a) (i) NaH (1.5 equiv.), DMF, rt, 0.5 h; (ii) R2Br (1.2 equiv.), 60 °C, 16 h, 85–99%; (b) ethyl chlorooxoacetate (5 equiv.), Et2O, 0 °C to rt, 16 h or (i) AlCl3 (5 equiv.), CH2Cl2, rt, 1 h; (ii) ethyl chlorooxoacetate (5 equiv.), Et2O, 0 °C to rt, 16 h, 31–83%; (c) benzofuran-3-yl-acetamide (1.1 equiv.), t-BuOK (2.5 equiv.), THF or DMF, 0 °C to rt, 16 h, 42–60%; (d) ethyl 2-(5-bromo-1-methyl-1H-indol-3-yl)-2-oxoacetate or ethyl 2-(5-methyl-5H-[1,3]dioxolo[4,5-f]indol-7-yl)-2-oxoacetate, t-BuOK (2.5 equiv.), THF or DMF, 0 °C to rt, 16 h, 30–40%; (e) Ph3PCH3Br (3 equiv.), t-BuOK (3 equiv.), THF, 0 °C to rt, 16 h, 80–90%; (f) (i) BH3.THF (1.1 equiv.), THF, rt, 2.5 h; (ii) 10% aq. NaOH (1.3 equiv.), 35% H2O2 (1.3 equiv.), rt, 3.5 h, 55–70%; (g) (i) NaH (1.5 equiv.), DMF, rt, 0.5 h; (ii) MeI (1.2 equiv.), 60 °C, 16 h, 90–99%; (h) tert-butyl 2,2,2-trichloroacetimidate (1.1 equiv.), cyclohexane, CH2Cl2, cat. BF3.OEt2, rt, 16 h, 53%.
To access target compounds 24, 25, 27–31 (Scheme 3), indole-7-carboxaldehyde 39 or 5-fluoroindole-7-carboxaldehyde 42 were initially subjected to a reductive amination protocol using ethanolamine and hydrogen gas with palladium(0) catalyst following a previously reported literature protocol.34 Sequential Boc protection of the secondary amine, tosylation of the hydroxyl group and sodium hydride-assisted cyclization34 gave the intermediary 1,2,3,4-tetrahydro-[1,4]diazepino[6,7,1-hi]indoles 43a and 43b. Glyoxylation with ethyl chlorooxoacetate, condensation with benzofuran-3-yl-acetamide and subsequent Boc deprotection afforded the maleimides 24 and 25. Amide coupling of the maleimides with the appropriate carboxylic acids using N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC.HCl) and HOBt as the coupling reagent or treatment with carbamoyl chlorides gave the target compounds 27–31. For the bromo analog 26 (Scheme 4), 5-bromoindoline 44 was first alkylated with 2-chloroethylamine hydrochloride using potassium carbonate as a base in DMF in a pressure tube and treated with aqueous formaldehyde in acetic acid and sulfuric acid35, followed by Boc protection. The resultant (tert-butyl 9-bromo-3,4,6,7-tetrahydro-[1,4]diazepino[6,7,1-hi]indole-2(1H)-carboxylate) 45 was subsequently treated with DDQ, followed by glyoxylation, condensation with benzofuran-3-yl-acetamide and Boc deprotection to yield the final maleimide 26.
Scheme 3. Synthesis of Target Compounds 24, 25 and 27–31.a.

a Reagents and conditions: (a) (i) ethanolamine (2 equiv.), 10% Pd/C (cat.), MeOH, rt, 1 h; (ii) H2, 1 atm, rt, 3 h, 84–95%; (b) (Boc)2O (1.2 equiv.), THF, aq. K2CO3, 0 °C to rt, 5 h, 85–95%; (c) MsCl (1.2 equiv.), N,N-diisopropylethylamine, THF, 0 °C to rt, 16 h, 72–96%; (d) NaH (1.5 equiv.), DMF, 0 °C, 1 h, 84–90%; (e) ethyl chlorooxoacetate (5 equiv.), Et2O, 0 °C to rt, 16 h, 42–49%; (f) benzofuran-3-yl-acetamide (1.1 equiv.), t-BuOK (2.5 equiv.), THF or DMF, 0 °C to rt, 16 h, 30–40%; (g) trifluoroacetic acid, CHCl3, rt, 2 h; (h) R4CO2H (1.2 equiv.), EDC.HCl (1.3 equiv.), HOBt (1.3 equiv.), DMF, N-methylmorpholine (3 equiv.), rt, 16 h, 30–50%; (i) R4COCl (1.5 equiv.), N,N-diisopropylethylamine (3 equiv.), MeOH, rt, 3 h, 30–50%.
RESULTS AND DISCUSSION
GSK-3 Inhibition by Maleimides 2–31
Testing of all the newly synthesized compounds for their ability to inhibit GSK-3β was carried out by Reaction Biology, Inc. using their standard protocol (Tables 1 and 2). The hydroxyethyl analogue 2 was found to have a similar potency compared to the parent maleimide 1 (IC50 value of 21 nM).36 Substitution of the bromine by a fluorine atom (2 vs 3) at the 5-position of the indole ring resulted in an approximately 4-fold increase in the potency, whereas lengthening of the hydroxyethyl to a hydroxypropyl moiety (3 vs 4) gave a 3-fold decrease in potency. Subsequent chemical modifications were carried out to improve upon the water solubility of these maleimides, which is commonly achieved by the addition of an ionizable nitrogen group. The aminoethyl substitution at the indole nitrogen (5 and 6) diminished the GSK-3β inhibition by approximately 6- to 8-fold in comparison with the hydroxyethyl group (compounds 2 and 3), with the 5-bromo analog 5 being 3-fold less potent than the 5-fluoro analog 6. This trend was also observed with the N-methylaminopropyl analogs 7 and 8. As a primary hydroxyl group generally poses a potential metabolic liability that arises from oxidation and bioconjugation processes, the hydroxyethyl moiety in analogs 2 and 3 was replaced with a 2-methoxyethyl group (compounds 9–12). Within this series, the unsubstituted indole analog 9 was identified as the most potent GSK-3β inhibitor with an IC50 value of 20 nM. The 5-fluoro and 5-bromo analogs 10 and 11 were found to be 2- and 4-fold less potent with IC50 values of 40 and 82 nM respectively. Direct comparison of the methoxy analogs 10 and 11 vs the hydroxyl analogs 3 and 2, respectively, showed that the hydroxyl analogs were approximately 4- to 8-fold more potent. The 5,6-difluoro analog 12 had a similar potency to the 5-fluoro analog with an IC50 value of 36 nM.
Table 1.
Inhibition of GSK-3β by Maleimides 2–20.

| ||||
|---|---|---|---|---|
| Cmpd. | R1 | X | R2 | IC50 (nM)a |
| 1 | 5-Br | CH | −CH3 | 21.1±1.8 |
| 2 | 5-Br | CH | −(CH2)2OH | 21.4±3.1 |
| 3 | 5-F | CH | −(CH2)2OH | 4.8±0.2 |
| 4 | 5-F | CH | −(CH2)3OH | 15.0±2.7 |
| 5 | 5-Br | CH | −(CH2)2NH2 | 116±9 |
| 6 | 5-F | CH | −(CH2)2NH2 | 41.1±0.7 |
| 7 | 5-Br | CH | −(CH2)3NHMe | 404±23 |
| 8 | 5-F | CH | −(CH2)3NHMe | 202±5 |
| 9 | H | CH | −(CH2)2OMe | 19.8±5.8 |
| 10 | 5-F | CH | −(CH2)2OMe | 39.7±0.6 |
| 11 | 5-Br | CH | −(CH2)2OMe | 82.3±5.7 |
| 12 | 5,6-difluoro | CH | −(CH2)2OMe | 35.7±1.9 |
| 13 | 5-Br | N | Me | 5.3±0.8 |
| 14 | 5-Br | N | −(CH2)2OMe | 25.2±1.2 |
| 15 | H | N | Me | 17.6±1.0 |
| 16 | H | N | −(CH2)2OMe | 58.2±3.5 |
| 17 | - | - | Me | 65.9±3.6 |
| 18 | - | - | −(CH2)2OMe | 127±5 |
| 19 | 5,6-methylenedioxy | - | Me | 5.3±1.0 |
| 20 | 5-Br | - | Me | 0.87±0.11 |
The synthesized maleimides were evaluated for their ability to inhibit phosphorylation of primed substrate (YRRAAVPPSPSLSRHSSPHQ(pS)EDEEE; 20 μM) by human GSK-3β in the presence of 10 μM ATP concentration. These compounds were tested at Reaction Biology Inc.37 and IC50 values are presented as average of duplicates ± SEM.
Table 2.
Inhibition of GSK-3β by Maleimides 21–31.

| |||||
|---|---|---|---|---|---|
| Cmpd. | R1 | R2 | R3 | R4 | IC50 (nM)a |
| 21 | H | Me | Me | - | 11.2±3.1 |
| 22 | H | Me | t-Bu | - | 148±15 |
| 23 | H | -(CH2)2OMe | Me | - | 114±16 |
| 24 | H | - | - | - | 4.2±0.7 |
| 25 | F | - | - | - | 3.7±0.5 |
| 26 | Br | - | - | - | 11.5±2.2 |
| 27 | H | - | - | 2-pyrazinyl | 0.20±0.06 |
| 28 | H | - | - | 4-pyridylmethyl | 0.59±0.11 |
| 29 | F | - | - | 1-piperidinyl | 1.9±0.4 |
| 30 | F | - | - | 4-piperidinopiperidin1-yl | 0.53±0.01 |
| 31 | F | - | - | 4-morpholinyl | 0.013±0.005 |
The synthesized maleimides were evaluated for their ability to inhibit phosphorylation of primed substrate (YRRAAVPPSPSLSRHSSPHQ(pS)EDEEE; 20 μM) by human GSK-3β in the presence of 10 μM ATP concentration. These compounds were tested at Reaction Biology Inc.37 and IC50 values are presented as either average of duplicates ± SEM.
The effects of: i) introduction of an azaindole ring in place of the indole ring (analogs 13–18) and ii) replacement of the benzofuran ring with an imidazopyridine (analogs 19 and 20) on the GSK-3β inhibition were examined.31, 32 To our delight, the azaindole analog 13 had an IC50 value of 5 nM while the extension of the methyl group to 2-methoxyethyl (compound 14) resulted in a 5-fold reduction in potency compared to 13. Deletion of the 5-bromo group gave a further ~3-fold reduction in GSK-3β inhibitory activity (15, 16 vs 13, 14), while the 5-methoxy-6-azaindoles 17 and 18 had IC50 values of 66 and 127 nM, respectively. Replacement of the benzofuran ring with an imidazopyridine (19 and 20) also resulted in a further increase in potency (IC50 values of 5 and 0.9 nM respectively).
The next set of SAR modifications focused on the introduction of a 2-methoxyethyl group at 7-position of the indole ring. Previous SAR studies had shown that introduction of hydroxyalkyl groups at the 7-position resulted in single digit nanomolar GSK-3β inhibitors.29 Towards this end, the 2-methoxyethyl attached at the 7-position of the indole ring of analog 21 led to a potent inhibitor with an IC50 value of 11 nM (Table 2). Replacement of the methoxy group by a tert-butoxy was found to be deleterious for activity (22 vs 21). The bisalkylated analog 23 had similar potency to that of compound 22. Inspired by the diazepinoindole scaffold originally reported by Eli Lilly & Co. in 2004,34 cyclization of the two alkyl chains to a diazepinoindole scaffold exemplified by compounds 24–31, resulted in highly potent GSK-3β inhibitors. The diazepinoindole analogs 24–26 all possessed IC50 values between 4 and 12 nM and further extension of the diazepinoindole nitrogen atom to an amide moiety gave even more potent GSK-3β inhibitors 27–31. Of a particular note, compound 31 was identified as the most potent GSK-3β inhibitor from this series with an IC50 value of 13 pM, representing the most potent compound to date in our library of GSK-3 inhibitors. In addition, the aqueous solubility of compounds 5, 20 and 24 was determined to be 180, 35 and 240 mg/L using an HPLC-based method.38
Characterization of GSK-3β Inhibitors as Stimulators of Steroidogenesis
In order to test the hypothesis that GSK-3β inhibition could induce the production of neuroactive steroids thereby mediating the modulation of anxiety-like behavior, 17 of the most potent, newly-synthesized GSK-3β inhibitors (Tables 1 and 2) were screened for their ability to induce progesterone expression in MA-10 mouse tumor Leydig cells. This cell line is a hormone-responsive steroidogenic cell model which has been extensively used to study the regulation of steroidogenesis whereby the biosynthesis of progesterone, the main steroid formed, is measured.39 Initially, MA-10 cells were incubated in the presence of the GSK-3β inhibitors for 2 h at a concentration of 100 μM and at the end of the incubation period, progesterone formation was measured by radioimmunoassay (RIA). As shown in Figure 2A, 7 of the 17 tested GSK-3β inhibitors, namely analogs 1, 9, 12, 13, 14, 21, and 26, were bioactive in stimulating progesterone production. Dose-response experiments were next carried out for all the tested compounds ranging from 0.1 to 100 μM, in which a statistically significant induction of progesterone synthesis was observed starting at a concentration of approximately 50 μM (Figure 2B). As the high micromolar / low millimolar concentrations posed significant concern regarding cytotoxicity, and as steroid production can be facilitated by disruption of mitochondrial membrane integrity,40 additional cytotoxicity profiling was performed. MA-10 Leydig cells were exposed to GSK-3 inhibitors for 2 h at 100 μM concentration and cellular toxicity assessed by the MTT assay (Figure 2C). Some of the tested analogs showed low, but observable signs of cytotoxicity at the 100 μM concentrations, for example compounds 16 and 23, whereas maleimides 12, 25-28, 30, and 31 were found to be significantly more cytotoxic at the tested concentration. Dose dependency curves for the cytotoxicity were also obtained for all the tested compounds, ranging from 0.1 to 100 μM concentrations (Figure 2D). Examination of the overall progesterone production and cytotoxicity data presented in Figures 2A-D revealed that out of the seven compounds that stimulated progesterone production (Figure 2A), analogs 12 and 26 were also found to be toxic to the MA-10 cells. However, the remaining five GSK-3 inhibitors that stimulated steroidogenesis (compounds 1, 9, 13, 14, and 21) showed either low or no apparent toxicity at a concentration of 100 μM. To examine the time-dependency of these GSK-3β inhibitors in stimulating steroidogenesis, two of the most potent, non-toxic stimulators 1 and 9 were incubated with MA-10 cells at a concentration of 50 μM for 0–120 min, media collected, and progesterone production was then assessed by RIA (Figure 3). There appears to be a biphasic time response to these two compounds, with the first maximum steroid production appearing within 15 min and remaining until 60 min, followed by a linear increase in steroid production between 60 and 120 min. These data altogether demonstrated for the first time that some of the maleimide-based GSK-3β inhibitors, namely 1, 9, 13, 14, and 21, were able to stimulate progesterone production in MA-10 tumor Leydig cells in a dose-dependent manner with either low or no apparent signs of toxicity as assessed by the MTT assay.
Figure 2.
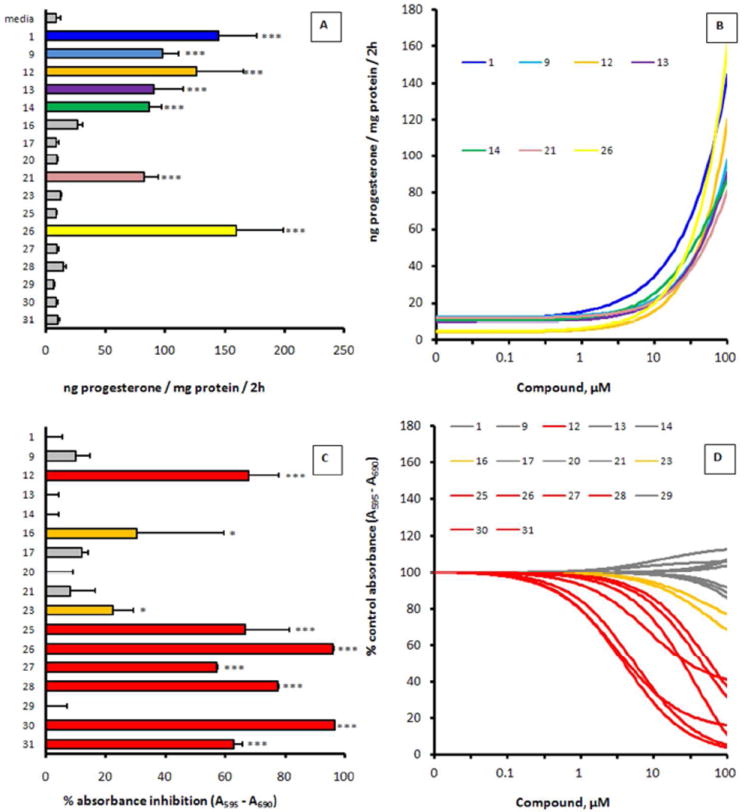
Steroidogenesis stimulation of maleimide-based GSK-3β inhibitors in MA-10 tumor Leydig cells. MA-10 tumor Leydig cells were incubated in the presence or absence of GSK-3β inhibitor, and at the end of incubation, steroid (progesterone) production and cellular toxicity were evaluated by RIA and MTT assay, respectively. (A). Steroid production by MA-10 cells exposed to 100 μM GSK-3 inhibitors. MA-10 Leydig cells were exposed to GSK-3 inhibitor for 2 h and steroid production assessed by RIA. ***, p<0.001 as assessed by one-way ANOVA with Newman-Keuls post-test. (B). Dose-dependent steroid production by MA-10 cells exposed to 0–100 μM GSK-3 inhibitors. MA-10 Leydig cells were exposed to GSK-3 inhibitor for 2 h and steroid production assessed by RIA. Only those that showed significant stimulation of steroid production are presented; the remaining tested compounds (16, 17, 20, 23, 25, 27–31) showed <30 ng progesterone/mg protein at all the tested concentrations after 2 h incubation. (C). Cellular toxicity of MA-10 cells exposed to 100 μM GSK-3 inhibitors. MA-10 Leydig cells were exposed to GSK-3 inhibitor for 2 h and cellular toxicity assessed by MTT assay. *, p<0.05 (orange); ***, p<0.001 (red) as assessed by one-way ANOVA with Newman-Keuls post-test. (D). Dose-dependent cellular toxicity of MA-10 cells exposed to 0–100 μM GSK-3 inhibitors. MA-10 Leydig cells were exposed to GSK-3 inhibitor for 2 h and cellular toxicity assessed by MTT assay (red = toxic, orange = slightly toxic, gray = non-toxic). Results are presented as the mean ± S.E.M. (n=3) for A and C, and presented as nonlinear regression curve fit of data points for B and D.
Figure 3.
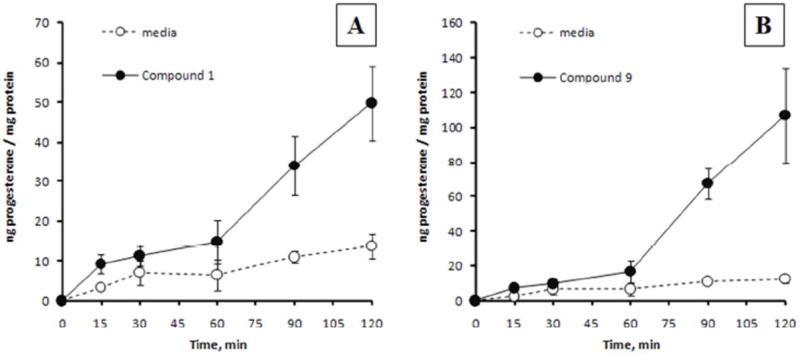
Steroidogenic time-response of GSK-3 inhibitors 1 and 9. MA-10 cells were cultured with 50 μM of 1 (panel A) or 9 (panel B) for 0–120 min and progesterone production was assessed by RIA. Results are presented as the mean ± S.E.M (n=2).
In vivo SmartCube® Assay for GSK-3 Inhibitors 1 and 9
Encouraged by the demonstrated ability of some of the newly synthesized GSK-3β inhibitors to stimulate steroidogenesis in vitro, two of the most potent, non-toxic stimulators 1 and 9 were administered to C57BL/6 mice in the proprietary SmartCube® system developed by Psychogenics, Inc.41, 42 This behavioral screening platform provides a sequence of challenges to mice and captures more than 1,400 features during the 45 min testing session. These features were analyzed with computer algorithms and data mining approaches to automate the study of mice behavior and compared to a database of behavioral signatures obtained using a set of diverse reference compounds, including antipsychotics, anxiolytics, and antidepressants.41 In this assay, mice were injected once a day with either compound 1 or 9 (50 mg/kg, i.p.) for 13 days, and the SmartCube® signatures were depicted in Figure 4. Neither compound demonstrated any classified behavioral activity after a single dose (15 min, day 1 test, Figure 4A). In contrast, a robust signature was obtained after chronic administration of both compounds over 13 days (Figure 4B). The signature classes for both compounds were found to be relatively similar, with the predominant portion of the signatures predicting anxiolytic-like activity, as well as some cognitive enhancer effects. In accordance with the ability of compounds 1 and 9 to stimulate steroidogenesis in MA-10 cells (Figure 2), the in vitro and in vivo data presented herein supported the hypothesis that GSK-3 inhibition could influence the formation of neuroactive steroids, which in turn mediate the anxiolytic-like behavior in living systems.
Figure 4.

Smartcube® signatures of GSK-3 inhibitors 1 and 9 after 1 (acute) or 13 days (chronic) of treatment, 15 min following one dose of each compound at 50 mg/kg i.p.
CONCLUSIONS
In this study, we presented our findings on the SAR exploration of the benzofuran-3-yl-(indol-3-yl)maleimides with the aim to improve their potency at GSK-3β inhibitory activity as well as their aqueous solubility. The replacement of indole ring in the original parent maleimide 1 with an azaindole or diazepinoindole moiety resulted in highly potent GSK-3β inhibitors with improved aqueous solubility. Compound 31 has been identified as the most potent GSK-3β inhibitor in our library thus far with an IC50 value of 13 pM. In order to test the hypothesis that GSK-3β inhibition could indeed stimulate steroid production thereby mediating the modulation of anxiety-like behavior in vivo, 17 newly synthesized analogs were screened for their ability to induce steroidogenesis in a steroidogenic cell model, the MA-10 mouse tumor Leydig cell lines. Following dose-response experiments measuring the progesterone induction and cytotoxicity profiles, compounds 1, 9, 13, 14, and 21 were identified for the first time to be the most potent steroidogenesis stimulators in this assay without any significant toxicity at the tested concentration of 100 μM. The two most potent, non-toxic steroidogenesis stimulators 1 and 9 were subsequently tested in a proprietary SmartCube® assay. Daily administration of each compound at 50 mg/kg intraperitoneally resulted in anxiolytic-like signatures after 13 days. Taken together, these data supported the hypothesis that GSK-3β inhibition could be involved in the induction of steroidogenesis and thus neuroactive steroid production, thereby mediating the modulation of anxiety-like behavior in vivo. Continued efforts are directed towards the elucidation of the role of GSK-3β inhibition on the stimulation of steroidogenesis.
EXPERIMENTAL SECTION
Chemical Synthesis
General Methods
Starting materials, reagents, and solvents were purchased from commercial suppliers and used without further purification unless otherwise stated. Anhydrous THF and CH2Cl2 were obtained by distillation over sodium wire or CaH2 respectively. All non-aqueous reactions were run under an argon atmosphere with exclusion of moisture from reagents. The progress of reactions was monitored by TLC on SiO2. For flash chromatography, 230–400 mesh particle size SiO2 was used. Melting points were determined on a Thomas-Hoover uni-melt apparatus and are uncorrected. Infrared absorption spectra were recorded on a Nicolet 6700 FT-IR spectrometer equipped with a Ge-on-KBr beamsplitter. 1H NMR and 13C NMR spectra were recorded on a Bruker spectrometer at 400 and 100 MHz, respectively, and were referenced to the residual peaks of CDCl3 at 7.26 ppm or DMSO-d6 at 2.50 ppm (1H NMR) and CDCl3 at 77.2 ppm or DMSO-d6 at 39.51 ppm (13C NMR). Chemical shifts were reported in parts per million (ppm) downfield of tetramethylsilane (TMS) and the following abbreviations used to denote coupling patterns: s = singlet; d = doublet, t = triplet, q = quartet, br = broad. 13C NMR chemical shifts are reported in ppm relative to the internal perdeuteriated solvent resonance and are specified as quarternary (C), tertiary (CH), secondary (CH2) or primary (CH3) where appropriate, using additional DEPT experiments. Mass spectra were measured in the ESI mode at an ionization potential of 70 eV with an LC-MS MSD (Hewlett-Packard). HRMS experiments were performed on a Micromass Q-TOF-2 tandem mass instrument. Purities of all final compounds (>96%) were established by analytical HPLC, which was carried out on an Agilent 1100 HPLC system with a Synergi 4μ Hydro-RP 80A column, with detection at 254 and 280 nm on a Shimadzu SPD-10A VP detector; flow rate = 1.4 mL/min; gradient of 0–100% acetonitrile in water (both containing 0.05 vol % of TFA) in 25 min. Final products were purified by preparative HPLC under the following conditions: ACE 5 AQ column, 150 mm × 21.2 mm; flow rate = 17 mL/min; all solvents containing 0.05 vol % TFA; UV detection at 254 and 280 nm; gradient I: 40–100% MeOH in water over 35 min; Gradient II: 8–100% MeOH in water over 40 min.
General Procedure for the Indole or Azaindole N-Alkylation (Method A)
To a solution of substituted indole or azaindole (1 equiv) in dry DMF (3 mL/mmol) cooled with an ice bath was added NaH (55% suspension in mineral oil, 1.5 equiv). After 30 min stirring at 0 to 5 °C, an alkyl bromide (1.2 equiv) or methyl iodide (1.2 equiv.) was added in one portion. The reaction mixture was then heated to 60 °C and stirred overnight. Upon completion of the reaction as indicated by TLC, the reaction mixture was poured into ice-water and extracted with EtOAc (3 × 20 mL). The combined organic phases were washed with water (50 mL), brine (50 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was either purified by flash chromatography (hexane to EtOAc-hexane; 1:3) or directly used for further reaction without additional purification.
General Procedure for the Indole or Azaindole Glyoxylation (Method B)
To a solution of the substituted N-alkylindole (1 mmol) in Et2O (5 mL/mmol) cooled with an ice bath was added ethyl chlorooxoacetate (5 equiv) dropwise. The reaction mixture was allowed to slowly warm to room temperature overnight until completion. Upon reaction completion as indicated by TLC, the reaction mixture was cooled with an ice bath and carefully quenched by slow addition of saturated aqueous NaHCO3. The reaction mixture was extracted with CH2Cl2 (3 × 20 mL) and the combined organic phases were washed with saturated aqueous NaHCO3 (2 × 40 mL), water (40 mL), brine (40 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (EtOAc-hexane; 1:2) to give the substituted glyoxalates. For N-alkylazaindoles, a solution of the azaindole (1 equiv.) in CH2Cl2 was treated with AlCl3 (5 equiv.) and stirred at ambient temperature for 1 h prior to the addition of ethyl chlorooxoacetate (5 equiv.). Quenching with MeOH and evaporation in vacuo gave the residue which was purified by flash chromatography (EtOAc-hexane; 2:3).
General Procedure for the Perkin-type Condensation to Give Maleimides (Method C)
To a suspension of indolyl-3-glyoxylate or azaindolyl-3-glyoxylate (1 equiv) and benzofuran-3-yl-acetamide (1.1 equiv) in dry THF or DMF (10 mL/mmol) at 0–5 °C was added a 1.0 M solution of t-BuOK in THF (2.5 equiv) in a dropwise manner. The reaction mixture was stirred at room temperature overnight. Upon completion of the reaction as ascertained by TLC, the reaction mixture was quenched with saturated aqueous NH4Cl and extracted with EtOAc (3 × 20 mL). The combined organic phases were washed with water (50 mL), brine (50 mL), dried over Na2SO4 and evaporated in vacuo. The residue was purified by flash chromatography (1:2 to 1:1 EtOAc-hexane eluent) prior to further HPLC purification.
General Procedure for the Deprotection of N-Boc Amines (Method D)
To a solution of the N-Boc protected precursor (1 mmol) in CHCl3 (5 mL) was added TFA (1 mL) at ambient temperature. The mixture was stirred for 2 h and concentrated in vacuo. The crude product was dissolved in DMF and filtered over a syringe filter (polytetrafluoroethylene, 17 mm diameter, 0.45 μm pore size) prior to the preparative HPLC.
General Procedure for the Wittig Reaction (Method E)
To a suspension of the indole carboxaldehyde (1 equiv.) and methyenetriphenylphosphorane (3 equiv.) in dry THF (15 mL/mmol) was slowly added a 1.0 M solution of t-BuOK (3 equiv.) at 0 to 5 °C. The reaction mixture was slowly warmed to room temperature overnight with stirring and concentrated in vacuo. The residue was purified by flash chromatography (1:4 to 1:2 EtOAc-hexane eluent).
General Procedure for the Hydroboration (Method F)
To a solution of the vinyl indole (1 equiv.) in dry THF (6 mL/mmol) was added a solution of borane-tetrahydrofuran complex (1.0 M in THF, 1.1 equiv.) in a dropwise manner at ambient temperature. After 2.5 h stirring, 10% aqueous NaOH (1.3 equiv.) was added in one portion (effervescence was observed), followed by a hydrogen peroxide solution (35%, 1.3 equiv.). After 3.5 h stirring at ambient temperature, the reaction mixture was quenched by the addition of saturated aqueous NH4Cl and diluted with water. Following extraction with EtOAc (3 × 50 mL), the combined organic phases were washed with brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (1:4 to 1:1 EtOAc-hexane eluent).
General Procedure for the tert-Butyl Ether (Method G)
To a solution of 2-(1-methyl-1H-indol-7-yl)ethanol (1 mmol) in CH2Cl2 (1 mL/mmol) and cyclohexane (2 mL/mmol) was added tert-butyltrichloroacetimidate (1.1 mmol) at ambient temperature in one portion, followed by 30 μL of boron trifluoride diethyletherate. After stirring overnight, the reaction mixture was diluted with CH2Cl2 and directly purified by flash chromatography (1:7 to 1:3 EtOAc-hexane eluent) to give the desired product.
General Procedure for the Amide Coupling (Method H)
To a solution of the free amine (1 equiv.), carboxylic acid (1.2 equiv.), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (1.3 equiv.) and HOBt (1.3 equiv.) in DMF (5 mL/mmol) was added N-methylmorpholine (3 equiv.) in one portion at ambient temperature. After overnight stirring, saturated aqueous NaHCO3 was added, followed by extraction with CH2Cl2 (3 × 15 mL). The combined organic phases were washed with brine (40 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (1:2 to 1:1 EtOAc-hexane or 3:97 MeOH:CH2Cl2 eluent) prior to further HPLC purification.
General Procedure for the Urea Formation (Method I)
To a solution of the free amine (1 equiv.) in MeOH (10 mL/mmol) was added carbamoyl chloride (1.5 equiv.) at ambient temperature, followed by N,N-diisopropylethylamine (3 equiv.) in one portion. After 3 h stirring, the reaction mixture was diluted with aqueous 1 M NaOH and extracted with EtOAc (3 × 15 mL). The combined organic phases were washed with brine (40 mL), dried over anhydrous Na2SO4 and concentrated in vacuo prior to purification by HPLC.
3-(Benzofuran-3-yl)-4-(5-bromo-1-(2-hydroxyethyl)-1H-indol-3-yl)-1H-pyrrole-2,5-dione (2)
The title compound was synthesized from commercially available 5-bromoindole (32a) and O-trityl-2-bromoethan-1-ol according to general procedures A–C, purified using gradient I preparative HPLC, and isolated as an orange solid in 15% yield over 3 steps. M.p. 152–157 °C. IR (ATR): νmax 3140, 3057, 2944, 1702, 1700, 1544, 1356, 1114, 744 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.20 (s, 1H), 8.29 (s, 1H), 7.94 (s, 1H), 7.63 (d, J = 8.4 Hz, 1H), 7.50 (d, J = 8.8 Hz, 1H), 7.25 (t, J = 7.2 Hz, 1H), 7.18 (dd, J = 8.8, 2.0 Hz, 1H), 7.06 (d, J = 2.0 Hz, 1H), 6.94 (t, J = 7.4 Hz, 1H), 6.85 (d, J = 7.6 Hz, 1H), 4.93–4.89 (m, 1H), 4.27 (t, J = 5.2 Hz, 2H), 3.69–3.65 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.1 (C=O), 171.8 (C=O), 154.4 (C), 147.2 (CH), 135.3 (C), 135.1 (CH), 132.3 (C), 127.2 (C), 125.2 (C), 125.0 (CH), 124.4 (CH), 123.5 (CH), 123.0 (C), 122.9 (CH), 121.7 (CH), 112.9 (CH), 112.7 (C), 111.5 (CH), 111.4 (C), 103.7 (C), 60.1 (CH2), 48.9 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C22H16BrN2O4: 451.0288, found: 451.0278. HPLC purity: 97.6%.
3-(Benzofuran-3-yl)-4-(5-fluoro-1-(2-hydroxyethyl)-1H-indol-3-yl)-1H-pyrrole-2,5-dione (3)
The title compound was synthesized from commercially available 5-fluoroindole (32b) and O-trityl-2-bromoethan-1-ol according to general procedures A–C, purified using gradient I preparative HPLC, and isolated as an orange solid in 15% yield over 3 steps. M.p. 107–112 °C. IR (ATR): νmax 3232, 1699, 1695, 1541, 1478, 1450, 1334, 741 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.19 (s, 1H), 8.28 (s, 1H), 7.98 (s, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.59–7.51 (m, 1H), 7.24 (t, J = 8.2 Hz, 1H), 6.97–6.85 (m, 3H), 6.58 (dd, J = 9.2, 2.0 Hz, 1H), 4.92 (br s, 1H), 4.29 (t, J = 5.2 Hz, 2H), 3.68 (t, J = 5.4 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.0 (C=O), 171.7 (C=O), 157.0 (d, 1JC–F = 232.6 Hz, C), 154.2 (C), 147.1 (CH), 135.4 (CH), 133.1 (C), 132.3 (C), 126.1 (d, 3JC–F = 11.0 Hz, C), 125.2 (C), 124.8 (CH), 122.8 (CH), 122.3 (C), 121.7 (CH), 112.0 (d, 3JC–F = 10.2 Hz, CH), 111.4 (CH), 111.3 (C), 110.0 (d, 2JC–F = 26.4 Hz, CH), 105.8 (d, 2JC–F = 24.9 Hz, CH), 104.0 (d, 4JC–F = 4.4 Hz, C), 60.0 (CH2), 48.9 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C22H16FN2O4: 391.1089, found: 391.1104. HPLC purity: 99.1%.
3-(Benzofuran-3-yl)-4-(5-fluoro-1-(3-hydroxypropyl)-1H-indol-3-yl)-1H-pyrrole-2,5-dione (4)
The title compound was synthesized from commercially available 5-fluoroindole (32b) and O-trityl-3-bromopropan-1-ol according to general procedures A–C, purified using gradient I preparative HPLC, and isolated as an orange solid in 25% yield over 3 steps. M.p. 101–106 °C. IR (ATR): νmax 3140, 2944, 1704, 1699, 1545, 1479, 1451, 1336, 742 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.20 (s, 1H), 8.32 (s, 1H), 7.93 (s, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.55–7.50 (m, 1H), 7.22 (t, J = 8.4 Hz, 1H), 6.97–6.85 (m, 2H), 6.78 (d, J = 7.6 Hz, 1H), 6.64 (dd, J = 10.4, 2.4 Hz, 1H), 4.63 (br s, 1H), 4.30 (t, J = 6.8 Hz, 2H), 3.34 (t, J = 6.0 Hz, 2H), 1.85–1.79 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.0 (C=O), 171.7 (C=O), 157.2 (d, 1JC–F = 233.5 Hz, C), 154.2 (C), 147.1 (CH), 134.8 (CH), 132.7 (C), 132.2 (C), 126.1 (d, 3JC–F = 11.0 Hz, C), 125.1 (C), 124.8 (CH), 122.8 (CH), 122.7 (C), 121.5 (CH), 111.7 (d, 3JC–F = 10.3 Hz, CH), 111.4 (CH), 111.2 (C), 110.2 (d, 2JC–F = 26.4 Hz, CH), 105.8 (d, 2JC–F = 26.4 Hz, CH), 104.1 (d, 4JC–F = 4.4 Hz, C), 57.4 (CH2), 43.1 (CH2), 32.7 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C23H18FN2O4: 405.1245, found: 405.1249. HPLC purity: 97.9%.
3-(1-(2-Aminoethyl)-5-bromo-1H-indol-3-yl)-4-(benzofuran-3-yl)-1H-pyrrole-2,5-dione trifluoroacetate (5)
The title compound was synthesized from commercially available 5-bromoindole (32a) and tert-butyl (2-bromoethyl)carbamate according to general procedures A–D, purified using gradient II preparative HPLC, and isolated as an orange solid in 12% yield over 3 steps. M.p. 153–157 °C. IR (ATR): νmax 3054, 1704, 1699, 1545, 1451, 1343, 1185, 1119, 743, 722 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.26 (s, 1H), 8.28 (s, 1H), 7.99 (br s, 3H), 7.94 (s, 1H), 7.65 (d, J = 8.0 Hz, 1H), 7.56 (d, J = 8.8 Hz, 1H), 7.31–7.24 (m, 2H), 7.18 (s, 1H), 6.98 (d, J = 7.6 Hz, 1H), 6.91 (d, J = 8.0 Hz, 1H), 4.44 (t, J = 6.4 Hz, 2H), 3.17 (t, J = 5.6 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) 171.9 (C=O), 171.6 (C=O), 158.3 (q, 2JC–F = 36.1 Hz, TFA), 154.4 (C), 147.4 (CH), 134.9 (C), 134.6 (CH), 131.8 (C), 127.3 (C), 125.0 (CH), 124.9 (CH), 123.9 (CH), 123.8 (C), 122.9 (CH), 121.8 (CH), 117.3 (d, 1JC–F = 298.2 Hz, TFA), 113.1 (C), 112.4 (CH), 111.5 (CH), 111.1 (C), 104.5 (C), 104.5 (C), 43.6 (CH2), 38.5 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C22H17BrN3O3: 450.0448, found: 450.0458. HPLC purity: 98.2%.
3-(1-(2-Aminoethyl)-5-fluoro-1H-indol-3-yl)-4-(benzofuran-3-yl)-1H-pyrrole-2,5-dione trifluoroacetate (6)
The title compound was synthesized from commercially available 5-fluoroindole (32b) and tert-butyl (2-bromoethyl)carbamate according to general procedures A–D, purified using gradient II preparative HPLC, and isolated as an orange solid in 16% yield over 3 steps. M.p. 144–148 °C. IR (ATR): νmax 3052, 1704, 1700, 1544, 1480, 1451, 1342, 1198, 1116, 742, 721 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.25 (s, 1H), 8.28 (s, 1H), 7.97 (s, 1H), 7.91 (br s, 3H), 7.64 (d, J = 8.0 Hz, 1H), 7.62–56 (m, 1H), 7.28 (t, J = 7.6 Hz, 1H), 7.05–6.91 (m, 3H), 6.69 (dd, J = 7.6, 2.5 Hz, 1H), 4.45 (t, J = 6.4 Hz, 2H), 3.19 (br s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.0 (C=O), 171.7 (C=O), 158.4 (q, 2JC–F = 35.9 Hz, TFA), 157.4 (d, 1JC–F = 233.5 Hz, C), 154.3 (C), 147.4 (CH), 135.1 (CH), 132.8 (C), 131.9 (C), 126.2 (d, 3JC–F = 10.6 Hz, C), 125.0 (CH), 125.0 (C), 123.3 (C), 122.9 (CH), 121.9 (CH), 117.1 (d, 1JC–F = 299.3 Hz, TFA), 111.6 (d, 3JC–F = 9.5 Hz, CH), 111.5 (CH), 111.2 (C), 110.6 (d, 2JC–F = 26.4 Hz, CH), 106.4 (d, 2JC–F = 24.9 Hz, CH), 105.0 (d, 4JC–F = 2.9 Hz, C), 43.7 (CH2), 38.5 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C22H17FN3O3: 390.1249, found: 390.1256. HPLC purity: 98.0%.
3-(Benzofuran-3-yl)-4-(5-bromo-1-(3-(methylamino)propyl)-1H-indol-3-yl)-1H-pyrrole-2,5-dione trifluoroacetate (7)
The title compound was synthesized from commercially available 5-bromoindole (32a) and tert-butyl (3-bromopropyl)(methyl)carbamate according to general procedures A–D, purified using gradient II preparative HPLC, and isolated as an orange solid in 14% yield over 4 steps. M.p. 126–129 °C. IR (ATR): νmax 3041, 1713, 1671, 1547, 1452, 1337, 1202, 1127, 742, 721 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.25 (s, 1H), 8.33 (s, 1H), 8.29 (br s, 2H), 7.95 (s, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.55 (d, J = 8.8 Hz, 1H), 7.29–7.21 (m, 2H), 7.13 (d, J = 1.2 Hz, 1H), 6.93 (t, J = 7.4 Hz, 1H), 6.80 (d, J = 8.0 Hz, 1H), 4.33 (t, J = 6.4 Hz, 2H), 2.82–2.78 (m, 2H), 2.54 (s, 3H), 2.04–2.00 (m, 2H). 13C NMR (100 MHz, DMSO-d6) 171.9 (C=O), 171.6 (C=O), 158.2 (q, 2JC–F = 36.1 Hz, TFA), 154.3 (C), 147.2 (CH), 134.6 (C), 134.1 (CH), 131.9 (C), 127.3 (C), 125.0 (C), 124.9 (CH), 124.7 (CH), 123.6 (CH), 123.6 (C), 122.8 (CH), 121.5 (CH), 117.2 (d, 1JC–F = 298.2 Hz, TFA), 112.9 (C), 112.5 (CH), 111.5 (CH), 111.1 (C), 104.0 (C), 45.6 (CH2), 43.2 (CH2), 32.5 (CH3), 26.2 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C24H21BrN3O3 : 478.0761, found: 478.0768. HPLC purity: 99.1%.
3-(Benzofuran-3-yl)-4-(5-fluoro-1-(3-(methylamino)propyl)-1H-indol-3-yl)-1H-pyrrole-2,5-dione trifluoroacetate (8)
The title compound was synthesized from commercially available 5-fluoroindole (32b) and tert-butyl (3-bromopropyl)(methyl)carbamate according to general procedures A–D, purified using gradient II preparative HPLC, and isolated as an orange solid in 16% yield over 4 steps. M.p. 120–125 °C. IR (ATR): νmax 3041, 1713, 1670, 1547, 1480, 1452, 1338, 1199, 1123, 745, 721 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.23 (s, 1H), 8.32 (s, 1H), 8.30 (br s, 2H), 7.99 (s, 1H), 7.65–7.56 (m, 2H), 7.25 (t, J = 8.2 Hz, 1H), 7.01–6.90 (m, 2H), 6.82 (d, J = 7.6 Hz, 1H), 6.65 (dd, J = 10.4, 2.6 Hz, 1H), 4.34 (t, J = 7.0 Hz, 2H), 2.85–2.79 (m, 2H), 2.54 (s, 3H), 2.07–2.01 (m, 2H). 13C NMR (100 MHz, DMSO-d6) 172.0 (C=O), 171.7 (C=O), 158.2 (q, 2JC–F = 31.5 Hz, TFA), 157.2 (d, 1JC–F = 234 Hz, C), 154.2 (C), 147.2 (CH), 134.6 (CH), 132.5 (C), 132.1 (C), 126.3 (d, 3JC–F = 10.5 Hz, C), 125.1 (C), 124.9 (CH), 123.1 (C), 122.8 (CH), 121.6 (CH), 117.2 (d, 1JC–F = 298.2 Hz, TFA), 111.8 (d, 3JC–F = 9.8 Hz, CH), 111.5 (CH), 111.1 (C), 110.4 (d, 2JC–F = 25.8Hz, CH), 106.1 (d, 2JC–F = 24.9 Hz, CH), 104.5 (d, 4JC–F = 4.4 Hz, C), 45.7 (CH2), 43.3 (CH2), 32.6 (CH3), 26.2 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C24H21FN3O3: 418.1561, found: 418.1570. HPLC purity: 98.4%.
3-(Benzofuran-3-yl)-4-(1-(2-methoxyethyl)-1H-indol-3-yl)-1H-pyrrole-2,5-dione (9)
The title compound was synthesized from commercially available indole (32c) and 2-bromoethyl methyl ether according to general procedures A–C, purified using gradient I preparative HPLC, and isolated as a yellow solid in 35% yield over 3 steps. M.p. 147–150 °C. IR (ATR): νmax 3144, 2928, 1710, 1700, 1548, 1451, 1326, 1099, 1072, 734 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.17 (s, 1H), 8.27 (s, 1H), 7.90 (s, 1H), 7.59 (d, J = 8.0 Hz, 1H), 7.52 (d, J = 8.0 Hz, 1H), 7.21 (t, J = 7.6 Hz, 1H), 7.06 (t, J = 7.8 Hz, 1H), 6.96–6.79 (m, 3H), 6.74 (t, J = 7.6 Hz, 1H), 4.41 (t, J = 5.2 Hz, 2H), 3.62 (t, J = 5.2 Hz, 2H), 3.19 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 172.1 (C=O), 171.8 (C=O), 154.2 (C), 147.0 (CH), 136.2 (C), 133.6 (CH), 132.6 (C), 125.6 (C), 125.3 (C), 124.7 (CH), 122.7 (CH), 122.6 (C), 122.1 (CH), 121.7 (CH), 120.9 (CH), 120.1 (CH), 111.4 (C), 111.4 (CH), 110.7 (CH), 104.2 (C), 70.7 (CH2), 58.2 (CH3), 45.8 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C23H19N2O4: 387.1339, found: 387.1351. HPLC purity: 98.4%.
3-(Benzofuran-3-yl)-4-(5-fluoro-1-(2-methoxyethyl)-1H-indol-3-yl)-1H-pyrrole-2,5-dione (10)
The title compound was synthesized from commercially available 5-fluoroindole (32b) and 2-bromoethyl methyl ether according to general procedures A–C, purified using gradient I preparative HPLC, and isolated as an orange solid in 36% yield over 3 steps. M.p. 83–88 °C. IR (ATR): νmax 3189, 2933, 1701, 1696, 1542, 1479, 1451, 1333, 1113, 742 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.19 (s, 1H), 8.31 (s, 1H), 7.92 (s, 1H), 7.62 (d, J = 8.4 Hz, 1H), 7.57–7.53 (m, 1H), 7.23 (t, J = 8.0 Hz, 1H), 6.96–6.87 (m, 2H), 6.80 (d, J = 8.0 Hz, 1H), 6.65 (dd, J = 9.2, 2.6 Hz, 1H), 4.40 (t, J = 4.8 Hz, 2H), 3.59 (t, J = 5.2 Hz, 2H), 3.18 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 172.0 (C=O), 171.7 (C=O), 157.3 (d, 1JC–F = 233.5 Hz, C), 154.2 (C), 147.1 (CH), 135.1 (CH), 133.0 (C), 132.2 (C), 126.1 (d, 3JC–F = 11.0 Hz, C), 125.1 (C), 124.9 (CH), 122.8 (CH), 122.7 (C), 121.6 (CH), 112.0 (d, 3JC–F = 9.5 Hz, CH), 111.4 (CH), 111.2 (C), 110.1 (d, 2JC–F = 25.9 Hz, CH), 105.8 (d, 2JC–F = 24.7 Hz, CH), 104.4 (d, 4JC–F = 4.4 Hz, C), 70.8 (CH2), 58.2 (CH3), 46.1 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C23H18FN2O4: 405.1245, found: 405.1264. HPLC purity: 98.6%.
3-(Benzofuran-3-yl)-4-(5-bromo-1-(2-methoxyethyl)-1H-indol-3-yl)-1H-pyrrole-2,5-dione (11)
The title compound was synthesized from commercially available 5-bromoindole (32a) and 2-bromoethyl methyl ether according to general procedures A–C, purified using gradient I preparative HPLC, and isolated as an orange solid in 30% yield over 3 steps. M.p. 92–96 °C. IR (ATR): νmax 3213, 2931, 1704, 1695, 1541, 1451, 1333, 1113, 742 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.21 (s, 1H), 8.30 (s, 1H), 7.87 (s, 1H), 7.61 (d, J = 8.4 Hz, 1H), 7.48 (d, J = 8.4 Hz, 1H), 7.23 (t, J = 7.8 Hz, 1H), 7.17 (dd, J = 8.8, 1.6 Hz, 1H), 7.13 (d, J = 1.6 Hz, 1H), 6.89 (t, J = 7.8 Hz, 1H), 6.76 (d, J = 7.6 Hz, 1H), 4.36 (t, J = 5.0 Hz, 2H), 3.56 (t, J = 5.0 Hz, 2H), 3.15 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 172.1 (C=O), 171.7 (C=O), 154.4 (C), 147.2 (CH), 135.1 (C), 134.7 (CH), 132.2 (C), 127.3 (C), 125.1 (C), 125.0 (CH), 124.6 (CH), 123.5 (CH), 123.4 (C), 122.8 (CH), 121.6 (CH), 112.8 (C), 112.8 (CH), 111.5 (CH), 111.3 (C), 103.9 (C), 70.8 (CH2), 58.2 (CH3), 46.1 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C23H18BrN2O4: 465.0444, found: 465.0458. HPLC purity: 99.2%.
3-(Benzofuran-3-yl)-4-(5,6-difluoro-1-(2-methoxyethyl)-1H-indol-3-yl)-1H-pyrrole-2,5- dione (12)
The title compound was synthesized from commercially available 5,6-difluoroindole (32d) and 2-bromoethyl methyl ether according to general procedures A–C, purified using gradient I preparative HPLC, and isolated as an orange solid in 37% yield over 3 steps. M.p. 98–103 °C. IR (ATR): νmax 3231, 3061, 2931, 1704, 1700, 1544, 1482, 1451, 1335, 1113, 854, 745 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.22 (s, 1H), 8.34 (s, 1H), 7.88 (s, 1H), 7.74–7.67 (m, 1H), 7.63 (d, J = 8.6 Hz, 1H), 7.24 (t, J = 8.3 Hz, 1H), 6.92–6.84 (m, 2H), 6.73 (d, J = 8.1 Hz, 1H), 4.37 (t, J = 5.1 Hz, 2H), 3.57 (t, J = 4.8 Hz, 2H), 3.17 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 171.9 (C=O), 171.6 (C=O), 154.2 (C), 147.3 (CH), 147.1 (dd, J = 237, 16.1 Hz, C), 144.8 (dd, J = 234, 14.6 Hz, C), 134.9 (C), 134.8 (CH), 131.8 (C), 131.7 (C), 124.9 (CH), 123.6 (C), 122.8 (CH), 121.5 (CH), 121.2 (d, 3JC–F = 8.8 Hz, C), 111.5 (CH), 111.0 (C), 107.6 (d, 2JC–F = 20.5 Hz, CH), 104.5 (d, 4JC–F = 3.7 Hz, C), 99.4 (d, 2JC–F = 22.0 Hz, CH), 70.7 (CH2), 58.1 (CH3), 46.2 (CH2). HRMS (ESI): m/z [M-H]- calculated for C23H15F2N2O4: 421.1005, found: 421.1010. HPLC purity: 96.5%.
3-(Benzofuran-3-yl)-4-(5-bromo-1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-1H-pyrrole-2,5-dione (13)
The title compound was synthesized from commercially available 5-bromo-7-azaindole (37a) according to general procedures A–C, purified using gradient I preparative HPLC, and isolated as an orange solid in 25% yield over 3 steps. M.p. 244–248 °C. IR (ATR): νmax 3136, 3039, 2751, 1707, 1538, 1452, 1338, 1120, 744 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.28 (s, 1H), 8.36 (s, 1H), 8.27 (s, 1H), 8.13 (s, 1H), 7.65 (d, J = 8.2 Hz, 1H), 7.41 (s, 1H), 7.26 (t, J = 7.7 Hz, 1H), 6.96 (t, J = 7.2 Hz, 1H), 6.83 (d, J = 7.8 Hz, 1H), 3.85 (s, 3H). 13C NMR (100 MHz, DMSO-d6) 171.8 (C=O), 171.5 (C=O), 154.3 (C), 147.4 (CH), 145.9 (C), 143.3 (CH), 135.6 (CH), 131.4 (CH), 131.4 (C), 125.0 (CH), 124.9 (C), 123.8 (C), 122.9 (CH), 121.4 (CH), 119.4 (C), 111.5 (CH), 111.5 (C), 110.9 (C), 102.0 (C), 31.6 (CH3). HRMS (ESI): m/z [M+H]+ calculated for C20H13BrN3O3: 422.0135, found: 422.0124. HPLC purity: 98.3%.
3-(Benzofuran-3-yl)-4-(5-bromo-1-(2-methoxyethyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)-1Hpyrrole-2,5-dione (14)
The title compound was synthesized from commercially available 5-bromo-7-azaindole (37a) and 2-bromoethyl methyl ether according to general procedures A–C, purified using gradient I preparative HPLC, and isolated as a yellow solid in 25% yield over 3 steps. M.p. 142–145 °C. IR (ATR): νmax 3133, 3040, 2752, 1714, 1547, 1455, 1335, 1108, 744 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.28 (s, 1H), 8.37 (s, 1H), 8.28 (d, J = 1.8 Hz, 1H), 8.02 (s, 1H), 7.65 (d, J = 8.1 Hz, 1H), 7.60 (d, J = 8.0 Hz, 1H), 7.26 (t, J = 8.1 Hz, 1H), 6.93 (t, J = 7.4 Hz, 1H), 6.75 (d, J = 7.8 Hz, 1H), 4.42 (t, J = 5.2 Hz, 2H), 3.60 (t, J = 5.2 Hz, 2H), 3.14 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 171.8 (C=O), 171.5 (C=O), 154.4 (C), 147.5 (CH), 145.6 (C), 143.3 (CH), 134.8 (CH), 131.6 (CH), 131.4 (C), 125.0 (CH), 124.6 (C), 124.3 (C), 122.9 (CH), 121.5 (CH), 119.6 (C), 111.6 (C), 111.5 (CH), 110.7 (C), 102.3 (C), 70.2 (CH2), 57.9 (CH3), 44.0 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C22H17BrN3O4: 466.0397, found: 466.0397. HPLC purity: 99.2%.
3-(Benzofuran-3-yl)-4-(1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-1H-pyrrole-2,5-dione (15)
The title compound was synthesized from commercially available 7-azaindole (37b) according to general procedures A–C, purified using gradient I preparative HPLC, and isolated as an orange solid in 14% yield over 3 steps. M.p. 219–223 °C. IR (ATR): νmax 3138, 3020, 2734, 1717, 1700, 1545, 1452, 1332, 1110, 978, 742 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.25 (s, 1H), 8.33 (s, 1H), 8.11 (d, J = 4.5 Hz, 1H), 8.13 (s, 1H), 7.62 (d, J = 8.3 Hz, 1H), 7.26–7.19 (m, 2H), 6.92 (t, J = 7.2 Hz, 1H), 6.87–6.78 (m, 2H), 3.88 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 172.0 (C=O), 171.7 (C=O), 154.2 (C), 147.4 (C), 147.3 (CH), 143.3 (CH), 134.3 (CH), 131.9 (C), 129.1 (CH), 125.2 (C), 124.9 (CH), 123.0 (C), 122.8 (CH), 121.6 (CH), 117.9 (C), 116.3 (CH), 111.5 (CH), 111.1 (C), 102.4 (C), 31.4 (CH3). HRMS (ESI): m/z [M+H]+ calculated for C20H14N3O3: 344.1030, found: 344.1041. HPLC purity: 99.9%.
3-(Benzofuran-3-yl)-4-(1-(2-methoxyethyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)-1H-pyrrole-2,5-dione (16)
The title compound was synthesized from commercially available 7-azaindole (37b) and 2-bromoethyl methyl ether according to general procedures A–C, purified using gradient I preparative HPLC, and isolated as an orange solid in 15% yield over 3 steps. M.p. 180–183 °C. IR (ATR): νmax 3136, 2993, 2731, 1712, 1547, 1331, 1109, 982, 744 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.25 (s, 1H), 8.35 (s, 1H), 8.19 (d, J = 4.8 Hz, 1H), 8.04 (s, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.37 (d, J = 8.1 Hz, 1H), 7.23 (t, J = 7.3 Hz, 1H), 6.93–6.84 (m, 2H), 6.77 (d, J = 7.9 Hz, 1H), 4.47 (t, J = 5.3 Hz, 2H), 3.65 (t, J = 5.3 Hz, 2H), 3.18 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 171.9 (C=O), 171.6 (C=O), 154.3 (C), 147.3 (CH), 147.2 (C), 143.3 (CH), 133.6 (CH), 131.9 (C), 129.3 (CH), 124.9 (CH), 124.9 (C), 123.6 (C), 122.8 (CH), 121.6 (CH), 118.0 (C), 116.5 (CH), 111.5 (CH), 110.9 (C), 102.7 (C), 70.3 (CH2), 58.0 (CH3), 43.8 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C22H18N3O4: 388.1292, found: 388.1298. HPLC purity: 98.6%.
3-(Benzofuran-3-yl)-4-(5-methoxy-1-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-pyrrole-2,5-dione (17)
The title compound was synthesized from commercially available 6-aza-5-methoxyindole according to general procedures A–C, purified using gradient I preparative HPLC, and isolated as an orange solid in 13% yield over 3 steps. M.p. 170–175 °C. IR (ATR): νmax 3039, 2732, 1717, 1678, 1652, 1547, 1186, 1119, 722 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.24 (s, 1H), 8.49 (s, 1H), 8.26 (s, 1H), 8.21 (s, 1H), 7.64 (d, J = 8.3 Hz, 1H), 7.28–7.24 (m, 1H), 7.00 (s, 2H), 6.03 (s, 1H), 3.94 (s, 3H), 3.49 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 171.9 (C=O), 171.6 (C=O), 156.6 (C), 154.2 (C), 147.3 (CH), 140.9 (CH), 135.4 (C), 131.5 (C), 130.4 (C), 128.6 (CH), 125.5 (CH), 125.0 (C), 123.1 (C), 123.0 (CH), 121.5 (CH), 111.5 (CH), 111.2 (C), 103.0 (C), 97.3 (CH), 54.2 (CH3), 33.7 (CH3). HRMS (ESI): m/z [M+H]+ calculated for C21H16N3O4: 374.1135, found: 374.1138. HPLC purity: 99.7%.
3-(Benzofuran-3-yl)-4-(5-methoxy-1-(2-methoxyethyl)-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-pyrrole-2,5-dione (18)
The title compound was synthesized from commercially available 6-aza-5-methoxyindole and 2-bromoethyl methyl ether according to general procedures A–C, purified using gradient I preparative HPLC, and isolated as an orange solid in 15% yield over 3 steps. M.p. 183–188 °C. IR (ATR): νmax 3046, 2941, 2724, 1712, 1695, 1649, 1549, 1451, 1338, 1179, 1115, 743 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.24 (s, 1H), 8.54 (s, 1H), 8.29 (s, 1H), 8.12 (s, 1H), 7.64 (d, J = 8.2 Hz, 1H), 7.26 (t, J = 8.2 Hz, 1H), 6.96–6.87 (m, 2H), 6.19 (s, 1H), 4.47 (d, J = 5.2 Hz, 2H), 3.63 (d, J = 5.2 Hz, 2H), 3.53 (s, 3H), 3.20 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 171.8 (C=O), 171.5 (C=O), 156.3 (C), 154.2 (C), 147.4 (CH), 140.5 (CH), 135.8 (C), 131.3 (C), 129.9 (C), 128.6 (CH), 125.2 (CH), 125.0 (C), 123.9 (C), 123.0 (CH), 121.4 (CH), 111.6 (CH), 111.0 (C), 103.6 (C), 97.4 (CH), 70.8 (CH2), 58.2 (CH3), 54.4 (CH3), 46.6 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C23H20N3O5: 418.1398, found: 418.1406. HPLC purity: 99.9%.
3-(Imidazo[1,2-a]pyridin-3-yl)-4-(5-methyl-5H-[1,3]dioxolo[4,5-f]indol-7-yl)-1H-pyrrole-2,5-dione trifluoroacetate (19)
The title compound was synthesized from 5,6-methylenedioxyindole and 2-(imidazo[1,2-a]pyridin-3-yl)acetamide 38 according to general procedures A–C, purified using gradient II preparative HPLC, and isolated as a red solid in 15% yield over 3 steps. M.p. 134–139 °C. IR (ATR): νmax 3129, 2725, 1711, 1671, 1471, 1334, 1182, 1100, 755 cm-1. 1H NMR (400 MHz, CDCl3/CD3OD) δ 11.25 (s, 1H), 8.16 (s, 1H), 7.91 (s, 1H), 7.80-7.84 (m, 2H), 7.70 (t, J = 7.8 Hz, 1H), 7.04 (s, 1H), 6.92 (t, J = 6.9 Hz, 1H), 5.73 (s, 2H), 5.54 (s, 1H), 3.74 (s, 3H). 13C NMR (100 MHz, CDCl3/CD3OD) δ 170.7 (C=O), 170.5 (C=O), 145.6 (C), 144.1 (C), 135.0 (C), 134.3 (CH), 132.3 (C), 131.8 (CH), 127.2 (CH), 126.7 (C), 119.3 (C), 117.5 (C), 115.7 (CH), 112.9 (CH), 111.9 (C), 104.6 (C), 100.7 (CH), 100.7 (CH2), 96.9 (CH), 91.2 (CH), 33.1 (CH3). HRMS (ESI): m/z [M+H]+ calculated for C21H15N4O4: 387.1088, found: 387.1087. HPLC purity: 99.0%.
3-(5-Bromo-1-methyl-1H-indol-3-yl)-4-(imidazo[1,2-a]pyridin-3-yl)-1H-pyrrole-2,5-dione trifluoroacetate (20)
The title compound was synthesized from 5-bromoindole and 2-(imidazo[1,2-a]pyridin-3-yl)acetamide 38 according to general procedures A–C, purified using gradient II preparative HPLC, and isolated as a yellow solid in 15% yield over 3 steps. M.p. 230–235 °C. IR (ATR): νmax 3163, 3081, 2940, 2708, 1717, 1656, 1552, 1330, 1201, 1178, 1126, 760 cm-1. 1H NMR (400 MHz, CD3OD/DMSO-d6) δ 11.24 (s, 1H), 8.33 (s, 1H), 8.17 (s, 1H), 7.92 (d, J = 9.0 Hz, 1H), 7.85 (d, J = 6.9 Hz, 1H), 7.70 (t, J = 8.0 Hz, 1H), 7.36 (d, J = 8.7 Hz, 1H), 7.13 (dd, J = 8.7, 1.7 Hz, 1H), 6.95 (t, J = 6.9 Hz, 1H), 6.21 (d, J = 1.7 Hz, 1H), 3.85 (s, 3H). 13C NMR (100 MHz, CD3OD/DMSO-d6) δ 171.7 (C=O), 171.3 (C=O), 158.2 (q, 2JC–F = 36.1 Hz, TFA), 141.7 (C), 137.9 (CH), 136.4 (CH), 135.5 (C), 133.0 (CH), 128.3 (CH), 128.1 (C), 127.6 (C), 125.9 (CH), 122.1 (CH), 117.8 (C), 117.2 (d, 1JC–F = 298.2 Hz, TFA), 116.6 (C), 115.0 (C), 114.5 (CH), 114.0 (CH), 113.4 (CH), 104.2 (C), 33.7 (CH3). HRMS (ESI): m/z [M+H]+ calculated for C20H14BrN4O2: 421.02946, found: 421.0300. HPLC purity: 96.8%.
3-(Benzofuran-3-yl)-4-(7-(2-methoxyethyl)-1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione (21)
The title compound was synthesized from alcohol 40a (generated from indole-7-carboxaldehyde 39 employing general methods E and F) and methyl iodide according to general procedures A–C, purified using gradient I preparative HPLC, and isolated as an orange solid in 26% yield over 3 steps. M.p. 91–96 °C. IR (ATR): νmax 3135, 2928, 1704, 1699, 1695, 1545, 1450, 1320, 1102, 743 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.17 (s, 1H), 8.22 (s, 1H), 7.85 (s, 1H), 7.58 (d, J = 8.4 Hz, 1H), 7.25–7.19 (m, 1H), 6.94–6.89 (m, 2H), 6.85 (d, J = 7.6 Hz, 1H), 6.70 (d, J = 8.0 Hz, 1H), 6.61 (t, J = 7.6 Hz, 1H), 4.11 (s, 3H), 3.59 (t, J = 7.0 Hz, 2H), 3.28 (t, J = 7.2 Hz, 2H), 3.24 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 172.1 (C=O), 171.9 (C=O), 154.1 (C), 146.9 (CH), 136.0 (CH), 134.9 (C), 132.4 (C), 126.9 (C), 125.5 (C), 124.7 (CH), 124.5 (CH), 123.3 (C), 122.7 (CH), 122.4 (C), 121.7 (CH), 120.1 (CH), 119.0 (CH), 111.6 (C), 111.3 (CH), 103.5 (C), 73.3 (CH2), 58.0 (CH3), 37.2 (CH3), 31.5 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C24H21N2O4: 401.1496, found: 401.1510. HPLC purity: 98.1%.
3-(Benzofuran-3-yl)-4-(7-(2-(tert-butoxy)ethyl)-1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione (22)
The title compound was synthesized from alcohol 40a (generated from indole-7-carboxaldehyde 39 employing general methods E and F) and tert-butyl 2,2,2-trichloroacetimidate (method G) according to general procedures A–C, purified using gradient I preparative HPLC, and isolated as an orange solid in 16% yield over 3 steps. M.p. 107–112 °C. IR (ATR): νmax 3211, 2971, 1704, 1699, 1545, 1451, 1322, 1106, 1077, 743 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.16 (s, 1H), 8.22 (s, 1H), 7.85 (s, 1H), 7.58 (d, J = 8.4 Hz, 1H), 7.24–7.18 (m, 1H), 6.93–6.88 (m, 2H), 6.85 (d, J = 7.6 Hz, 1H), 6.71 (dd, J = 8.0, 1.0 Hz, 1H), 6.61 (t, J = 7.6 Hz, 1H), 4.12 (s, 3H), 3.55 (t, J = 7.0 Hz, 2H), 3.18 (t, J = 6.8 Hz, 2H), 1.06 (s, 9H). 13C NMR (100 MHz, DMSO-d6) δ 172.2 (C=O), 171.9 (C=O), 154.1 (C), 146.9 (CH), 136.0 (CH), 135.0 (C), 132.4 (C), 126.9 (C), 125.5 (C), 124.7 (CH), 124.6 (CH), 123.7 (C), 122.7 (CH), 122.2 (C), 121.7 (CH), 120.1 (CH), 119.0 (CH), 111.5 (C), 111.3 (CH), 103.5 (C), 72.3 (CH2), 62.9 (C), 37.1 (CH3), 32.6 (CH2), 27.2 (CH3). HRMS (ESI): m/z [M+H]+ calculated for C27H27N2O4: 443.1965, found: 443.1983. HPLC purity: 96.9%.
3-(Benzofuran-3-yl)-4-(1,7-bis(2-methoxyethyl)-1H-indol-3-yl)-1H-pyrrole-2,5-dione (23)
The title compound was synthesized from alcohol 40b (generated from indole-7-carboxaldehyde 39 employing general methods E and F) and 2-bromoethyl methyl ether according to general procedures A–C, purified using gradient I preparative HPLC, and isolated as an orange solid in 16% yield over 3 steps. M.p. 178–182 °C. IR (ATR): νmax 3146, 2892, 1697, 1559, 1337, 1109, 747 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.19 (s, 1H), 8.29 (s, 1H), 7.78 (s, 1H), 7.57 (d, J = 8.4 Hz, 1H), 7.20 (t, J = 7.6 Hz, 1H), 6.89–6.81 (m, 3H), 6.77 (d, J = 8.0 Hz, 1H), 6.67 (t, J = 7.6 Hz, 1H), 4.55 (t, J = 5.0 Hz, 2H), 3.63–3.54 (m, 4H), 3.25–3.15 (m, 8H). 13C NMR (100 MHz, DMSO-d6) δ 172.0 (C=O), 171.8 (C=O), 154.1 (C), 147.1 (CH), 135.1 (CH), 134.2 (C), 132.2 (C), 127.3 (C), 125.2 (C), 124.8 (CH), 124.7 (CH), 123.4 (C), 122.8 (C), 122.6 (CH), 121.7 (CH), 120.1 (CH), 119.2 (CH), 111.4 (CH), 111.3 (C), 104.3 (C), 72.8 (CH2), 71.7 (CH2), 58.3 (CH3), 58.1 (CH3), 48.2 (CH2), 32.0 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C26H25N2O5: 445.1758, found: 445.1777. HPLC purity: 99.4%.
3-(Benzofuran-3-yl)-4-(1,2,3,4-tetrahydro-[1,4]diazepino[6,7,1-hi]indol-7-yl)-1H-pyrrole-2,5-dione trifluoroacetate (24)
The title compound was synthesized from diazepinoindole 43a (generated from indole-7-carboxaldehyde 39) according to general procedures B–D, purified using gradient II preparative HPLC, and isolated as an orange solid in 12% yield over 3 steps. M.p. 160–165 °C. IR (ATR): νmax 3038, 2731, 1706, 1695, 1669, 1545, 1451, 1340, 1180, 1116, 745 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.26 (s, 1H), 9.44 (br s, 2H), 8.26 (s, 1H), 7.97 (s, 1H), 7.61 (d, J = 8.4 Hz, 1H), 7.25 (t, J = 8.0 Hz, 1H), 7.05 (d, J = 7.2 Hz, 1H), 7.02–6.86 (m, 3H), 6.77 (t, J = 8.0 Hz, 1H), 4.68 (br s, 4H), 3.72 (br s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.0 (C=O), 171.7 (C=O), 158.2 (q, 2JC–F = 36.1 Hz, TFA), 154.2 (C), 147.3 (CH), 135.3 (C), 134.4 (CH), 131.9 (C), 126.8 (C), 125.3 (C), 124.9 (CH), 123.9 (C), 122.9 (CH), 122.8 (CH), 121.8 (CH), 121.3 (CH), 120.5 (CH), 118.0 (C), 117.2 (d, 1JC–F = 298.2 Hz, TFA), 111.5 (CH), 111.3 (C), 104.9 (C), 46.9 (CH2), 46.6 (CH2), 45.2 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C23H18N3O3: 384.1343, found: 384.1362. HPLC purity: 98.2%.
3-(Benzofuran-3-yl)-4-(9-fluoro-1,2,3,4-tetrahydro-[1,4]diazepino[6,7,1-hi]indol-7-yl)-1H-pyrrole-2,5-dione trifluoroacetate (25)
The title compound was synthesized from diazepinoindole 43b (generated from 5-fluoroindole-7-carboxaldehyde 42)34 according to general procedures B–D, purified using gradient II preparative HPLC, and isolated as an orange solid in 14% yield over 3 steps. M.p. 152–157 °C. IR (ATR): νmax 3136, 3050, 2738, 1710, 1670, 1547, 1452, 1340, 1181, 1120, 746 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.29 (s, 1H), 9.65 (br s, 2H), 8.30 (s, 1H), 7.99 (s, 1H), 7.63 (d, J = 8.4 Hz, 1H), 7.27 (t, J = 7.6 Hz, 1H), 7.04–6.94 (m, 2H), 6.90 (d, J = 7.6 Hz, 1H), 6.63 (d, J = 9.2 Hz, 1H), 4.66 (br s, 4H), 3.71 (br s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.9 (C=O), 171.6 (C=O), 158.2 (q, 2JC–F = 36.1 Hz, TFA), 156.6 (d, 1JC–F = 233.4 Hz, C), 154.2 (C), 147.4 (CH), 135.9 (CH), 132.1 (C), 131.6 (C), 127.5 (d, 3JC–F = 10.6 Hz, C), 125.1 (C), 125.0 (CH), 124.2 (C), 123.0 (CH), 121.6 (CH), 119.8 (d, 3JC–F = 9.6 Hz, C), 117.3 (d, 1JC–F = 298.2 Hz, TFA), 111.6 (CH), 111.1 (C), 110.9 (d, 2JC–F = 27 Hz, CH), 105.9 (d, 2JC–F = 24.4 Hz, CH), 105.1 (d, 4JC–F = 4.4 Hz, C), 46.7 (CH2), 46.6 (CH2), 45.6 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C23H17FN3O3: 402.1248, found: 402.1260. HPLC purity: 97.9%.
3-(Benzofuran-3-yl)-4-(9-bromo-1,2,3,4-tetrahydro-[1,4]diazepino[6,7,1-hi]indol-7-yl)-1H-pyrrole-2,5-dione trifluoroacetate (26)
The title compound was synthesized from 5-bromoindoline 45 (see Supplementary Information), purified using gradient II preparative HPLC, and isolated as an orange solid. M.p. 243–248 °C. IR (ATR): νmax 3138, 3050, 1709, 1700, 1695, 1623, 1545, 1336, 1179, 746 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.29 (s, 1H), 9.55 (br s, 2H), 8.31 (s, 1H), 7.96 (s, 1H), 7.65 (d, J = 7.8 Hz, 1H), 7.30–7.25 (m, 2H), 7.08 (s, 1H), 6.99 (t, J = 7.6 Hz, 1H), 6.88 (d, J = 8.8 Hz, 1H), 4.66 (br s, 4H), 3.70 (br s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.8 (C=O), 171.5 (C=O), 158.2 (q, 2JC–F = 30.9 Hz, TFA), 154.4 (C), 147.4 (CH), 135.4 (CH), 134.1 (C), 131.5 (C), 128.4 (C), 125.0 (CH), 124.9 (C), 124.9 (CH), 124.6 (C), 123.6 (CH), 123.0 (CH), 121.6 (CH), 120.1 (C), 116.3 (d, 1JC–F = 294.3 Hz, TFA), 112.5 (C), 111.5 (CH), 111.1 (C), 104.6 (C), 46.5 (CH2), 46.3 (CH2), 45.3 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C23H17BrN3O3: 462.0448, found: 462.0448. HPLC purity: 98.9%.
3-(Benzofuran-3-yl)-4-(9-fluoro-2-(2-(pyridin-4-yl)acetyl)-1,2,3,4-tetrahydro-[1,4]diazepino[6,7,1-hi]indol-7-yl)-1H-pyrrole-2,5-dione (27)
The title compound was synthesized from maleimide 24 and pyrazine-2-carboxylic acid according to general procedure H, purified using gradient II preparative HPLC, and isolated as an orange solid (30%). M.p. 167–172 °C. IR (ATR): νmax 3138, 3053, 1708, 1627, 1545, 1451, 1332, 1017, 744 cm-1. 1H NMR (400 MHz, DMSO-d6, two rotamers observed) δ 11.22 (s, 1H), 11.21 (s, 1H), 8.87 (d, J = 1.5 Hz, 1H), 8.77 (d, J = 2.5 Hz, 1H), 8.75 (d, J = 2.5 Hz, 1H), 8.69 (dd, J = 2.5, 1.5 Hz, 1H), 8.66 (dd, J = 2.5, 1.5 Hz, 1H), 8.59 (d, J = 1.5 Hz, 1H), 8.25 (s, 2H), 7.96 (s, 1H), 7.85 (s, 1H), 7.61 (s, 1H), 7.59 (s, 1H), 7.27–7.21 (m, 2H), 7.03 (d, J = 6.8 Hz, 1H), 6.97–6.93 (m, 4H), 6.82–6.69 (m, 3H), 6.60 (t, J = 7.6 Hz, 1H), 6.52 (d, J = 6.8 Hz, 1H), 5.17 (s, 2H), 4.98 (s, 2H), 4.73–4.69 (m, 2H), 4.61–4.57 (m, 2H), 4.23–4.18 (m, 2H), 4.10–4.05 (m, 2H). 13C NMR (100 MHz, DMSO-d6, two rotamers observed) δ 172.0 (C=O), 171.8 (C=O), 166.1 (C=O), 165.9 (C=O), 154.2 (C), 149.3 (C), 149.2 (C), 147.2 (CH), 145.7 (CH), 145.6 (CH), 144.6 (CH), 143.8 (CH), 143.2 (CH), 143.0 (CH), 135.6 (C), 135.2 (C), 134.4 (CH), 134.3 (CH), 132.2 (C), 126.7 (C), 125.3 (C), 125.3 (C), 124.8 (CH), 123.3 (C), 123.3 (C), 123.2 (C), 122.8 (CH), 121.8 (CH), 121.7 (CH), 120.6 (CH), 120.3 (CH), 120.1 (CH), 120.0 (CH), 111.4 (CH), 104.7 (C), 104.5 (C), 50.3 (CH2), 50.1 (CH2), 48.7 (CH2), 47.3 (CH2), 46.9 (CH2), 40.5 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C28H20N5O4: 490.1510, found: 490.1523. HPLC purity: 98.2%.
3-(Benzofuran-3-yl)-4-(9-fluoro-2-(2-(pyridin-4-yl)acetyl)-1,2,3,4-tetrahydro-[1,4]diazepino[6,7,1-hi]indol-7-yl)-1H-pyrrole-2,5-dione trifluoroacetate (28)
The title compound was synthesized from maleimide 24 and 2-(pyridin-4-yl)acetic acid according to general procedure H, purified using gradient II preparative HPLC, and isolated as an orange solid (30%). M.p. 142–147 °C. IR (ATR): νmax 3063, 2725, 1711, 1645, 1548, 1451, 1338, 1180, 1127, 748 cm-1. 1H NMR (400 MHz, DMSO-d6, two rotamers observed) δ 11.23 (s, 1H), 11.23 (s, 1H), 8.80 (s, 1H), 8.78 (s, 1H), 8.72 (s, 1H), 8.71 (s, 1H), 8.25 (s, 1H), 8.22 (s, 1H), 7.99 (s, 1H), 7.90 (s, 1H), 7.82 (s, 1H), 7.81 (s, 1H), 7.71 (s, 1H), 7.70 (s, 1H), 7.61 (s, 1H), 7.59 (s, 1H), 7.29–7.21 (m, 2H), 7.05–6.60 (m, 12H), 5.09 (s, 2H), 5.03 (s, 2H), 4.76 (br s, 2H), 4.60 (br s, 2H), 4.22–4.15 (m, 4H), 4.11 (s, 2H), 4.03 (s, 2H). 13C NMR (100 MHz, DMSO-d6, two rotamers observed) δ 172.1 (C=O), 172.1 (C=O), 171.8 (C=O), 171.8 (C=O), 168.0 (C=O), 167.9 (C=O), 158.5 (q, 2JC–F = 35.1 Hz, TFA), 155.2 (C), 154.3 (C), 154.2 (C), 154.2 (C), 147.2 (CH), 147.1 (CH), 142.5 (CH), 142.1 (CH), 135.6 (C), 135.3 (C), 134.3 (CH), 132.3 (C), 132.2 (C), 128.2 (C), 127.7 (CH), 126.8 (CH), 126.5 (C), 125.5 (C), 125.3 (C), 124.8 (CH), 123.7 (C), 123.4 (C), 123.3 (C), 123.0 (C), 122.9 (CH), 122.7 (CH), 121.8 (CH), 121.4 (CH), 120.8 (CH), 120.2 (CH), 120.2 (CH), 119.9 (CH), 116.2 (d, 1JC–F = 294.3 Hz, TFA), 111.4 (CH), 111.4 (C), 104.6 (C), 104.5 (C), 49.2 (CH2), 48.7 (CH2), 48.1 (CH2), 47.3 (CH2), 47.0 (CH2), 45.8 (CH2), 39.4 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C30H23N4O4: 503.1714, found: 503.1727. HPLC purity: 99.4%.
3-(Benzofuran-3-yl)-4-(9-fluoro-2-(piperidine-1-carbonyl)-1,2,3,4-tetrahydro-[1,4]diazepino[6,7,1-hi]indol-7-yl)-1H-pyrrole-2,5-dione (29)
The title compound was synthesized from maleimide 25 and piperidine-1-carbonyl chloride according to general procedure I, purified using gradient II preparative HPLC, and isolated as an orange solid (40%). M.p. 129–134 °C. IR (ATR): νmax 3136, 3053, 2934, 2855, 2731, 1714, 1614, 1548, 1451, 1336, 1116, 746 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.21 (s, 1H), 8.28 (s, 1H), 7.90 (s, 1H), 7.61 (d, J = 7.8 Hz, 1H), 7.25 (t, J = 8.0 Hz, 1H), 6.93 (t, J = 7.2 Hz, 1H), 6.89–6.82 (m, 2H), 6.45 (d, J = 8.4 Hz, 1H), 4.65 (s, 2H), 4.50–4.44 (m, 2H), 3.87–3.81 (m, 2H), 3.05–2.97 (m, 4H), 1.51–1.43 (m, 6H). 13C NMR (100 MHz, DMSO-d6) δ 172.0 (C=O), 171.7 (C=O), 163.3 (C=O), 156.8 (d, 1JC–F = 245.9 Hz, C), 154.3 (C), 147.3 (CH), 135.7 (CH), 132.3 (C), 132.0 (C), 127.2 (d, 3JC–F = 10.7 Hz, C), 126.9 (d, 3JC–F = 8.6 Hz, C), 125.2 (C), 125.0 (CH), 123.3 (C), 122.8 (CH), 121.7 (CH), 111.5 (CH), 111.3 (C), 109.3 (d, 2JC–F = 30.0 Hz, CH), 104.8 (d, 4JC–F = 4.4 Hz, C), 104.2 (d, 2JC–F = 22.0 Hz, CH), 50.9 (CH2), 49.1 (CH2), 49.0 (CH2), 47.7 (CH2), 25.2 (CH2), 24.2 (CH2). HRMS (ESI): m/z [M+H]+ calculated for C29H26FN4O4: 513.1933, found: 513.1955. HPLC purity: 97.3%.
3-(2-([1,4’-Bipiperidine]-1’-carbonyl)-9-fluoro-1,2,3,4-tetrahydro-[1,4]diazepino[6,7,1-hi]indol-7-yl)-4-(benzofuran-3-yl)-1H-pyrrole-2,5-dione trifluoroacetate (30)
The title compound was synthesized from maleimide 25 and 4-piperidino-1-piperidine-carbonyl chloride according to general procedure I, purified using gradient II preparative HPLC, and isolated as a yellow solid (40%). M.p. 190–195 °C. IR (ATR): νmax 3138, 3046, 2946, 2725, 1712, 1695, 1616, 1451, 1338, 1180, 1112, 748 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.23 (s, 1H), 8.97 (br s, 1H), 8.29 (s, 1H), 7.93 (s, 1H), 7.63 (d, J = 8.1 Hz, 1H), 7.26 (t, J = 6.3 Hz, 1H), 6.98–6.85 (m, 3H), 6.47 (d, J = 9.9 Hz, 1H), 4.71 (s, 2H), 4.53–4.49 (m, 2H), 3.90–3.86 (m, 2H), 3.60–3.46 (m, 5H), 2.96–2.89 (m, 2H), 2.77–2.67 (m, 2H), 1.95–1.33 (m, 10H). 13C NMR (100 MHz, DMSO-d6) δ 172.0 (C=O), 171.7 (C=O), 162.6 (C=O), 158.2 (q, 2JC–F = 31.6 Hz, TFA), 156.7 (d, 1JC–F = 233.4 Hz, C), 154.2 (C), 147.2 (CH), 135.8 (CH), 132.2 (C), 131.9 (C), 127.3 (d, 3JC–F = 10.8 Hz, C), 126.6 (d, 3JC–F = 8.6 Hz, C), 125.2 (C), 124.9 (CH), 123.4 (C), 122.8 (CH), 121.7 (CH), 117.0 (d, 1JC–F = 294.8 Hz, TFA), 111.4 (CH), 111.2 (C), 109.3 (d, 2JC–F = 25.6 Hz, CH), 104.8 (d, 4JC–F = 4.3 Hz, C), 104.4 (d, 2JC–F = 23.4 Hz, CH), 62.6 (CH2), 50.7 (CH2), 49.0 (CH2), 48.9 (CH2), 48.9 (CH2), 45.4 (CH2), 25.5 (CH2), 22.8 (CH2), 21.4 (CH2). HRMS (ESI): m/z [M-H]- calculated for C34H33FN5O4: 594.2522, found: 594.2531. HPLC purity: 99.0%.
3-(Benzofuran-3-yl)-4-(9-fluoro-2-(morpholine-4-carbonyl)-1,2,3,4-tetrahydro-[1,4]diazepino[6,7,1-hi]indol-7-yl)-1H-pyrrole-2,5-dione (31)
The title compound was synthesized from maleimide 25 and morpholine-4-carbonyl chloride according to general procedure I, purified using gradient I preparative HPLC, and isolated as an orange solid (50%). M.p. 157–160 °C. IR (ATR): νmax 3139, 3058, 2962, 2856, 1711, 1700, 1615, 1547, 1458, 1334, 1111, 746 cm-1. 1H NMR (400 MHz, DMSO-d6) δ 11.22 (s, 1H), 8.28 (s, 1H), 7.91 (s, 1H), 7.62 (d, J = 8.1 Hz, 1H), 7.26 (t, J = 8.3 Hz, 1H), 6.94 (t, J = 7.3 Hz, 1H), 6.89–6.85 (m, 2H), 6.46 (d, J = 9.4 Hz, 1H), 4.71 (s, 2H), 4.53–4.49 (m, 2H), 3.90–3.86 (m, 2H), 3.60–3.54 (m, 4H), 3.09–3.03 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 171.9 (C=O), 171.7 (C=O), 163.0 (C=O), 156.8 (d, 1JC–F = 234.9 Hz, C), 154.2 (C), 147.2 (CH), 135.7 (CH), 132.2 (C), 131.9 (C), 127.2 (d, 3JC–F = 10.7 Hz, C), 126.6 (d, 3JC–F = 8.6 Hz, C), 125.2 (C), 124.9 (CH), 123.3 (C), 122.7 (CH), 121.7 (CH), 111.4 (CH), 111.2 (C), 109.5 (d, 2JC–F = 26.4 Hz, CH), 104.7 (d, 4JC–F = 4.4 Hz, C), 104.3 (d, 2JC–F = 24.9 Hz, CH), 65.7 (CH2), 50.3 (CH2), 49.0 (CH2), 48.8 (CH2), 47.2 (CH2). HRMS (ESI): m/z [M-H]- calculated for C28H22FN4O5: 513.1580, found: 513.1605. HPLC purity: 97.3%.
Steroidogenesis assay
Cell culture
MA-10 mouse Leydig tumor cells were a gift from Dr. Mario Ascoli (University of Iowa). Cells were cultured in 75 cm2-cell culture flasks with Dulbecco’s modified Eagle’s medium (DMEM)/nutrient mixture F-12 Ham supplemented with 5% fetal bovine serum and 2.5% heat-inactivated horse serum, as previously described.43
Steroid biosynthesis
For steroid synthesis experiments in the presence of medium with the compounds under investigation, MA-10 cells were plated into 96-well plates at 2.5 × 104 cells per well. After allowing cells to adhere for 18 h, the cells were washed with PBS and exposed to the respective treatment (0–100 μM tested compound in medium). All media conditions were serum-free. Compounds were prepared as DMSO stock solutions and used at a final DMSO concentration of <0.02% in media. Controls contained the same amount of DMSO. At the end of the incubation, culture medium was collected and tested for progesterone production using RIA with anti-progesterone antisera (MP Biomedicals, Solon, OH), following the manufacturer’s recommended conditions. Progesterone production was normalized for the amount of protein in each well. RIA data were analyzed using Prism 5.03 from GraphPad.
Protein measurement
Proteins were quantified using the dye-binding assay of Bradford44 with bovine serum albumin (BSA) as the standard.
SmartCube® assay
Drugs were injected 15 min before the test, during which multiple challenges were presented over the course of the test session. At least 12 mice were used in each treatment group. Digital videos of the subjects were processed with computer vision algorithms to extract over 1,400 dependent measures including frequency and duration of behavioral states such as grooming and rearing and many other features obtained during the test session. These data were compared against a standard database of therapeutic class signatures obtained with the same experimental protocol. The database comprises 14 classes of drugs with some of the major classes, such as the antidepressant class, comprising several subclasses with representatives of most of the drugs on the market. Using machine learning techniques, the reference database was combed to find reference drug class signatures. The best performing techniques were chosen from our evaluation tests and used to build classifiers that capture the therapeutic class signatures. The behavioral signatures of the test compound were then assessed quantitatively with these classifiers to predict total and specific potential therapeutic utility.41, 42
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Mental Health (R01MH079400 to A.K.), the Canadian Institutes of Health Research (MOP102647to V.P.) and a Canada Research Chair in Biochemical Pharmacology (V.P.). A.M. was supported in part by a postdoctoral fellowship from the Le Fonds de la Recherche du Québec-Santé. The Research Institute of MUHC was supported by a Center grant from Le Fonds de la recherche du Québec-Santé. The funding agencies had no role in the study design, collection, analysis and interpretation of data, writing the manuscript, and decision to submit the manuscript for publication.
ABBREVIATIONS
- GSK-3
glycogen synthase kinase 3
- HPA
hypothalamus pituitary adrenal
- SAR
structure-activity relationship
- TLC
thin layer chromatography
- HPLC
high performance liquid chromatography
- rt
room temperature
- TFA
trifluoroacetic acid
- RIA
radioimmunoassay
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- ATR
attenuated total reflectance
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. Experimental procedures and analytical data for all the compounds not included in the main text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.Kovacsics CE, Gottesman II, Gould TD. Lithium’s antisuicidal efficacy: elucidation of neurobiological targets using endophenotype strategies. Annu Rev Pharmacol Toxicol. 2009;49:175–198. doi: 10.1146/annurev.pharmtox.011008.145557. [DOI] [PubMed] [Google Scholar]
- 2.Wada A. Lithium and neuropsychiatric therapeutics: neuroplasticity via glycogen synthase kinase-3beta, beta-catenin, and neurotrophin cascades. J Pharmacol Sci. 2009;110:14–28. doi: 10.1254/jphs.09r02cr. [DOI] [PubMed] [Google Scholar]
- 3.Phiel CJ, Klein PS. Molecular targets of lithium action. Annu Rev Pharmacol Toxicol. 2001;41:789–813. doi: 10.1146/annurev.pharmtox.41.1.789. [DOI] [PubMed] [Google Scholar]
- 4.Marmol F. Lithium: bipolar disorder and neurodegenerative diseases Possible cellular mechanisms of the therapeutic effects of lithium. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:1761–1771. doi: 10.1016/j.pnpbp.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 5.Spencer CM, Jahng JW, Ryu V, Houpt TA. Lithium-induced gene expression of inducible cyclic adenosine monophosphate early repressor in the rat adrenal gland. J Neurosci Res. 2005;82:273–282. doi: 10.1002/jnr.20617. [DOI] [PubMed] [Google Scholar]
- 6.Beaulieu JM, Marion S, Rodriguiz RM, Medvedev IO, Sotnikova TD, Ghisi V, Wetsel WC, Lefkowitz RJ, Gainetdinov RR, Caron MG. A beta-arrestin 2 signaling complex mediates lithium action on behavior. Cell. 2008;132:125–136. doi: 10.1016/j.cell.2007.11.041. [DOI] [PubMed] [Google Scholar]
- 7.Jope RS. Glycogen synthase kinase-3 in the etiology and treatment of mood disorders. Front Mol Neurosci. 2011;4(16):1–11. doi: 10.3389/fnmol.2011.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valvezan AJ, Klein PS. GSK-3 and Wnt Signaling in Neurogenesis and Bipolar Disorder. Front Mol Neurosci. 2012;5(1):1–13. doi: 10.3389/fnmol.2012.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Brien WT, Huang J, Buccafusca R, Garskof J, Valvezan AJ, Berry GT, Klein PS. Glycogen synthase kinase-3 is essential for beta-arrestin-2 complex formation and lithium-sensitive behaviors in mice. J Clin Invest. 2011;121:3756–3762. doi: 10.1172/JCI45194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gould TD, Manji HK. The Wnt signaling pathway in bipolar disorder. Neuroscientist. 2002;8:497–511. doi: 10.1177/107385802237176. [DOI] [PubMed] [Google Scholar]
- 11.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem Res. 2007;32:577–595. doi: 10.1007/s11064-006-9128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boulahjar R, Ouach A, Matteo C, Bourg S, Ravache M, le Guevel R, Marionneau S, Oullier T, Lozach O, Meijer L, Guguen-Guillouzo C, Lazar S, Akssira M, Troin Y, Guillaumet G, Routier S. Novel Tetrahydropyrido[1,2-a]isoindolone Derivatives (Valmerins): Potent Cyclin-Dependent Kinase/Glycogen Synthase Kinase 3 Inhibitors with Antiproliferative Activities and Antitumor Effects in Human Tumor Xenografts. J Med Chem. 2012;55:9589–9606. doi: 10.1021/jm3008536. [DOI] [PubMed] [Google Scholar]
- 14.Fugel W, Oberholzer AE, Gschloessl B, Dzikowski R, Pressburger N, Preu L, Pearl LH, Baratte B, Ratin M, Okun I, Doerig C, Kruggel S, Lemcke T, Meijer L, Kunick C. 3,6-Diamino-4-(2-halophenyl)-2-benzoylthieno[2,3-b]pyridine-5-carbonitriles Are Selective Inhibitors of Plasmodium falciparum Glycogen Synthase Kinase-3. J Med Chem. 2013;56:264–275. doi: 10.1021/jm301575n. [DOI] [PubMed] [Google Scholar]
- 15.Sutton LP, Rushlow WJ. The effects of neuropsychiatric drugs on glycogen synthase kinase-3 signaling. Neuroscience. 2011;199:116–124. doi: 10.1016/j.neuroscience.2011.09.056. [DOI] [PubMed] [Google Scholar]
- 16.Beaulieu JM, Gainetdinov RR, Caron MG. Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol. 2009;49:327–347. doi: 10.1146/annurev.pharmtox.011008.145634. [DOI] [PubMed] [Google Scholar]
- 17.Beaulieu JM. A role for Akt and glycogen synthase kinase-3 as integrators of dopamine and serotonin neurotransmission in mental health. J Psychiatry Neurosci. 2012;37:7–16. doi: 10.1503/jpn.110011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beurel E, Song L, Jope RS. Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol Psychiatry. 2011;16:1068–1070. doi: 10.1038/mp.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Latapy C, Rioux V, Guitton MJ, Beaulieu JM. Selective deletion of forebrain glycogen synthase kinase 3beta reveals a central role in serotonin-sensitive anxiety and social behaviour. Philos Trans R Soc Lond B Biol Sci. 2012;367:2460–2474. doi: 10.1098/rstb.2012.0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mines MA, Yuskaitis CJ, King MK, Beurel E, Jope RS. GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS One. 2010;5(e9706):1–12. doi: 10.1371/journal.pone.0009706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mines MA, Jope RS. Glycogen synthase kinase-3: a promising therapeutic target for fragile x syndrome. Front Mol Neurosci. 2011;4(35):1–8. doi: 10.3389/fnmol.2011.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu ZH, Chuang DM, Smith CB. Lithium ameliorates phenotypic deficits in a mouse model of fragile X syndrome. Int J Neuropsychopharmacol. 2011;14:618–630. doi: 10.1017/S1461145710000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chiu CT, Liu G, Leeds P, Chuang DM. Combined treatment with the mood stabilizers lithium and valproate produces multiple beneficial effects in transgenic mouse models of Huntington’s disease. Neuropsychopharmacology. 2011;36:2406–2421. doi: 10.1038/npp.2011.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan XB, Wang SS, Hou HL, Ji R, Zhou JN. Lithium improves the behavioral disorder in rats subjected to transient global cerebral ischemia. Behav Brain Res. 2007;177:282–289. doi: 10.1016/j.bbr.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 25.Schule C, Eser D, Baghai TC, Nothdurfter C, Kessler JS, Rupprecht R. Neuroactive steroids in affective disorders: target for novel antidepressant or anxiolytic drugs? Neuroscience. 2011;191:55–77. doi: 10.1016/j.neuroscience.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 26.Wirth MM. Beyond the HPA Axis: Progesterone-Derived Neuroactive Steroids in Human Stress and Emotion. Front Endocrinol (Lausanne) 2011;2(19):1–14. doi: 10.3389/fendo.2011.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roy L, McDonald CA, Jiang C, Maroni D, Zeleznik AJ, Wyatt TA, Hou X, Davis JS. Convergence of 3’,5’-cyclic adenosine 5’-monophosphate/protein kinase A and glycogen synthase kinase-3beta/beta-catenin signaling in corpus luteum progesterone synthesis. Endocrinology. 2009;150:5036–5045. doi: 10.1210/en.2009-0771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kozikowski AP, Gaisina IN, Yuan H, Petukhov PA, Blond SY, Fedolak A, Caldarone B, McGonigle P. Structure-based design leads to the identification of lithium mimetics that block mania-like effects in rodents. possible new GSK-3beta therapies for bipolar disorders. J Am Chem Soc. 2007;129:8328–8332. doi: 10.1021/ja068969w. [DOI] [PubMed] [Google Scholar]
- 29.Gaisina IN, Gallier F, Ougolkov AV, Kim KH, Kurome T, Guo S, Holzle D, Luchini DN, Blond SY, Billadeau DD, Kozikowski AP. From a natural product lead to the identification of potent and selective benzofuran-3-yl-(indol-3-yl)maleimides as glycogen synthase kinase 3beta inhibitors that suppress proliferation and survival of pancreatic cancer cells. J Med Chem. 2009;52:1853–1863. doi: 10.1021/jm801317h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Faul MM, Winneroski LL, Krumrich CA. A new one step synthesis of maleimides by condensation of glyoxylate esters with acetamides. Tetrahedron Lett. 1999;40:1109–1112. [Google Scholar]
- 31.Routier S, Ayerbe N, Merour JY, Coudert G, Bailly C, Pierre A, Pfeiffer B, Caignard DH, Renard P. Synthesis and biological evaluation of 7-azaindolocarbazoles. Tetrahedron. 2002;58:6621–6630. [Google Scholar]
- 32.O’Neill DJ, Shen L, Prouty C, Conway BR, Westover L, Xu JZ, Zhang HC, Maryanoff BE, Murray WV, Demarest KT, Kuo GH. Design, synthesis, and biological evaluation of novel 7-azaindolyl-heteroaryl-maleimides as potent and selective glycogen synthase kinase-3 beta (GSK-3 beta) inhibitors. Bioorg Med Chem. 2004;12:3167–3185. doi: 10.1016/j.bmc.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 33.Chedid M. Hematopoietic neoplasm chemotherapy. US20100261712A1. 2010
- 34.Engler TA, Henry JR, Malhotra S, Cunningham B, Furness K, Brozinick J, Burkholder TP, Clay MP, Clayton J, Diefenbacher C, Hawkins E, Iversen PW, Li Y, Lindstrom TD, Marquart AL, McLean J, Mendel D, Misener E, Briere D, O’Toole JC, Porter WJ, Queener S, Reel JK, Owens RA, Brier RA, Eessalu TE, Wagner JR, Campbell RM, Vaughn R. Substituted 3-imidazo[1,2-a]pyridin-3-yl-4-(1,2,3,4-tetrahydro-[1,4]diazepino-[6,7,1-hi]indol-7-yl)pyrrole-2,5-diones as highly selective and potent inhibitors of glycogen synthase kinase-3. J Med Chem. 2004;47:3934–3937. doi: 10.1021/jm049768a. [DOI] [PubMed] [Google Scholar]
- 35.Magnus NA, Ley CP, Pollock PM, Wepsiec JP. Pictet-Spengler based synthesis of a bisarylmaleimide glycogen synthase kinase-3 inhibitor. Org Lett. 2010;12:3700–3703. doi: 10.1021/ol101405g. [DOI] [PubMed] [Google Scholar]
- 36.Chen PC, Gaisina IN, El-Khodor BF, Ramboz S, Makhortova NR, Rubin LL, Kozikowski AP. Identification of a Maleimide-Based Glycogen Synthase Kinase-3 (GSK-3) Inhibitor, BIP-135, that Prolongs the Median Survival Time of Δ7 SMA KO Mouse Model of Spinal Muscular Atrophy. ACS Chem Neurosci. 2012;3:5–11. doi: 10.1021/cn200085z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.http://www.reactionbiology.com/.
- 38.Braeuning A, Buchmann A. The Glycogen Synthase Kinase Inhibitor 3-(2,4-Dichlorophenyl)-4-(1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione (SB216763) Is a Partial Agonist of the Aryl Hydrocarbon Receptor. Drug Metab Dispos. 2009;37:1576–1580. doi: 10.1124/dmd.109.027821. [DOI] [PubMed] [Google Scholar]
- 39.Diemer T, Allen JA, Hales KH, Hales DB. Reactive oxygen disrupts mitochondria in MA-10 tumor Leydig cells and inhibits steroidogenic acute regulatory (StAR) protein and steroidogenesis. Endocrinology. 2003;144:2882–2891. doi: 10.1210/en.2002-0090. [DOI] [PubMed] [Google Scholar]
- 40.Privalle CT, McNamara BC, Dhariwal MS, Jefcoate CR. ACTH control of cholesterol side-chain cleavage at adrenal mitochondrial cytochrome P-450scc. Regulation of intramitochondrial cholesterol transfer. Mol Cell Endocrinol. 1987;53:87–101. doi: 10.1016/0303-7207(87)90195-x. [DOI] [PubMed] [Google Scholar]
- 41.Roberds SL, Filippov I, Alexandrov V, Hanania T, Brunner D. Rapid, computer vision-enabled murine screening system identifies neuropharmacological potential of two new mechanisms. Front Neurosci. 2011;5(103):1–4. doi: 10.3389/fnins.2011.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang HK, Eaton JB, Yu LF, Nys M, Mazzolari A, van Elk R, Smit AB, Alexandrov V, Hanania T, Sabath E, Fedolak A, Brunner D, Lukas RJ, Vistoli G, Ulens C, Kozikowski AP. Insights into the structural determinants required for high-affinity binding of chiral cyclopropane-containing ligands to alpha4beta2-nicotinic acetylcholine receptors: an integrated approach to behaviorally active nicotinic ligands. J Med Chem. 2012;55:8028–8037. doi: 10.1021/jm3008739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hauet T, Yao ZX, Bose HS, Wall CT, Han Z, Li W, Hales DB, Miller WL, Culty M, Papadopoulos V. Peripheral-type benzodiazepine receptor-mediated action of steroidogenic acute regulatory protein on cholesterol entry into leydig cell mitochondria. Mol Endocrinol. 2005;19:540–554. doi: 10.1210/me.2004-0307. [DOI] [PubMed] [Google Scholar]
- 44.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.