

Abstract
The pregnane X receptor (PXR) regulates drug metabolism by regulating the expression of drug-metabolizing enzymes such as cytochrome P450 3A4 (CYP3A4), which is involved in the metabolism of >50% of clinically prescribed drugs. The activity of PXR can be controlled by the binding of small molecule agonists or antagonists. Because of its unique ligand binding pocket, PXR binds promiscuously to structurally diverse chemicals. To study the structure-activity relationship, novel modulators for PXR are needed. Here we report the virtual screening of ~25,000 natural product derivatives from the ZINC database using the Molecular Operating Environment docking software tool against the PXR-rifampicin complex x-ray crystal structure. Our screening resulted in identification of compounds based on the lowest S score, which measures Gibbs free energy. Interestingly, we found that the compounds that bind directly to PXR, as revealed in an intrinsic tryptophan fluorescence assay, modulate CYP3A4 promoter activity differentially in HepG2 cells. Mutational analysis and docking studies showed that these compounds bind broadly in the ligand binding pocket but interact with different amino acid residues. We further investigated the mechanism of binding by analyzing the functional groups that are important for distinguishing agonists from antagonists. The approach we used to identify novel modulators that bind to PXR can be useful for finding novel modulators of PXR.
Keywords: PXR, drug metabolism, molecular docking, natural product derivative
1. Introduction
Nuclear receptors are ligand-activated transcription factors involved in regulating many physiologic and pathologic processes [1]. Ligand binding to nuclear receptors leads to dissociation of co-repressors, recruitment of co-activators, and subsequent activation of gene expression [2]. Because of their associations with many human diseases, nuclear receptors are therapeutic targets for pharmaceutical development [2]. A typical nuclear receptor consists of an NH2–terminal ligand independent activation function 1 domain (AF-1), a highly conserved DNA binding domain (DBD), and a C-terminal ligand binding domain (LBD), followed by an activation function 2 domain (AF-2) [3]. Nuclear receptors can form homodimers or heterodimers with retinoic X receptor (RXR) through the amino acid sequences within the DBD and LBD.
The pregnane X receptor (PXR, NR1I2) belongs to the nuclear receptor family [3]. PXR regulates the expression of proteins such as drug metabolizing enzyme cytochrome P450 3A4 (CYP3A4), efflux transporter P-glycoprotein, and other multidrug resistance proteins, which are involved in metabolism and elimination of potentially harmful chemicals [2, 4–9]. PXR has been detected in various tissues including kidney, colon, brain capillaries, small intestine, and predominantly in liver [4], and it can be activated by various ligands that bind to its LBD. PXR forms a heterodimeric complex with RXR to activate gene transcription. Many agonists have been reported for PXR, including the antibiotics rifampicin, clotrimazole, and ritonavir; the antineoplastic drugs cyclophosphamide, cyproterone acetate, taxol, tamoxifen, and RU486; the anti-inflammatory agent dexamethasone; the anti-type 2 diabetes drug troglitazone; the antihypertensive drugs nifedipine and spironolactone; and the sedatives glutethimide and phenobarbital [5, 10]. In addition, some commonly used herbal medicines contain components that activate PXR, such as hyperforin from St. John’s wort [11]. The crystal structures of the LBD of human PXR in ligand-free and ligand-bound forms have been solved by x-ray crystallography, with ligands including PXR agonists hyperforin, colupulone (from hops), 17β-estradiol, SR12813, T1317, and rifampicin [12–16]. Analysis of those structures shows that the PXR LBD is promiscuous, highly hydrophobic, and flexible. Because of its promiscuous nature, it allows molecules of differing sizes to bind in multiple orientations [17]. Therefore, novel agonists are still valuable to investigate the regulation of PXR [18]. A limited number of PXR antagonists have also been reported [19–21], such as ET-743 [22], ketoconazole [23], sulforaphane [24], A-792611 [25], coumestrol [26], camptothecin [27], sesamine [28], and SPB00574 [29]. More recently, milk thistle’s active components silybin and isosilybin are shown to be inhibitors of PXR-mediated CYP3A4 induction [30]. Efforts have been made to develop PXR antagonists for potential use in overcoming drug resistance [19, 20]. Computational approaches led to the identification of key pharmacophores and binding regions in PXR for ketoconazole and its azoles derivatives [31]. Interestingly, another study identified a new class of compounds with similar structures but opposing activities [27]. In this study, camptothecin was found to be a potent inhibitor of PXR. In contrast, one of the analogues of camptothecin, irinotecan, was found to be a PXR agonist. Further structure-activity studies are needed to understand the molecular mechanism responsible for the receptor-compound interactions, leading to either activation or inhibition of PXR [27]. The lack of well-characterized PXR antagonists and the lack of knowledge of the crystal structure for a PXR-antagonist complex hinder the structure-activity studies of PXR and its role in drug metabolism [32].
In this study, we focused on identifying and characterizing novel modulators for PXR. We performed a virtual screening of natural product derivatives from the ZINC database using the Molecular Operating Environment (MOE) docking software tool against the x-ray crystal structure of the PXR-rifampicin complex. Putative PXR binders were identified based on the lowest S score, which measures Gibbs free energy, and synthesized to characterize their biological activities against PXR. Our structure-activity relationship (SAR) data suggest that the modulators behave differently based on the functional group present in the compounds. Intrinsic tryptophan fluorescence data suggest that PXR binds physically with these compounds. The structural basis of these compounds, molecular mode of interaction/mechanism of binding with PXR, and the functional groups that are important for distinguishing agonists/antagonists were also explored. PXR carrying mutations in residues important for ligand binding were also used to investigate the binding of PXR modulators. The method reported here can be used to find novel agonists or antagonists for PXR.
2. Materials and Methods
2.1 Materials and chemicals
HepG2 liver carcinoma cells and LS 174T human intestinal cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA). Cell culture reagents were obtained from Invitrogen (Carlsbad, CA). Anti-FLAG M2 antibody, anti-β-actin antibody, DMSO, and rifampicin were obtained from Sigma Aldrich (St. Louis, MO). Charcoal/dextran-treated FBS was purchased from Hyclone (Logan, UT); blocking buffer, anti-mouse, and anti-rabbit IR Dye secondary antibodies were from LI-COR Biosciences (Lincoln, NE). All small molecules and analogues used in this study were purchased from Ambinter (Orleans, France).
2.2 Virtual screening and molecular docking by MOE
The Diversity Set of the ZINC natural product derivatives database consisting of ~25,000 small molecules was selected for virtual screening using MOE (MOE 2010.10; http://www.chemcomp.com). Receptor files, ligands, and docking parameter files were prepared using MOE. The coordinates of the PXR-LBD-rifampicin x-ray crystal structure were taken from the Protein Data Bank (PDB: 1SKX) [15]. The crystal structure contained two PXR LBDs and two rifampicin molecules because PXR forms dimers in solution. One PXR-LBD-rifampicin complex was used for the virtual screening. All hydrogen atoms and partial charges were added to the protein using protonate 3D. The energy of the PXR-LBD-rifampicin complex molecule was minimized using an energy minimization algorithm that uses the MMFF94x force field. The energy-minimized structure was used as the template for the virtual screening studies. We chose the rifampicin binding site (active site) as our pharmacophore for virtual screening. The placement of the small molecule ligand was determined by pharmacophore and followed by rescoring using London DG. The placements of the ligands were refined again by force field. Finally, a three-dimensional pharmacophore model was generated using MOE. The ZINC dataset of ~25,000 small molecules was obtained in mol2 format. Compounds were selected based on scoring function (binding energy). The best conformation for each ligand was isolated based on the S score, which measures interactions. The compounds with the lowest S scores were chosen for biological evaluation.
2.3 Cell culture, plasmids, and transfection
Both HepG2 and LS 174T cells were maintained in modified Eagle’s minimal essential medium (ATCC) with 10% FBS, 2 mM L-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37°C in a humidified atmosphere containing 5% CO2. The pcDNA3-FLAG-hPXR construct and the CYP3A4-luciferase reporter were described previously [33]. CYP2B6 promoter reporter (CYP2B6pro-Luc, or CYP2B6-PBREM/XREM) was reported previously [34, 35]. pcDNA3-FLAG-hPXR mutants containing various mutations (H327A, R410A, M246A, V211A, Y306A, W299A, Q285A, M243A, and F288A) were generated by Codex BioSolutions, Inc. (Gaithersburg, MD). Mutations were verified through nucleotide sequencing. Transfections were performed using FuGENE 6 (Roche Diagnostics, Indianapolis, IN) according to the manufacturer’s instructions.
2.4 Transient transfection and luciferase reporter gene assays
The methods were described previously [33]. Briefly, for transient transfection, HepG2 cells in a T-25 tissue culture flask containing ~2 million cells were transfected with 3 μg of total plasmids. To prepare the plasmid mix for CYP3A4 promoter assay, 0.6 μg CMV-Renilla, 2.1 μg CYP3A4-luc, and 0.3 μg of FLAG-hPXR were mixed with 9 μl of Fugene 6, diluted in 250 μl of serum-free medium, and transferred to a T-25 flask to transfect HepG2 or LS 174T cells. For CYP2B6 promoter assay, 0.6 μg CMV-Renilla, 1.8 μg CYP2B6-luc (CYP2B6pro-Luc), and 0.6 μg of FLAG-hPXR were used to prepare the plasmid mix and added to 6-well plates containing 5 ×105 cells/well. After 24 h, cells were seeded in 384-well plates (5000 cells/well) in phenol red–free medium containing 5% charcoal/dextran-treated FBS and incubated for another 24 h before compound treatment. Compounds were transferred using pin tools. The cells were incubated with compounds for 24 h before processing using the Dual-Glo Luciferase Assay System (Promega, Madison, WI). Renilla luciferase activity was used to normalize the firefly luciferase activity. CYP3A4 promoter activity was determined as described previously [33]. Rifampicin (5 μM) and DMSO were used as positive and negative controls, respectively. Curve-fitting software (GraphPad Prism 4.0; GraphPad Software, La Jolla, CA) was used to generate the curves and to determine the EC50 or IC50 values.
2.5 Cell viability assay
HepG2 cells were transiently transfected with FLAG-hPXR and treated with compounds as described in section 2.4 before CellTiter-Glo (Promega) was used to measure cell viability. CellTiter-Glo reagent was added to the wells and incubated at room temperature for 10 min protected from light. Luminescence was recorded using an Envision plate reader (PerkinElmer Life Sciences). DMSO was used as a control. Values of viability of treated cells were expressed as a percentage of DMSO.
2.6 Mammalian two-hybrid assay
The mammalian two-hybrid assay was performed as described previously [36]. The mammalian two-hybrid system (Promega) consists of VP16-hPXR, Gal4-SRC-1, and a luciferase reporter pG5-luc co-transfected into HepG2 cells. The Gal4 vector (pBIND) also constitutively expresses Renilla luciferase, which was used as an internal transfection control. The Dual-Glo Luciferase Assay (Promega) was used to measure luciferase activity, which is an indicator of protein-protein interactions. The relative luciferase activity for pG5-luc was determined by normalizing firefly luciferase activity with Renilla luciferase activity.
2.7 Western blot analysis
All cell extracts were harvested in 1× RIPA buffer from Cell Signaling Technology, Inc. (Danvers, MA), and samples were centrifuged at 12,000 × g at 4°C for 25 min. The samples were then boiled in sample loading buffer (Invitrogen) containing SDS, and equal amounts of samples were resolved on 4–12% SDS-PAGE gradient gel and then transferred onto a nitrocellulose membrane. The membrane was blocked and incubated with the indicated antibodies overnight at 4°C. All Western blot analyses were performed on the Odyssey Infrared Imaging system (LI-COR Biosciences, Lincoln, NE).
2.8 Protein expression and purification
The recombinant pETDuet1-hPXR-LBD/mSRC-1 plasmid (Medicilon, Shanghai, China) was transformed into Escherichia coli BL21 DE3 cells for protein expression. Saturated LB-ampicillin starter culture was diluted (1:25, v/v) in LB media and grown at 17°C to an A600 of 0.6. Expression was induced with 0.8 mM IPTG, and cells were harvested by centrifugation after incubation for 18 h. The bacterial cells were suspended in buffer A (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10% glycerol, and Cocktail [EDTA-free protein inhibitor, Roche]). The clarified cell lysates were incubated with His-Select Ni resin (Qiagen, Gaithersburg, MD) equilibrated in buffer A for 1 h at 4°C. The resin was subsequently washed with 100 mL of buffer B (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10% glycerol, and 50–100 mM imidazole). PXR-LBD was eluted using buffer C (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10% glycerol, and 200 mM imidazole). Column fractions were pooled and subjected to an SP cation exchange column (Bio-Rad Laboratories, Inc., Hercules, CA) pre-equilibrated with buffer containing 20 mM Tris-HCl (pH 7.8), 50 mM NaCl, 5 mM DTT, pH 7.5, and 5% glycerol. The column was washed with the same buffer, and fractions containing the PXR LBD were eluted using 1 M NaCl gradient and pooled. The protein was concentrated and buffer exchanged (20 mM phosphate buffer and 1 mM DTT, pH 7.4) using Centri-prep 10K (Millipore Corp., Billerica, MA) units just before further experiments. Protein concentrations were measured using the Bradford method.
2.9 Intrinsic tryptophan fluorescence studies
Intrinsic tryptophan fluorescence of freshly prepared PXR-LBD in 20 mM phosphate buffer, 1 mM DTT, pH 7.4, and compounds was measured by FluoroLog-3 spectrofluorometer (Jobin-Yvon, Inc.) using a quartz cuvette of 10 mm path length at a constant temperature of 25°C, equipped with a constant temperature water circulator bath. Baseline corrections were made by subtracting the spectra of the PXR LBD and the PXR LBD with compounds from that of compounds in buffer alone. The protein was excited at 280 nm, and the emission spectra were recorded from 300 nm to 400 nm with an emission maximum at 340 nm. The spectrum of the PXR LBD showed a maximum at 340 nm, indicating the presence of tryptophan residue. Emission from the controls was corrected by recording subtraction spectra between sample and control probes. The decrease in the fluorescence intensity was calculated as (F0 − FC)/(F0 − Fmin), where F0 is the initial fluorescence intensity of free PXR LBD, FC is the corrected fluorescence intensity at a ligand concentration [C], and Fmin is the fluorescence intensity at the highest concentration of the protein. The data were fitted to a nonlinear regression of the plot of (F0 − FC)/(F0 − Fmin) against [C] with the equation corresponding to a single binding site using GraphPad Prism software (GraphPad Software).
2.10. Statistical analysis
Results are expressed as the mean ± standard deviation of at least 3 independent experiments, and error bars indicate the standard deviation. Statistical analyses were performed using Student’s t-test. Differences were considered statistically significant for p ≤0.05 (*).
3. Results
3.1 Virtual screening identifies novel putative modulators for PXR
The ZINC natural product derivatives database consisting of ~25,000 small molecules was selected for the virtual screening to identify novel putative PXR modulators, using a work flow scheme shown in Figure 1. Based on the lowest S score, which measures Gibbs free energy, 9 compounds (S score ≤ −33.0 Kcal/mol) were selected as putative PXR modulators (Figure 2). These putative PXR modulators have scaffolds that differ from those in previously published [12, 21, 22, 37–42].
Figure 1. Work flow for identifying novel modulators for PXR.

Schematic representations of the virtual screening method and SAR.
Figure 2. Compounds selected after virtual screening based on S values, which measure binding energy.

Structures of these compounds and corresponding S values are provided.
3.2 Functional characterization of the putative PXR modulators and analogues leads to identification of novel PXR agonists and antagonists
We used HepG2 transfected with FLAG-hPXR, CYP3A4-luc (with luciferase expression controlled by the PXR-regulated CYP3A4 promoter), and CMV-Renilla (as a transfection control) to evaluate the agonistic or antagonistic (in the presence of 5 μM rifampicin) activity of the 9 putative PXR modulators. Only compound 1 affected the activity of PXR as an agonist (Figure 3). To investigate the SAR, seven analogues of compound 1, namely compounds 2, 3, 4, 5, 6, 7, and 8 (Figure 4), were obtained and evaluated for their agonistic and antagonistic effects on PXR. Among the analogues of compound 1, compounds 2, 3, 4, and 7 were agonists, with estimated EC50 values in the range of 0.1–10.0 μM (Figure 5 and Table 1). Compounds 1, 2, and 7 were more potent than compounds 3 and 4. Interestingly, compounds 5, 6, and 8 displayed antagonistic effects on PXR with estimated IC50 values in the 2–6 μM range (Figure 6A–C and Table 1). Compounds 5, 6, and 8 alone slightly increased luciferase activity, suggesting that these compounds have weak agonistic effects in the absence of a potent agonist (Figure 6D–F). We used the CellTiter Glo cell viability assay to evaluate the compound toxicity in HepG2 cells treated with compounds for 24 h, the same treatment time used in the transactivation assay. As shown in Figure 7, whereas the maximal cytotoxicity at the highest compound concentration (56 μM) was less than 40%, the CYP3A4-luc reporter activity was completely inhibited. At 1 μM, no apparent cytotoxicity was observed; however, the CYP3A4-luc activity was inhibited by > 40%. These data indicated that the antagonistic effects of compounds 5, 6, and 8 were not due to compound cytotoxicity. Among the antagonists, compound 8 was the least toxic and showed the least agonistic activity compared with compounds 5 and 6. To evaluate the effects of agonist and antagonist on CYP3A4 promoter in a different cellular background, we used an intestinal cell line LS 174T. Both compound 1 and rifampicin activated CYP3A4 promoter activity in LS 174T cells (EC50=0.63 μM and 0.3 μrespectively) (Figure 8A). However, compound 6 only showed weak antagonistic effect in LS 174T cells (IC50=13.57 μM) (Figure 8B). To evaluate the effects of agonist and antagonist on a different PXR-regulated promoter in HepG2 cells, we used CYP2B6pro-Luc. Whereas both compound 1 and rifampicin showed agonistic effect on CYP2B6 promoter (EC50=0.88 μM and 6.45 μrespectively) (Figure 9), no significant antagonistic effect on CYP2B6 promoter was observed for compound 6 (Data not shown).
Figure 3. Compound 1 activates PXR-regulated CYP3A4 promoter.

HepG2 transiently transfected with hPXR, CYP3A4-luc, and CMV-Renilla were treated for 24 h with indicated concentrations of rifampicin or compound 1 prior to luciferase assay.
Figure 4. Analogues of compound 1.

Compounds 1, 2, 4, and 7 are agonists, whereas compounds 5, 6, and 8 are antagonists.
Figure 5. Compounds 1, 2, 4, and 7 are PXR agonists.

HepG2 cells transiently transfected with hPXR, CYP3A4-luc, and CMV-Renilla were treated for 24 h with indicated concentrations of compounds prior to luciferase assay.
Table 1.
Comparison of compound activities from luciferase reporter assays in HepG2 cells.
| Compounds | EC50(μM) |
|---|---|
| Rifampicin | 1.7 |
| Compound 1 | 0.11 |
| Compound 2 | 0.42 |
| Compound 3 | 10.07 |
| Compound 4 | 3.69 |
| Compound 7 | 0.36 |
| Compounds | IC50 (μM) |
|---|---|
| Compound 5 | 1.86 |
| Compound 6 | 2.25 |
| Compound 8 | 5.29 |
Figure 6. Compounds 5, 6, and 8 are PXR antagonists.

HepG2 cells transiently transfected with hPXR, CYP3A4-luc, and CMV-Renilla were treated for 24 h with 5 μM of rifampicin and indicated concentrations of compound 5, 6, or 8 prior to luciferase assay (A–C). HepG2 cells transiently transfected with hPXR, CYP3A4-luc, and CMV-Renilla were treated for 24 h with indicated concentrations of compounds prior to luciferase assay (D–F).
Figure 7. Cytotoxicity of compounds in HepG2 cells.

Cells were treated with increasing concentrations (1.7 nM to 56 μM) of compounds.
Figure 8. Compounds 1 and 6 modulate PXR-regulated CYP3A4 promoter activity in LS 174T cells.

(A) LS 174T cells transiently transfected with hPXR, CYP3A4-luc, and CMV-Renilla were treated for 24 h with indicated concentrations of rifampicin or compound 1 prior to luciferase assay. (B) LS 174T cells transiently transfected with hPXR, CYP3A4-luc, and CMV-Renilla were treated for 24 h with 5 μM of rifampicin and indicated concentrations of compound 6 prior to luciferase assay.
Figure 9. Compound 1 activate PXR-regulated CYP2B6 promoter activity in HepG2 cells.
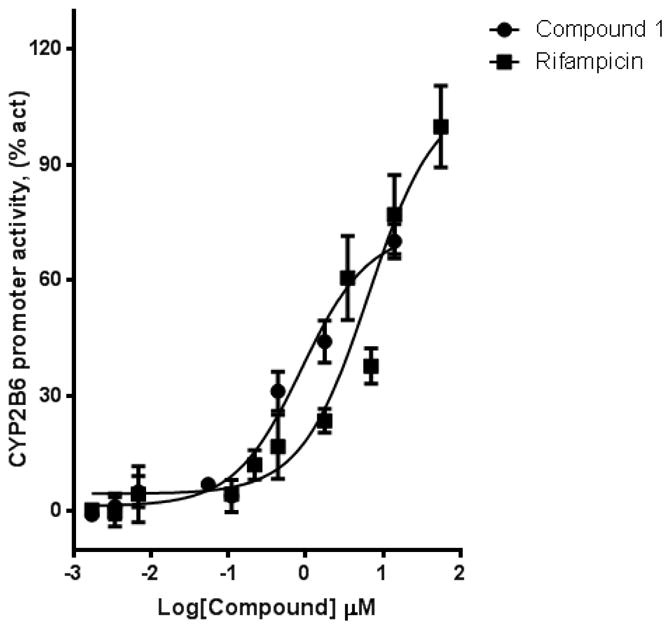
HepG2 cells transiently transfected with hPXR, CYP2B6-luc, and CMV-Renilla were treated for 24 h with indicated concentrations of rifampicin or compound 1 prior to luciferase assay.
Co-activator SRC-1 has been shown to mediate the ligand-induced activation of PXR [38]. To correlate with the luciferase data, we used a mammalian two-hybrid assay as previously described [36] to evaluate the effect of the newly identified PXR agonists and antagonists on the interaction between PXR and SRC-1. As shown in Figure 10A, rifampicin and compounds 1, 2, and 7, which are potent agonists (Figure 5), significantly induced the interaction between hPXR and SRC-1. Compounds 3 and 4, which are weak agonists (Figure 5), did not induce significant interaction between PXR and SRC-1. Compounds 5, 6, and 8 significantly inhibited the rifampicin-inducible interaction of PXR with SRC-1 (Figure 10B). Together, these data suggest that compounds 1, 2, and 7 induce PXR to recruit SRC-1 to activate the CYP3A4 promoter, and compounds 5, 6, and 8 inhibit the rifampicin-induced PXR/SRC-1 interaction.
Figure 10. Mammalian two-hybrid assay confirms that compounds affect hPXR interaction with SRC-1.

Mammalian two-hybrid assays were performed in HepG2 cells transiently cotransfected with plasmids encoding Gal4-SRC-1 and the reporter gene pG5-luc, along with empty vector pACT as indicated. (A) The cells were treated with DMSO, 5 μM rifampicin, or 3 μM compound 1, 2, 3, 4, or 7. * Indicates p < 0.05 (comparisons were made between compound 1, 2, 3, 4, or 7 and DMSO control for samples transfected with pACT-hPXR). (B) Cells were treated with DMSO, 5 μM rifampicin, or 10 μM compound 5, 6, or 8 in the presence of 5 μM rifampicin (Rif) 24 h after transfection. Luciferase assays were performed 24 h after the compound treatment. The relative luminescence for pG5-luc was determined by normalizing firefly luciferase activity with Renilla luciferase activity. The values represent the means of five independent experiments, and the bars denote the S.D. * Indicates p < 0.05 for samples transfected with pACT-hPXR (samples co-treated with rifampicin and either compound 5, 6, or 8 were compared to rifampicin control; the difference between rifampicin and DMSO is also statistically significant with p < 0.05).
3.3 Biophysical characterization and direct interaction of ligands with PXR LBD
To understand the mechanism of action responsible for the regulation of PXR by the agonists and antagonists, it is important to investigate changes in the structural properties of PXR upon ligand binding. The cell-based assays discussed above suggest that the compounds modulate the activity of PXR. To investigate whether these compounds physically bind to the PXR LBD, we performed an intrinsic tryptophan fluorescence assay, which is commonly used to examine protein-ligand interactions. In the intrinsic tryptophan fluorescence quenching assays, binding of a compound to the PXR LBD will cause a change in the conformation of the protein, resulting in a decrease in fluorescence intensity. The protein was excited at 280 nm, and emission maximum was observed at 340 nm, which is typical for tryptophan-containing proteins and did not shift during titration. The fluorescence maxima gradually decreased upon ligand binding in a concentration-dependent manner. The change in fluorescence intensity was fitted to a non-linear, single-site binding equation to generate a dose-response curve (Figure 11). The dissociation constant ranged from 0.02 to 0. 1 μM (Table 2). The dissociation constant values, which are in the same range as that of rifampicin, indicated that compounds 1, 2, 5, 6, 7, and 8 bind to PXR with moderate affinity. Compound 3, which did not show significant agonistic activity, did not show fluorescence quenching at a detectable level; therefore, no dissociation constant could be determined (“ND” in Table 2). Together, these data indicate that the compounds identified in this study bind to PXR to affect its function.
Figure 11. Compounds physically binds to PXR LBD.

Decrease in the fluorescence intensities of PXR LBD samples upon addition of compounds as indicated. Solid lines represent the fit of the data to the single-binding site equation for each compound.
Table 2.
Dissociation constants (KD) for the binding of various ligands to PXR LBD.
| Compounds | Dissociation constants (KD) (μM) |
|---|---|
| Compound 1 | 0.10 |
| Compound 2 | 0.05 |
| Compound 3 | ND |
| Compound 5 | 0.03 |
| Compound 6 | 0.02 |
| Compound 7 | 0.04 |
| Compound 8 | 0.07 |
| Rifampicin | 0.08 |
3.4 SAR and selectivity of the ligands
SAR studies were performed to correlate small molecule structure and its role in modulating PXR. We found that the various substituents of the ligands and the position of their phenyl ring of the sulfonamide moiety influenced activity and binding. Compounds with aryl sulfonamides (compounds 1, 2, 4, 5, 6, 7, and 8) were biologically active. In contrast, compound 3 (Figure 4), a methyl sulfonamide derivative lacking an aromatic moiety, did not show significant activity in transactivation or in the binding assays. This can be explained by the small methyl group, which is not optimal for binding to PXR. Thus, our SAR study showed that changes in functional groups can be important for determining the character of the ligand for PXR (Figure 4). We also found that substitutions at the ortho position of aryl sulfonamides (compounds 1, 2, 4, and 7) led to an agonistic effect on PXR, whereas para-substituted aryl sulfonamides led to antagonistic activity (compounds 5, 6, and 8). From the SAR point of view, the agonistic/antagonistic activity is dependent on the position of the substitution on the aryl sulfonamides.
3.5 Predicted binding mode and molecular docking of the compounds into the binding site of the PXR LBD
With the aim of understanding the interactions between PXR modulators and the target, molecular modeling of these compounds into the rifampicin binding pocket of the PXR LBD was performed (Figure 12). Our molecular modeling studies showed that the mode of binding is slightly different in the cases of agonists and antagonists, assuming both are binding at the same place. The ligands fit into the hydrophobic binding pocket created by mostly the aromatic amino acid residues. Structural investigation showed that hydrophobic amino acid residues present at the binding pocket are important for ligand binding (Figure 12). Residues that are most important for interactions are Arg 410, His 407, Ile 414, Met 246, Ser 247, Met 243, Gln 285, His 327, Phe 288, Met 323, Tyr 306, Trp 299, and Val 211. The computational binding studies provided a better understanding of the binding of these compounds to the active site of the PXR LBD. From the compound-PXR interaction analysis, it became evident that Phe 288, Tyr 306, His 327, and Trp 299 are important for interaction with the sulfonamide moiety (Figure 13). When the aryl sulfonamide is replaced by methyl sulfonamide, no interaction was observed. It was found that in the case of antagonists, para substitution of aryl sulfonamide pointed outward from the ligand binding site, in contrast to the binding of agonists, in which ortho substitution of aryl sulfonamide pointed inward toward the binding site. Docking mode 3D interaction schemes of predicted binding poses of various compounds are shown (Figure 14).
Figure 12. Binding modes of different compounds to PXR LBD.

Docking mode two-dimensional (2D) interaction schemes of predicted binding poses of compound 1 (A), compound 2 (B), compound 7 (C), compound 5 (D), compound 6 (E), and compound 8 (F) at the PXR LBD binding site.
Figure 13. Comparison of the binding modes of compound 1 and compound 3 to PXR LBD.
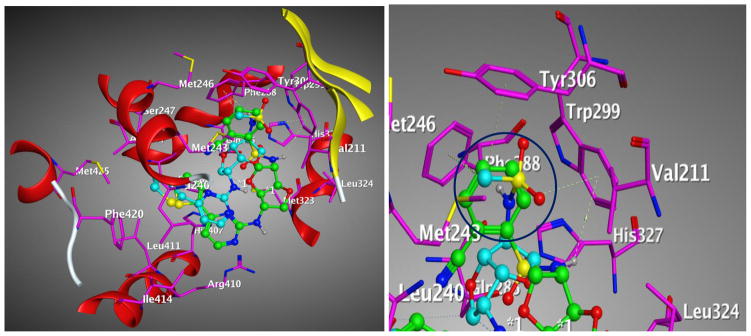
Main interacting side chains identified through docking of compound 1 (green) and compound 3 (cyan) with the PXR LBD. The aryl group of compound 1 can form pi-pi interactions but not the methyl group of compound 3 shown as circle (blue).
Figure 14. Binding modes of different compounds to PXR LBD.

Docking mode three-dimensional (3D) interaction schemes of predicted binding orientations of compound 1 (A), compound 2 (B), compound 7 (C), compound 5 (D), compound 6 (E), and compound 8 (F) at the PXR LBD binding site.
Based on modeling predictions of the PXR LBD contact residues with compound 1, the following residues of hPXR were mutated to alanine: H327A, R410A, M246A, V211A, Y306A, W299A, Q285A, M243A, and F288A. In HepG2 cells, the wild-type and all the PXR mutants were activated by 5 μM of compound 1. Interestingly, H327A, M246A, V211A, W299A, and M243A mutants showed significantly increased CYP3A4 promoter activity by compound 1 (Figure 15A). In contrast, R410A and G285A showed decreased activity in the presence of compound 1. Western blot analysis showed that all the PXR mutants except W299A and H327A were expressed at a level similar to that of the wild-type PXR in HepG2 cells in the presence of 5 μM compound 1 (Figure 15B). These data suggest that different residues of the PXR LBD are involved in the interaction with compound 1 in different modes, confirming the direct interaction between compound 1 and the PXR LBD.
Figure 15. Comparison of hPXR and hPXR mutants upon 5 μM compound 1 treatment.

(A) HepG2 cells transiently transfected with hPXR or hPXR mutants, CYP3A4-luc, and CMV-Renilla were treated for 24 h with indicated concentrations of compound 1 prior to luciferase assay. The relative luciferase units (a.u.) were determined by normalizing with the Renilla luciferase control. (B) The expression of hPXR or hPXR mutants upon compound 1 treatment. Actin expression level was used to verify equal loading of lysates. The values represent the means of three independent experiments, and the bars denote the S.D. * Indicates p < 0.05 (comparisons were made between PXR mutant and WT hPXR).
4. Discussion
PXR is a ligand-activated transcription factor involved in regulating the expression of biologically important genes that are involved in various physiological processes [43, 44]. PXR functions as a xenobiotic sensor mainly due to its ability to accommodate structurally diverse xenobiotics. Agonist binding disrupts the interaction of PXR with co-repressors such as SMRT and NCoR and recruitment of co-activators such as SRC-1 and SRC-3 [38, 45, 46]. Identification of compounds that directly bind to PXR to modulate its function will help understand the SAR, and facilitate the prediction of xenobiotics that might bind to PXR to affect drug metabolism and clearance. Additionally, limited PXR antagonists have been reported; PXR antagonists might have clinical utilization in PXR-mediated drug resistance [19, 21]. We report here a new class of natural product derivatives that act as modulators of hPXR. Our findings show that based on the functional groups present, compounds with similar scaffold can act as an agonist, an antagonist, or even a non-binder. The various substituents, and the position of substitution on the aryl sulfonamides, may affect protein binding and the resulting activity (agonistic vs. antagonistic effect).
We focused on compound 1, an agonist identified from the virtual screen and confirmed in cell-based assays. We also obtained and evaluated 7 analogues of compound 1 containing different functional groups at different positions. Interestingly, whereas some analogues (e.g., compounds 1, 2, and 7) acted as agonists, others (e.g., compounds 5, 6, and 8) acted as antagonists and reduced the effect of rifampicin, a well-known agonist for hPXR, in a dose-dependent manner. It has also been noted that compounds 5, 6, and 8 slightly increased luciferase activity at low concentrations. The phenomenon that compounds act as potent PXR antagonists in the presence of a potent agonist but act as a weak to moderate agonist in the absence of a potent agonist was observed previously [47]. However, compound 3 did not significantly activate PXR. It is worth noting that a small change in the functional group has a significant effect on protein binding properties and biological activity. The lack of agonistic effect of compound 3 is in agreement with its lack of activity in inducing the interaction between PXR and SRC-1. It has also been found that compound 3 does not bind to the PXR LBD, as determined by fluorescence assay. As expected, all potent agonists induced significant PXR/SRC-1 interactions. In the presence of rifampicin, all the antagonists (compounds 5, 6, and 8) reduced the effect of rifampicin in activating CYP3A4 promoter and in recruiting SRC-1 to PXR. In the absence of rifampicin, these antagonists weakly or moderately activated CYP3A4 promoter. Since all the agonists and antagonists are analogues that bind to PXR, it becomes an interesting question why binding of a compound to PXR could lead to different biological outcomes.
SAR studies showed that the substituents over the phenyl ring affect the biological activities of the compounds. For example, compounds 1 and 7 with a cyano group at the ortho position exhibited agonistic activity. Changing the thiophene to furan on the pyrimidine ring did not alter biological activity. Methyl and acetamide substitutions on the para position of the phenyl ring led to antagonistic activity. A five-member heteroaromatic sulfonamide (compound 5) was found to be an antagonist. A bicyclic aryl sulfonamide (compound 2) was found to be an agonist, whereas methyl sulfonamide (compound 3) was inactive; however, a benzyl sulfonamide (compound 4) was found to be better in terms of biological activity than compound 3. Fluorescence studies for all the biologically active compounds showed consistent quenching of intrinsic tryptophan uorescence intensity upon binding, which is indicative of a change in the environment of the uorophore. The binding of the ligand to the protein may directly affect the uorescence of a tryptophan residue with the ligand acting as a quencher or by physically interacting with the uorophore and thereby changing the polarity of its environment or its accessibility to the solvent. Because aromatic amino acid residues of the LBD are important for compound binding, it is possible that these compounds bind to the PXR LBD physically. Rifampicin, a well-known PXR ligand, was used to validate the fluorescence intensity assay. Interestingly, compound 3 did not bind to PXR, as we did not see significant fluorescence quenching upon ligand binding, which is consistent with the lack of activity of compound 3 in either activating the CYP3A4 promoter or recruiting SRC-1 to PXR. Thus, the intrinsic tryptophan uorescence quenching assay could be useful in differentiating binders and non-binders of PXR. Although agonist-PXR co-crystal structures have been reported for multiple agonists, no PXR-antagonist co-crystal structure is available. Therefore, only molecular modeling studies can be useful in explaining the ligand binding. It may be that all the compounds bind at the same site. From the interaction analysis, it is evident that Phe 288, Tyr 306, His 327, and Trp 299 are important for interaction with the sulfonamide moiety. These residues are probably involved in pi-pi interaction with the aryl sulfonamides. However, in the case of compound 3, aryl sulfonamide is replaced by methyl sulfonamide, which caused a loss of pi-pi interactions and resulted in significantly reduced bioactivity. In the case of compound 4, which has a flexible –CH2 group attached to the aryl, it somehow accommodates the aryl moiety with decreased activity. Thus, from the molecular modeling analysis, it is evident that aryl sulfonamide is important for interacting with the LBD. Our docking studies predicted the residues critical for ligand interactions, which were confirmed by mutagenesis and CYP3A4-luc reporter assay. Therefore, docking studies combined with mutagenesis and functional analyses could facilitate mapping the ligand binding site of the PXR LBD.
In summary, we identified compounds representing a new class of small molecule modulators for hPXR. These compounds were able to activate or inhibit CYP3A4 promoter activity by physically binding to PXR. Compounds 1 and 6 affect CYP3A4 promoter in both HepG2 and LS 174T cells. However, no significant antagonistic effect on CYP2B6 promoter was observed for compound 6, suggesting that the antagonistic effect of compound 6 might be promoter specific. Both CYP3A4 promoter and CYP2B6 promoter can be activated by rifampicin in a PXR-dependent manner [35]. However, CYP3A4 promoter is preferably regulated by PXR while CYP2B6 promoter is preferably regulated by CAR [35]. SAR studies showed that the various functional groups with different electronic natures on aryl sulfonamide influenced the activity and were important for determining the character of the modulators of PXR. The crystal structures of the LBD of human PXR in both ligand-free and ligand-bound forms have been solved by x-ray crystallography, with ligands including PXR agonists hyperforin, colupulone (from hops), 17β-estradiol, SR12813, T1317, and rifampicin [12–16]. However, no crystal structure is available for antagonist-bound PXR LBD. Although many agonists are available, there are very few antagonists. The newly discovered antagonists reported here are different structurally from the previously reported antagonists [22–29, 31]. Ketoconazole is a general inhibitor of activated PXR that represses the coordinated activation of genes involved in drug metabolism by disrupting co-activator binding to hPXR without affecting DNA binding, ligand binding or receptor dimerization [23]. By using computational approaches and a novel pharmacophore for PXR antagonists, Ekins et al. discovered several new PXR antagonists with in vitro activity [29]. Their data suggested that these new PXR antagonists, and most of the known PXR antagonists, such as coumestrol and sulforaphane, could interact on the outer surface of PXR at the AF-2 domain [29]. More recently, Li et al. used a novel yeast-based strategy and molecular docking analysis to show that ketoconazole interacts with specific PXR surface residues such as Ser-208, which is on the opposite side of the protein from the AF-2 region critical for receptor regulation [48].
Further investigation is needed to develop better antagonists for PXR by modifying the functional groups of our recently discovered antagonists and reduce their cytotoxicity. Further modification and optimization of the chemical structures would be worth pursuing to generate new analogues with improved potency and selectivity against hPXR. The approach described here can be useful for finding novel modulators for PXR.
Acknowledgments
This work was supported by the American Lebanese Syrian Associated Charities (ALSAC), St. Jude Children’s Research Hospital, National Institutes of Health National Institute of General Medical Sciences [Grant GM086415], and National Institutes of Health National Cancer Institute [Grant P30-CA21765]. We thank Jing Wu and Dr. Rajendra Tangallaphy for many valuable suggestions and discussions and David Galloway for editing the manuscript.
Footnotes
Conflict of interest
The authors have no conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Olefsky JM. Nuclear receptor minireview series. J Biol Chem. 2001;276:36863–4. doi: 10.1074/jbc.R100047200. [DOI] [PubMed] [Google Scholar]
- 2.Krasowski MD, Ni A, Hagey LR, Ekins S. Evolution of promiscuous nuclear hormone receptors: LXR, FXR, VDR, PXR, and CAR. Mol Cell Endocrinol. 2011;334:39–48. doi: 10.1016/j.mce.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ihunnah CA, Jiang M, Xie W. Nuclear receptor PXR, transcriptional circuits and metabolic relevance. Biochim Biophys Acta. 2011;1812:956–63. doi: 10.1016/j.bbadis.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang TK. Activation of pregnane X receptor (PXR) and constitutive androstane receptor (CAR) by herbal medicines. AAPS J. 2009;11:590–601. doi: 10.1208/s12248-009-9135-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matter H, Anger LT, Giegerich C, Gussregen S, Hessler G, Baringhaus KH. Development of in silico filters to predict activation of the pregnane X receptor (PXR) by structurally diverse drug-like molecules. Bioorg Med Chem. 2012;20:5352–65. doi: 10.1016/j.bmc.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 6.Rosenfeld JM, Vargas R, Jr, Xie W, Evans RM. Genetic profiling defines the xenobiotic gene network controlled by the nuclear receptor pregnane X receptor. Mol Endocrinol. 2003;17:1268–82. doi: 10.1210/me.2002-0421. [DOI] [PubMed] [Google Scholar]
- 7.Ekins S, Erickson JA. A pharmacophore for human pregnane X receptor ligands. Drug Metab Dispos. 2002;30:96–9. doi: 10.1124/dmd.30.1.96. [DOI] [PubMed] [Google Scholar]
- 8.Xie W, Uppal H, Saini SP, Mu Y, Little JM, Radominska-Pandya A, et al. Orphan nuclear receptor-mediated xenobiotic regulation in drug metabolism. Drug Discov Today. 2004;9:442–9. doi: 10.1016/S1359-6446(04)03061-2. [DOI] [PubMed] [Google Scholar]
- 9.Banerjee M, Robbins D, Chen T. Modulation of Xenobiotic Receptors by Steroids. Molecules. 2013;18:7389–406. doi: 10.3390/molecules18077389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.di Masi A, De Marinis E, Ascenzi P, Marino M. Nuclear receptors CAR and PXR: Molecular, functional, and biomedical aspects. Mol Aspects Med. 2009;30:297–343. doi: 10.1016/j.mam.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 11.Staudinger JL, Ding X, Lichti K. Pregnane X receptor and natural products: beyond drug-drug interactions. Expert Opin Drug Metab Toxicol. 2006;2:847–57. doi: 10.1517/17425255.2.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, et al. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science. 2001;292:2329–33. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- 13.Watkins RE, Maglich JM, Moore LB, Wisely GB, Noble SM, Davis-Searles PR, et al. 2.1 A crystal structure of human PXR in complex with the St. John’s wort compound hyperforin. Biochemistry. 2003;42:1430–8. doi: 10.1021/bi0268753. [DOI] [PubMed] [Google Scholar]
- 14.Watkins RE, Davis-Searles PR, Lambert MH, Redinbo MR. Coactivator binding promotes the specific interaction between ligand and the pregnane X receptor. J Mol Biol. 2003;331:815–28. doi: 10.1016/s0022-2836(03)00795-2. [DOI] [PubMed] [Google Scholar]
- 15.Chrencik JE, Orans J, Moore LB, Xue Y, Peng L, Collins JL, et al. Structural disorder in the complex of human pregnane X receptor and the macrolide antibiotic rifampicin. Mol Endocrinol. 2005;19:1125–34. doi: 10.1210/me.2004-0346. [DOI] [PubMed] [Google Scholar]
- 16.Xue Y, Moore LB, Orans J, Peng L, Bencharit S, Kliewer SA, et al. Crystal structure of the pregnane X receptor-estradiol complex provides insights into endobiotic recognition. Mol Endocrinol. 2007;21:1028–38. doi: 10.1210/me.2006-0323. [DOI] [PubMed] [Google Scholar]
- 17.Liu YH, Mo SL, Bi HC, Hu BF, Li CG, Wang YT, et al. Regulation of human pregnane X receptor and its target gene cytochrome P450 3A4 by Chinese herbal compounds and a molecular docking study. Xenobiotica. 2011;41:259–80. doi: 10.3109/00498254.2010.537395. [DOI] [PubMed] [Google Scholar]
- 18.Wang YM, Ong SS, Chai SC, Chen T. Role of CAR and PXR in xenobiotic sensing and metabolism. Expert Opin Drug Metab Toxicol. 2012;8:803–17. doi: 10.1517/17425255.2012.685237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen T. Overcoming drug resistance by regulating nuclear receptors. Adv Drug Deliv Rev. 2010;62:1257–64. doi: 10.1016/j.addr.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Venkatesh M, Wang H, Cayer J, Leroux M, Salvail D, Das B, et al. In vivo and in vitro characterization of a first-in-class novel azole analog that targets pregnane X receptor activation. Mol Pharmacol. 2011;80:124–35. doi: 10.1124/mol.111.071787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mani S, Dou W, Redinbo MR. PXR antagonists and implication in drug metabolism. Drug Metab Rev. 2013;45:60–72. doi: 10.3109/03602532.2012.746363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Synold TW, Dussault I, Forman BM. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat Med. 2001;7:584–90. doi: 10.1038/87912. [DOI] [PubMed] [Google Scholar]
- 23.Huang H, Wang H, Sinz M, Zoeckler M, Staudinger J, Redinbo MR, et al. Inhibition of drug metabolism by blocking the activation of nuclear receptors by ketoconazole. Oncogene. 2007;26:258–68. doi: 10.1038/sj.onc.1209788. [DOI] [PubMed] [Google Scholar]
- 24.Zhou C, Poulton EJ, Grun F, Bammler TK, Blumberg B, Thummel KE, et al. The dietary isothiocyanate sulforaphane is an antagonist of the human steroid and xenobiotic nuclear receptor. Mol Pharmacol. 2007;71:220–9. doi: 10.1124/mol.106.029264. [DOI] [PubMed] [Google Scholar]
- 25.Healan-Greenberg C, Waring JF, Kempf DJ, Blomme EA, Tirona RG, Kim RB. A human immunodeficiency virus protease inhibitor is a novel functional inhibitor of human pregnane X receptor. Drug Metab Dispos. 2008;36:500–7. doi: 10.1124/dmd.107.019547. [DOI] [PubMed] [Google Scholar]
- 26.Wang H, Li H, Moore LB, Johnson MD, Maglich JM, Goodwin B, et al. The phytoestrogen coumestrol is a naturally occurring antagonist of the human pregnane X receptor. Mol Endocrinol. 2008;22:838–57. doi: 10.1210/me.2007-0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y, Tang Y, Robbins GT, Nie D. Camptothecin attenuates cytochrome P450 3A4 induction by blocking the activation of human pregnane X receptor. J Pharmacol Exp Ther. 2010;334:999–1008. doi: 10.1124/jpet.110.168294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lim YP, Ma CY, Liu CL, Lin YH, Hu ML, Chen JJ, et al. Sesamin: A Naturally Occurring Lignan Inhibits CYP3A4 by Antagonizing the Pregnane X Receptor Activation. Evid Based Complement Alternat Med. 2012;2012:242810. doi: 10.1155/2012/242810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ekins S, Kholodovych V, Ai N, Sinz M, Gal J, Gera L, et al. Computational discovery of novel low micromolar human pregnane X receptor antagonists. Mol Pharmacol. 2008;74:662–72. doi: 10.1124/mol.108.049437. [DOI] [PubMed] [Google Scholar]
- 30.Mooiman KD, Maas-Bakker RF, Moret EE, Beijnen JH, Schellens JH, Meijerman I. Milk Thistle’s Active Components Silybin and Isosilybin: Novel Inhibitors of PXR-Mediated CYP3A4 Induction. Drug Metab Dispos. 2013;41:1494–504. doi: 10.1124/dmd.113.050971. [DOI] [PubMed] [Google Scholar]
- 31.Ekins S, Chang C, Mani S, Krasowski MD, Reschly EJ, Iyer M, et al. Human pregnane X receptor antagonists and agonists define molecular requirements for different binding sites. Mol Pharmacol. 2007;72:592–603. doi: 10.1124/mol.107.038398. [DOI] [PubMed] [Google Scholar]
- 32.Shukla SJ, Nguyen DT, Macarthur R, Simeonov A, Frazee WJ, Hallis TM, et al. Identification of pregnane X receptor ligands using time-resolved fluorescence resonance energy transfer and quantitative high-throughput screening. Assay Drug Dev Technol. 2009;7:143–69. doi: 10.1089/adt.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin W, Wu J, Dong H, Bouck D, Zeng FY, Chen T. Cyclin-dependent kinase 2 negatively regulates human pregnane X receptor-mediated CYP3A4 gene expression in HepG2 liver carcinoma cells. J Biol Chem. 2008;283:30650–7. doi: 10.1074/jbc.M806132200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H, Faucette S, Sueyoshi T, Moore R, Ferguson S, Negishi M, et al. A novel distal enhancer module regulated by pregnane X receptor/constitutive androstane receptor is essential for the maximal induction of CYP2B6 gene expression. J Biol Chem. 2003;278:14146–52. doi: 10.1074/jbc.M212482200. [DOI] [PubMed] [Google Scholar]
- 35.Faucette SR, Sueyoshi T, Smith CM, Negishi M, Lecluyse EL, Wang H. Differential regulation of hepatic CYP2B6 and CYP3A4 genes by constitutive androstane receptor but not pregnane X receptor. J Pharmacol Exp Ther. 2006;317:1200–9. doi: 10.1124/jpet.105.098160. [DOI] [PubMed] [Google Scholar]
- 36.Pondugula SR, Brimer-Cline C, Wu J, Schuetz EG, Tyagi RK, Chen T. A phosphomimetic mutation at threonine-57 abolishes transactivation activity and alters nuclear localization pattern of human pregnane x receptor. Drug Metab Dispos. 2009;37:719–30. doi: 10.1124/dmd.108.024695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hernandez JP, Mota LC, Baldwin WS. Activation of CAR and PXR by Dietary, Environmental and Occupational Chemicals Alters Drug Metabolism, Intermediary Metabolism, and Cell Proliferation. Curr Pharmacogenomics Person Med. 2009;7:81–105. doi: 10.2174/187569209788654005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, et al. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 39.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102:1016–23. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Desai PB, Nallani SC, Sane RS, Moore LB, Goodwin BJ, Buckley DJ, et al. Induction of cytochrome P450 3A4 in primary human hepatocytes and activation of the human pregnane X receptor by tamoxifen and 4-hydroxytamoxifen. Drug Metab Dispos. 2002;30:608–12. doi: 10.1124/dmd.30.5.608. [DOI] [PubMed] [Google Scholar]
- 41.Mani S, Huang HY, Sundarababu S, Li WJ, Kalpana G, Smith AB, et al. Activation of the steroid and xenobiotic receptor (Human pregnane X receptor) by nontaxane microtubule-stabilizing agents. Clin Cancer Res. 2005;11:6359–69. doi: 10.1158/1078-0432.CCR-05-0252. [DOI] [PubMed] [Google Scholar]
- 42.Sui YP, Ai N, Park SH, Rios-Pilier J, Perkins JT, Welsh WJ, et al. Bisphenol A and Its Analogues Activate Human Pregnane X Receptor. Environ Health Perspect. 2012;120:399–405. doi: 10.1289/ehp.1104426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao J, Xie W. Targeting xenobiotic receptors PXR and CAR for metabolic diseases. Trends Pharmacol Sci. 2012;33:552–8. doi: 10.1016/j.tips.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharma D, Lau AJ, Sherman MA, Chang TK. Agonism of human pregnane X receptor by rilpivirine and etravirine: comparison with first generation non-nucleoside reverse transcriptase inhibitors. Biochem Pharmacol. 2013;85:1700–11. doi: 10.1016/j.bcp.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 45.Johnson DR, Li CW, Chen LY, Ghosh JC, Chen JD. Regulation and binding of pregnane X receptor by nuclear receptor corepressor silencing mediator of retinoid and thyroid hormone receptors (SMRT) Mol Pharmacol. 2006;69:99–108. doi: 10.1124/mol.105.013375. [DOI] [PubMed] [Google Scholar]
- 46.Ding X, Staudinger JL. Repression of PXR-mediated induction of hepatic CYP3A gene expression by protein kinase C. Biochem Pharmacol. 2005;69:867–73. doi: 10.1016/j.bcp.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 47.Wang H, Huang H, Li H, Teotico DG, Sinz M, Baker SD, et al. Activated pregnenolone X-receptor is a target for ketoconazole and its analogs. Clin Cancer Res. 2007;13:2488–95. doi: 10.1158/1078-0432.CCR-06-1592. [DOI] [PubMed] [Google Scholar]
- 48.Li H, Redinbo MR, Venkatesh M, Ekins S, Chaudhry A, Bloch N, et al. Novel yeast-based strategy unveils antagonist binding regions on the nuclear xenobiotic receptor PXR. J Biol Chem. 2013;288:13655–68. doi: 10.1074/jbc.M113.455485. [DOI] [PMC free article] [PubMed] [Google Scholar]