

Abstract
Background
The human SLC25A13 gene encodes citrin, the liver-type mitochondrial aspartate/glutamate carrier isoform 2 (AGC2), and SLC25A13 mutations cause citrin deficiency (CD), a disease entity that encompasses different age-dependant clinical phenotypes such as Adult-onset Citrullinemia Type II (CTLN2) and Neonatal Intrahepatic Cholestasis caused by Citrin Deficiency (NICCD). The analyses of SLC25A13 gene and its protein/mRNA products remain reliable tools for the definitive diagnoses of CD patients, and so far, the SLC25A13 mutation spectrum in Chinese CD patients has not been well-characterized yet.
Methods and Results
By means of direct DNA sequencing, cDNA cloning and SNP analyses, 16 novel pathogenic mutations, including 9 missense, 4 nonsense, 1 splice-site, 1 deletion and 1 large transposal insertion IVS4ins6kb (GenBank accession number KF425758), were identified in CTLN2 or NICCD patients from China, Japan and Malaysia, respectively, making the SLC25A13 variations worldwide reach the total number of 81. A large NICCD cohort of 116 Chinese cases was also established, and the 4 high-frequency mutations contributed a much larger proportion of the mutated alleles in the patients from south China than in those from the north (χ2 = 14.93, P<0.01), with the latitude of 30°N as the geographic dividing line in mainland China.
Conclusions
This paper further enriched the SLC25A13 variation spectrum worldwide, and formed a substantial contribution to the in-depth understanding of the genotypic feature of Chinese CD patients.
Introduction
The term CITRIN was designated in 1999 to stand for the protein product encoded by SLC25A13 gene, which was localized to chromosome 7q21.3 and cloned as the causative gene for Adult-onset Citrullinemia Type 2 (CTLN2, OMIM #603471) [1]. SLC25A13 mutations result in citrin deficiency (CD), and CTLN2 was the firstly-described CD phenotype, which occurs in adolescents or adults and the prognosis is usually not benign [2]. Subsequently, CD was found to be associated with intrahepatic cholestasis in neonates or infants [3]–[5], and these findings led to the introduction of a new nomenclature of Neonatal Intrahepatic Cholestasis caused by Citrin Deficiency (NICCD, OMIM #605814) [6]–[8]. NICCD usually demonstrates satisfactory clinical outcomes, with the clinical and laboratory presentations resolving in the first year of life. It has been traditionally-assumed for years that there is a “silent” or “apparently-healthy” stage after the NICCD period but before CTLN2 onset. However, this concept has been challenged by accumulating clinical, laboratory and even behavioral evidences [9]–[11], and actually, an additional CD phenotype, Failure to Thrive and Dyslipidemia caused by Citrin Deficiency (FTTDCD), has been put forward recently [2], [12]–[14].
The phenotypic features of CD patients are really complicated indeed, involving numerous clinical, biochemical, imaging, hepatohistological and metabolomic alterations [13], [15]–[18]. However, none of these changes are pathognomonic, and SLC25A13 genetic analysis has been recognized as a reliable method for the definitive diagnosis of CD. In the case of unknown mutation after the conventional genetic analyses such as PCR-RFLP and direct DNA sequencing, detection of the protein and mRNA product of SLC25A13 gene could be considered as the alternative diagnostic tools. However, cultured fibroblasts or biopsied/autopsied liver specimens are usually needed in such analyses [1], [19]–[23], and obtaining these invasive samples may not always be feasible. To address this issue, citrin protein analysis using peripheral blood lymphocytes (PBLs) had been tried [24]. More recently, it was proved that SLC25A13 cDNA cloning analysis using human PBLs could be taken as a less invasive and more feasible tool to identify aberrant SLC25A13 transcripts for the molecular diagnosis of CD [14]. However, more experience should be accumulated on the application of this new approach in identifying unknown SLC25A13 mutations.
Owing to the establishment of the molecular diagnostic tools, increasing number of CD patients were diagnosed in Asia [25]–[34], North America [20], [21], [35], and Europe [36]–[38]. Nevertheless, up to now, the majority of the reported CD patients were from East Asian countries, especially Japan [1], [3]–[5], [8], [9], [11], [15], [16], [22], [24], [39]–[43]. China is a vast country with a huge population. According to the latest official data (http://www.chinapop.gov.cn/xwzx/rkxw/), the resident population in China is more than 1.3 billion, with 472 millions of them in the south area of Yangtze River. The carrier rates of SLC25A13 mutations had been documented to be 1/63 in China, and particularly 1/48 in its south area [44], [45]. Therefore, if calculated according to Hardy-Weinberg principle, the number of CD patients in China would be approximately 85 700, over 51 300 of whom are living in the South China. However, the officially-reported CD patients from China, including those from the south, were rather limited in number [13], [14], [17], [23], [30], [32], [33], and the distribution of SLC25A13 mutations in Chinese patients remains far from being completely elucidated.
In this paper, novel SLC25A13 mutations in 16 Asian CD patients were identified, including a large transposal insertion that gave rise to exon 5 skipping in SLC25A13 transcripts. We established a large pediatric cohort of Chinese CD patients, and then explored the mutation spectrum and compared the distribution in different geographical areas. We herein reported the findings.
Subjects and Methods
Subjects
The research subjects in this paper encompassed 68 new CD patients comprising 60 Chinese, 7 Japanese and 1 Malaysian. In order to explore the SLC25A13 mutation spectrum and the distribution feature in different areas of China, 56 CD patients reported previously by our group were also enrolled in this paper to establish a large pediatric CD cohort.
Ethics Statement
This study has been approved by the Committee for Ethics of Kagoshima University Faculty of Medicine, Japan, and by the Medical Ethical Committee, the First Affiliated Hospital, Jinan University, China, and adheres to the World Medical Association Declaration of Helsinki (WMADH 2008), which was adopted by the 59th WMA General Assembly, Seoul, in October 2008. SLC25A13 analyses were conducted with the written informed consents from the patients or their guardians.
Mutation Screening and Direct DNA Sequencing
All patients were initially screened for the frequently recurring SLC25A13 mutations. Thirteen SLC25A13 mutations were included in the screening panel for the 7 Japanese patients in Kagoshima University, Japan, as in reference [22]. And, 4 mutations were screened in the remaining 61 novel subjects in Jinan University, China. In the subjects whose initial screening only revealed one mutated allele, the 18 exons and their flanking sequences in SLC25A13 gene were analyzed by direct DNA sequencing, using the approach as described in our previous publications [13], [14], [17], [33].
Pathogenicity Analysis of the Novel Mutations
According to the HUGO mutation database initiative/Human Genome Variation Society (Instruction for Authors [DB/OL]. New Jersey (IL): John Wiley & Sons, Inc. 2008 [2008-03-24]. http://www3.interscience.wiley.com/journal/38515/home/ForAuthors.html), proof of pathogenicity in this study was defined by at least one of the following criteria: (1) a mutation presenting at the frequency of <1% in at least 50 control individuals, (2) a mutation with co-segregation in a family; (3) alteration of an evolutionary conserved amino acid residue, and (4) nonsense and deletion variation in the coding sequence of SLC25A13 gene. Moreover, the function effect of the novel missense mutations identified in this study was predicted with the software PolyPhen-2 at http://genetics.bwh.harvard.edu/pph2/, and a mutation is classified as “probably damaging” if its probabilistic score is above 0.85 while as “possibly damaging” with the score above 0.15 [46].
The frequency of 1 novel splice-site and 4 missense mutations in at least 50 control individuals was investigated by newly-developed PCR-RFLP approaches. The PCR primer pairs were Ex-4F: 5′-TGGACAGACCACAATTCATCAA-3′ and IVS5B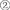 :5′-GACGGAGTCTCGCTCTTTCA-3′, IVS5NF: 5′-TGAGGGCTTGTTAGATCAAGAT-3′ and IVS6NB:5′-TTACCCAGACAACAAATTAACCT-3′, IVS1NF:5′-TTTATGCACTGGGGCAACATG-3′ and Ex2Bm
:5′-GACGGAGTCTCGCTCTTTCA-3′, IVS5NF: 5′-TGAGGGCTTGTTAGATCAAGAT-3′ and IVS6NB:5′-TTACCCAGACAACAAATTAACCT-3′, IVS1NF:5′-TTTATGCACTGGGGCAACATG-3′ and Ex2Bm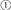 :5′-TGCTCTCTTGGTTAAAGCCACT-3′, IVS13F:5′-GGATGTCACAGGCAGAGTTC-3′ and IVS14B:5′-CTCATCTGCCAGAATGAAGATT-3′, and IVS10F:5′-GGACTGATGCGGCTGTTAGA-3′ and SLCE12A:5′-TGGTGAGTTCCCCTGCTTTC-3′. The restriction endonucleases (REs) used for the RFLP analyses were Hin1 II (Fermentas), BseNI (Fermentas), PsiI (New England Biolabs), Taq I (Fermentas) and Hae III (Takara) for RFLP analysis, respectively. And, the frequency of the remaining missense mutations was explored by using direct DNA sequencing due to the lack of REs for their PCR-RFLP analysis. If parental DNA samples were available in some families, mutation co-segregation analyses were conducted by the PCR-RFLP or DNA sequencing approach described above. Moreover, by using a comparative alignment software of Genetyx® version 7.1 (Genetyx company, Tokyo, Japan), the amino acid sequences of human citrin and aralar were aligned with those in the homologous proteins from 9 different eukaryotic species, including chimpanzee, dog, mouse, rat, chicken, xenopus tropicalis, caenorhabditis elegans, opossum and cow. The amino acid sequences for the proteins were collected from the database ENSEMBL at http://www.ensembl.org/index.html.
:5′-TGCTCTCTTGGTTAAAGCCACT-3′, IVS13F:5′-GGATGTCACAGGCAGAGTTC-3′ and IVS14B:5′-CTCATCTGCCAGAATGAAGATT-3′, and IVS10F:5′-GGACTGATGCGGCTGTTAGA-3′ and SLCE12A:5′-TGGTGAGTTCCCCTGCTTTC-3′. The restriction endonucleases (REs) used for the RFLP analyses were Hin1 II (Fermentas), BseNI (Fermentas), PsiI (New England Biolabs), Taq I (Fermentas) and Hae III (Takara) for RFLP analysis, respectively. And, the frequency of the remaining missense mutations was explored by using direct DNA sequencing due to the lack of REs for their PCR-RFLP analysis. If parental DNA samples were available in some families, mutation co-segregation analyses were conducted by the PCR-RFLP or DNA sequencing approach described above. Moreover, by using a comparative alignment software of Genetyx® version 7.1 (Genetyx company, Tokyo, Japan), the amino acid sequences of human citrin and aralar were aligned with those in the homologous proteins from 9 different eukaryotic species, including chimpanzee, dog, mouse, rat, chicken, xenopus tropicalis, caenorhabditis elegans, opossum and cow. The amino acid sequences for the proteins were collected from the database ENSEMBL at http://www.ensembl.org/index.html.
Total RNA Extraction and RT-nested PCR
In the patient C0054, only one paternally-inherited c.1399C>T mutation was revealed by routine screening and direct sequencing. The maternally-inherited mutation was further explored by mRNA analysis. Extraction of the total RNA and RT-nested PCR were performed as previously described [14], [33]. Briefly, PBLs were collected from heparinized venous blood of the patient, and the total RNA was extracted with RNAiso Plus (Takara). After that, 1 µg of RNA product was reverse-transcribed in the presence of 1 µg of oligo-(dT)18 and 200 U MMLV reverse transcriptase (Promega), and nested PCR was then performed with two pairs of primers, i.e. RAS2: 5′-AACGCACGCTGCCTGGCCGTATC-3′ and RACEA1: 5′-CCACCTTC ACAAATTCATGCGCC-3′ for the first PCR, while RAS3: 5′-GCCGCCGGGACTAGAAGTGAGC-3′ and Ex18R: 5′-TGCTTCATTCCCAGGAGGGA-3′ for the second. The amplicon contained the entire SLC25A13 ORF, with a deduced size of 2191 bp.
cDNA Cloning Analysis
After the mRNA extraction and RT-nested PCR, cDNA cloning was carried out in the patient C0054 to identify the aberrant mRNA molecule originated from the maternal allele that harbored an unknown mutation. In accord with the approach that was developed very recently [14], the nested PCR products were then separated by electrophoresis and the fragments of interest were excised and purified by gel extraction kit (Omega). Then the purified cDNA products were cloned into pSIMPLE-18 EcoR V/BAP Vector (Takara) and transformed into DH5α Escherichia coli competent cells by means of heat shock. The positive clones were screened by means of “white-blue spot selection” and tested by PCR with the primer pair RAS3 and Ex18R. The positive clones were then sequenced and comparatively aligned with the SLC25A13 mRNA sequence. The alternative splicing variants (ASVs) identified by cDNA cloning analysis were described based on the nomenclature guidelines [47]. Chi square test was adopted to compare the proportion difference of the ASVs in the patient C0054 and 8 healthy volunteers, with P<0.05 as the significant criterion.
SNP Analysis
The cDNA cloning analysis in the patient C0054 identified an aberrant transcript that was strongly suggestive of a large insertion/deletion mutation within the DNA fragment spanning from intron 4 to intron 5 in the SLC25A13 gene. Subsequently, seven SNPs within this span, i.e. rs6465494(A/G), rs4727337(A/G) and rs67843496 (AA/−) in intron 4, and rs6943325(C/T), rs13307036(C/T), rs6465490 (C/A) and rs4273779(G/A) in intron 5, were analyzed by means of PCR amplification and direct sequencing. DNA fragments with heterozygous SNPs were not suspected to carry large insertion/deletion. On the contrary, if an amplicon was found with a homozygous SNP, a DNA fragment larger than the amplicon in size would be amplified by Long-range PCR (LA-PCR) using Takara LA Taq™ polymerase (Takara) according to the manufacturer’s instructions. The product of unexpected size was excised, purified and sequenced to explicit the nature of the likely large insertion/deletion in the patient C0054. The large transposal insertion identified in this patient was then deposited in GenBank (Accession number: KF425758).
Update of the SLC25A13 Mutations
It was mainly by using Pubmed, the well-recognized information retrieval system that comprises more than 22 million citations for biomedical literature from MEDLINE, life science journals, and online books (http://www.ncbi.nlm.nih.gov/pubmed/), that we collected all the references on CD since the year 1999 when the SLC25A13 gene was cloned. The SLC25A13 mutations identified in CTLN2 or NICCD patients, including their locations, types, and the DNA, cDNA and amino acid changes were updated and listed into a table.
Mutation Spectrum in the Large Pediatric Cohort in China
The genotypic and phenotypic features of 60 new NICCD patients in China were described in this study. These new patients, along with the 56 subjects reported previously by our group [13], [14], [17], [33], [48], constituted a large Pediatric cohort of 116 CD patients in China. The SLC25A13 mutation spectrum in this cohort was then summarized, and the proportion of every mutation in the whole spectrum was calculated and listed into a table. By using Chi square test and P<0.05 as the significant criterion, the distribution feature of SLC25A13 mutations in South and North China were compared, with the latitude of 30°N as the dividing line, the most likely boundary between northern and southern Chinese who originated from distinct ancestors [49].
Results
Novel Point/Deletion Mutations
As shown in Table 1, 15 novel mutations, including 9 missense (c.443A>G, c.1498T>G, c.527G>T, c.1048G>A, c.1063C>G, c.415G>A, c.1364G>T, c.1215G>T and c.1775A>C), 4 nonsense (c.448G>T, c.1736G>A, c.1645C>T and c.72T>A), 1 splice-site (c.16-2A>T) and 1 deletion (c.265delG), were identified by direct DNA sequencing analysis of the 18 exons and their flanking sequences in SLC25A13 gene of the 15 CD patients in East Asia, everyone of whom harbored a previously-reported mutation that had been uncovered by routine mutation screening. Figure S1 demonstrated the segmental sequencing results of the 15 mutations.
Table 1. Fifteen novel mutations in the SLC25A13 gene of citrin-deficient patients from different countries.
| Patient | Gender | Origin | Phenotype | SLC25A13 Genotypes | Effects | Frequency | ||
| Patient | Father | Mother | ||||||
| P111 | Male | Japan | CTLN2 | c.448G>T/IVS11+1G>A | – | – | p.E150X | – |
| P530 | Male | Japan | NICCD | c.1736G>A/IVS16ins3kb | – | – | p.W579X | – |
| P645 | Female | Japan | CTLN2 | c.443A>G/c.851_854del | – | – | p.Y148C | <1%△ |
| P874 | Female | Japan | NICCD | c.1498T>G/IVS11+1G>A | IVS11+1G>A/N | c.1498T>G/N | p.Y500D | <1%△ |
| P917 | Female | Japan | NICCD | c.16-2A>T/IVS11+1G>A | IVS11+1G>A/N | c.16-2A>T/N | IVS1-2A>T | <1%△ |
| P1075 | Male | Japan | NICCD | c.527G>T/c.674C>A | – | – | p.G176V | <1%△ |
| P1508 | Male | Japan | CTLN2 | c.1645C>T/c.674C>A | c.674C>A/N | c.1645C>T/N | p.Q549X | – |
| C0060 | Male | China | NICCD | c.72T>A/c.775C>T | c.775C>T/N | c.72A>T/N | p.Y24X | – |
| C0079 | Male | China | NICCD | c.1048G>A/IVS16ins3kb | IVS16ins3kb/N | c.1048G>A/N | p.D350N | <1%▴ |
| C0098 | Male | China | NICCD | c.1063C>G/c.1638_1660dup | c.1638_1660dup/N | c.1063C>G/N | p.R355G | <1%▴ |
| C0106 | Male | Malaysia | NICCD | c.415G>A/IVS16ins3kb | – | – | p.G139R | <1%▴ |
| C0108 | Female | China | NICCD | c.1364G>T/c.1638_1660dup | c.1364G>T/N | c.1638_1660dup/N | p.R455L | <1%▴ |
| C0157 | Male | China | NICCD | c.265delG/c.1638_1660dup | c.1638_1660dup/N | c.265delG/N | p.D89fs94X | – |
| C0168 | Male | China | NICCD | c.1215G>T/c.851_854del | c.1215G>T/N | c.851_854del/N | p.K405N | <1%△ |
| C0170 | Female | China | NICCD | c.1775A>C/c.851_854del | c.851_854del/N | c.1775A>C/N | p.Q592P | <1%▴ |
The bold black letters denote the novel mutations, “N” indicates the normal SLC25A13 allele, while “–” means not analyzed. The effects and frequency refer to the corresponding novel mutations. The frequency was calculated based on the screening result by PCR-RFLP (△) or direct DNA sequencing approach (▴), respectively.
Pathogenicity
The frequency of the missense and splice-site mutations were investigated in controls by the newly-developed PCR-RFLP, as illustrated in Figure 1, or sequencing approach, and all of them proved less than 1% (Table 1). Moreover, on family linkage analysis, the novel mutations demonstrated different parental sources with the previously-reported mutations in the same family (Table 1). In addition, comparative alignment of the homologous proteins in different eukaryotic species with human citrin and aralar further documented the conservative properties of the amino acids involved by the 9 missense mutations (Figure 2). And, on PolyPhen2 analysis of their function effect, Q592P was “possibly damaging” with a score 0.529 whilst the remaining 8 ones “probably damaging”, all with the score above 0.99. These evidences documented that the novel splice-site/missense mutations identified in this study were all disease-causing.
Figure 1. PCR-RFLP approaches for the carrier rate investigation of the 5 novel missense mutations.

The figures  to
to 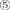 were representative gel electrophoresis of the RE-digested PCR products of the mutations c.443A>G, c.527G>T, c.16-2A>T, c.1498T>G and c.1215G>T, while the figures
were representative gel electrophoresis of the RE-digested PCR products of the mutations c.443A>G, c.527G>T, c.16-2A>T, c.1498T>G and c.1215G>T, while the figures 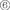 to
to 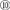 illustrated schematically the PCR-RFLP procedures for the 5 mutations, respectively. In
illustrated schematically the PCR-RFLP procedures for the 5 mutations, respectively. In  , both the patient P917 and one of her brothers B1 harbored the maternally-inherited novel mutation c.16-2A>T.
, both the patient P917 and one of her brothers B1 harbored the maternally-inherited novel mutation c.16-2A>T.
Figure 2. Comparative alignment of the homologous proteins.

The homologous proteins include human citrin (Homo sapiens) and aralar, and the others from 9 different eukaryotic species, i.e. Chimpanzee, Dog, Mouse, Rat, Chicken, Xenopus Tropicalis (X.Tropicalis), Caenorhabditis elegans (C. elegans), Opossum and Cow, respectively. The closed brown boxes in this figure represented the EF-hand or TM helices in citrin protein, as clarified in the reference by Kobayashi et al, 1999.
Results of cDNA Cloning Analysis
The cDNA cloning analysis using PBLs in the patient C0054 uncovered a variety of ASVs from the maternally-inherited SLC25A13 allele, and it was noteworthy that, as shown in Table 2, 95.8% (23/24) of the ASVs demonstrated exon 5 skipping (r.329_468del). However, SLC25A13 cDNA cloning analysis in 8 healthy volunteers documented that the ASV harboring r.329_468del only made up a proportion of 0.9% (1/116, as shown in Table S1). The difference between the two proportions proved significant, with χ2 = 119.7 and P<0.01 (Table S2). The common feature of exon 5 skipping in the maternally-inherited ASVs in the patient C0054 indicated that the unknown mutation of maternal origin had affected the splicing mechanism around exon 5, strongly suggestive of a large insertion or deletion within the DNA segment spanning from intron 4 to intron 5 in the SLC25A13 allele.
Table 2. The SLC25A13 ASVs detected by cDNA analysis in the patient C0054.
| Alleles | Name | ASVs | Remarks | Clones | % | ||
| Maternally-inherited | M-01 | r.213_468del | Exon 4,5 skipping | 8 | 33.3 | ||
| M-02 | r.70_468del | Exon 3,4,5 skipping | 4 | 16.7 | |||
| M-03 | r.213_468del; r.1453_1591del | Exon 4,5,15 skipping | 3 | 12.5 | |||
| M-04 | r.329_468del | Exon 5 skipping | 2 | 8.3 | |||
| M-05 | r.16_69del; r.213_468del | Exon 2,4,5 skipping | 1 | 4.2 | |||
| M-06 | r.70_468del; r.69_469ins212+6499_212+6611;r.1018_1019ins1018+1_1018+469 | Exon 3,4,5 skipping with intron3 and intron10fragment retention | 1 | 4.2 | |||
| M-07 | r.70_468del; r.1452_1453ins1452+12639_1452+12773 | Exon 3,4,5 skipping with intron14 fragment retention | 1 | 4.2 | |||
| M-08 | r.70_468del; r.1750_1751ins1750+1_1750+93 | Exon 3,4,5 skipping with intron16 retention | 1 | 4.2 | |||
| M-09 | r.213_468del; r.755_848del | Exon 4,5,8 skipping | 1 | 4.2 | |||
| M-10 | r.213_468del; r.69_70ins69+12147_69+12282 | Exon 4,5 skipping with intron2 fragment retention | 1 | 4.2 | |||
| M-11 | r.213_328del | Exon 4 skipping | 1 | 4.2 | |||
| In total | 24 | 100 | |||||
| Paternally-inherited | P-01 | r.213_328del; r.1399C>T | Exon 4 skipping | 1 | 33.3 | ||
| P-02 | r.213_468del; r.1399C>T | Exon 4,5 skipping | 1 | 33.3 | |||
| P-03 | r.213_328del;r.755_848del;r.1399C>T; r.1750_1751ins1750+1_1750+93 | Exon 4,8 skipping with intron16 retention | 1 | 33.3 | |||
| In total | 3 | 100 | |||||
In this table, all the maternally-inherited ASVs but M-11 harbored r.329_468del. The nucleotide numbering was based on SLC25A13 cDNA sequence (GenBank: NM_014251), with +1 indicating the A of the ATG-translation initiation codon.
Identification of a Large Transposal Insertion
Based on the cDNA cloning results, SNPs analysis was performed within the above DNA span of interest. The SNPs rs6465494, rs4727337, rs6943325, rs13307036, rs6465490 and rs4273779, as illustrated in Figure 3, all demonstrated heterozygous status. However, rs67843496 was heterozygous in the PCR product amplified with the primers set A, but homozygous with set B. This finding led to the design of the primers set C, and LA-PCR amplification with this primer set yielded an unexpected product of 7.5 kb of maternal origin, which was proved by segmental sequencing to be a transposal insertion of 6072 bp in length (IVS4ins6 kb, GenBank accession number: KF425758), with two repetitive sequences of 15 bp on the both sides of a 6057 bp large insertion translocated from chromosome 16p11.2. One of the two repetitive sequences is the fixed DNA sequence within intron 4.
Figure 3. Identification of the large transposal insertion IVS4ins6kb (GenBank accession number: KF425758).

Seven SNPs within the introns 4 and 5 were analyzed, and all of them were heterozygous but rs67843496 (The schematic overhead), an SNP detected heterozygously when amplified by the primers set A ( ) while homozygously with set B (
) while homozygously with set B (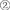 ). LA-PCR with the primers set C yielded an unexpected band of 7.5 kb inherited from the mother C0054M besides the expected 1439 bp product from the father C0054F (
). LA-PCR with the primers set C yielded an unexpected band of 7.5 kb inherited from the mother C0054M besides the expected 1439 bp product from the father C0054F ( ). Segmental sequencing of the 7.5 bp product revealed a 6057 bp insertion from 16p11.2 (The sequence in green) along with two repetitive sequences of 15 bp at both sides (Shaded boxes), as illustrated in
). Segmental sequencing of the 7.5 bp product revealed a 6057 bp insertion from 16p11.2 (The sequence in green) along with two repetitive sequences of 15 bp at both sides (Shaded boxes), as illustrated in 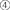 . Underlined were the positions of the primers IVS5F and IVS5A4 (Set C) for LA-PCR.
. Underlined were the positions of the primers IVS5F and IVS5A4 (Set C) for LA-PCR.
Update of SLC25A13 Mutations
To date, a total of 81 SLC25A13 sequence variations in CD patients have been reported worldwide (Table 3). The variations in the list demonstrated marked heterogeneity, comprising 32 missense (including 3 neutral), 18 nonsense, 14 splice-site (including c.1311C>T), 9 deletion, 3 insertion, 2 duplication, 1 indel (g./c.1610–1612delTAGinsAT), 1 aberrant transcript (r.16_212dup) with unknown mutation, and 1 pathogenic SNP (c.2T>C). Most (63%, 51/81) of the variations involved the C-terminal half of citrin protein that covers the 6 domains of transmembrane (TM) helices. In particular, 20 mutations were located within a DNA segment of 344 bp in size, spanning from exon 16 to exon 17 of the SLC25A13 gene.
Table 3. Update of SLC25A13 mutations in patients with citrin deficiency.
| No. | Locations | Mutations | Types | DNA/cDNA changes | Amino acids | References |
| 1 | Ex1 | c.2T>C | Pathogenic SNP | g./c.2T>C | p.Met1_Phe34del | 14,34 |
| 2 | Ex1 | g.Ex1-1G>A | Splice-site | g./c.15G>A | Unclear | 22 |
| 3 | IVS1 | IVS1-2A>T | Splice-site | g.16-2A>T | Unclear | This report |
| 4 | Ex2 | E16X | Nonsense | g./c.46G>T | p.E16X | 22 |
| 5 | Ex3 | Y24X | Nonsense | g./c.72T>A | p.Y24X | This report |
| 6 | Ex3 | p.A25E | Missense | g./c.74C>A | p.A25E | 21 |
| 7 | Ex3 | p.R43X | Nonsense | g./c.127C>T | p.R43X | 21 |
| 8 | Ex3 | c.172_173delGT | Deletion | g./c.172_173delGT | p.V58Gfs81X | 21 |
| 9 | IVS2_IVS3 | g./c.70-862_212+3527del4532 | Deletion | c.70_212del | Unclear | 20,35 |
| 10 | Ex2_3 | Unknown | Aberrant transcript | r.16_212dup (Ex2_3dup) | Unclear | 14 |
| 11 | Ex4 | L85P | Missense | g./c.254T>C | p.L85P | 23 |
| 12 | Ex4 | c.265delG | Deletion | g./c.265delG | p.D89fs94X | This report |
| 13 | IVS4 | IVS4ins6kb | Insertion | r.329_468del(Ex5 del) | p.E110fs127X | This report |
| 14 | Ex5 | G139R | Missense | g./c.415G>A | p.G139R | This report |
| 15 | Ex5 | Y148C | Missense | g./c.443A>G | p.Y148C | This report |
| 16 | Ex5 | E150X | Nonsense | g./c.448G>T | p.E150X | This report |
| 17 | Ex6 | Q159X | Nonsense | g./c.475C>T | p.Q159X | 31 |
| 18 | Ex6 | G176V | Missense | g./c.527G>T | p.G176V | This report |
| 19 | Ex6 | R184X | Nonsense | g./(c.)550C>T | p.R184X | 44 |
| 20 | IVS6 | g.IVS6+1G>C | Splice-site | c.IVS6(1789 bp)ins | p.A206fs212X | 44 |
| 21 | IVS6 | IVS6+1G>A | Splice-site | g./c.615+1G>A | Unclear | 23 |
| 22 | IVS6 | g.IVS6+5G>A | Splice-site | c. not detectable | Unclear | 44 |
| 23 | Ex7 | S225X | Nonsense | g./c.674C>A | p.S225X | 1 |
| 24 | Ex7 | Ex7-1G>A | Splice-site | g./c.754G>A/r.616_848del | p.A206fs213X | 33 |
| 25 | IVS7 | IVS7-2A>G | Splice-site | g./c.755-2A>G | Unclear | 28 |
| 26 | Ex8 | Q259X | Nonsense | g./c.775C>T | p.Q259X | 32 |
| 27 | Ex8 | G283X | Nonsense | g./c.847G>T | p.G283X | 13 |
| 28 | IVS8 | g.IVS8+3A>C | Splice-site | g./c.848+3A>C | Unclear | 35 |
| 29 | Ex9 | 851del4 | Deletion | g./c.851_854delGTAT | p.R284fs286X | 1 |
| 30 | Ex9 | Ex9-1G>A | Splice-site | c.933G>A | Unclear | 16 |
| 31 | Ex10 | R319X | Nonsense | g./c.955C>T | p.R319X | 28 |
| 32 | Ex10 | G333D | Missense | g./c.998G>A | p.G333D | 17 |
| 33 | Ex11 | D350N | Missense | g./c.1048G>A | p.D350N | This report |
| 34 | Ex11 | R355G | Missense | g./c.1063C>G | p.R355G | This report |
| 35 | Ex11 | p.R355X | Nonsense | g./c.1063C>T | p.R355X | 21 |
| 36 | Ex11 | R360X | Nonsense | g./c.1078C>T | p.R360X | 22 |
| 37 | Ex11 | 1092_1095delT | Deletion | g./c.1092_1095delT | p.F365fs407X | 23 |
| 38 | Ex11 | 1146delA | Deletion | g./c.1146delA | p.R383fs407X | 22 |
| 39 | Ex11 | G386V | Missense | g./c.1157G>T | p.G386V | 30 |
| 40 | Ex11 | G393S | Missense | g./(c.)1177G>A | p.G393S | 27 |
| 41 | IVS11 | g.IVS11+1G>A | Splice-site | c.1019_1177del | p.340_392del | 1 |
| 42 | Ex12 | Q397X | Nonsense | g./c.1189C>T | p.Q397X | 26 |
| 43 | Ex12 | 1192-1193delT | Deletion | g./c.1192_1193delT | p.L398fs407X | 30 |
| 44 | Ex12 | c.1215G>T | Missense | g./c.1215G>T | p.K405N | This report |
| 45 | Ex13 | c.1231G>A | Missense | g./c.1231G>A | p.V411M | 13 |
| 46 | Ex13 | G436E | Missense | g./c.1307-1308delGCinsAA | p.G436E | 38 |
| 47 | Ex13 | c.1311C>T | Splice-site? | g./c.1311C>T | p.C437C | 32 |
| 48 | IVS13 | g.IVS13+1G>A | Splice-site | c.1231_1311del | p.411_437del | 1 |
| 49 | IVS13 | g.IVS13+2T>G | Splice-site | c. not detectable | Unclear | 22 |
| 50 | Ex14 | T446P | Missense | g./c.1336A>C | p.T446P | 22 |
| 51 | Ex14 | K453R | Missense | g./c.1358A>G | p.K453R | 30 |
| 52 | Ex14 | R455L | Missense | g./c.1364G>T | p.R455L | This report |
| 53 | Ex14 | 1374_1375delG | Deletion | g./c.1374 or 1375delG | p.A459fs507X | 22 |
| 54 | Ex14 | R467X | Nonsense | g./c.1399C>T | p.R467X | 30 |
| 55 | IVS14_15 | g.Ex15dup | Duplication | c.1453_1591dup | p.M532fs560X | 19 |
| 56 | Ex15 | C489R | Missense | g./c.T1465C | p.C489R | 37 |
| 57 | Ex15 | D493G | Missense | g./c.1478A>G | p.D493G | 43 |
| 58 | Ex15 | Y500D | Missense | g./c.1498T>G | p.Y500D | This report |
| 59 | Ex15 | P502L | Neutral | g./c.1505C>T | p.P502L | 32,34 |
| 60 | IVS15 | g.IVS15+1G>T | Splice-site | c.1453_1591del | p.G485fs491X | 22 |
| 61 | Ex16 | Q549X | Nonsense | g./c.1645C>T | p.Q549X | This report |
| 62 | Ex16 | W579X | Nonsense | g./c.1736G>A | p.W579X | This report |
| 63 | Ex16 | G531D | Missense | g.1592G>A | p.G531D | 22 |
| 64 | Ex16 | c.1610_1612delTAGinsAT | Indel | g./c.1610_1612delTAGinsAT | p.L537fs538X | 36 |
| 65 | Ex16 | A541D | Missense | g./c.1622C>A | p.A541D | 28 |
| 66 | Ex16 | p.T546R | Missense | g./c.1637C>G | p.T546R | 21 |
| 67 | Ex16 | T546M | Missense | g./c.1637C>T | p.T546M | 22 |
| 68 | Ex16 | 1638ins23 | Duplication | g./c.1638_1660dup | p.A554fs570X | 1 |
| 69 | Ex16 | R553Q | Missense | g./c.1658G>A | p.R553Q | 31 |
| 70 | Ex16_IVS17 | g.Ex16+74_IVS17-32del516 | Deletion | c. aberrant RNA | p.Q556fs565X | 40 |
| 71 | IVS16 | g.IVS16ins3kb | Insertion | c. aberrant RNA | p.A584fs585X | 22 |
| 72 | Ex17 | R588Q | Missense | g./c.1763G>A | p.R588Q | 22 |
| 73 | Ex17 | L598R | Missense | g./c.1793T>G | p.L598R | 25 |
| 74 | Ex17 | R585H | Missense | g./c.1754G>A | p.R585H | 23 |
| 75 | Ex17 | Q592P | Missense | g./c.1775A>C | p.Q592P | This report |
| 76 | Ex17 | 1800ins1 | Insertion | g./c.1799–1800insA | p.Y600X | 39 |
| 77 | Ex17 | R605Q | Neutral | c.1814G>A | p.R605Q | 34 |
| 78 | Ex17 | R605X | Nonsense | g./c.1813C>T | p.R605X | 39 |
| 79 | Ex17 | E601X | Nonsense | g./(c.)1801G>T | p.E601X | 8 |
| 80 | Ex17 | E601K | Missense | g./(c.)1801G>A | p.E601K | 8 |
| 81 | Ex18 | P632L | Neutral | g./c.1895C>T | p.P632L | 22,34 |
New Chinese Patients with NICCD
We reported 60 new CD patients in this study (Table 4). All the 60 novel cases presented with clinical features of NICCD at the point of referral to our hospital. During their most recent follow-up, the NICCD manifestations had improved in 22 patients below 1 year of age. Among the 38 subjects beyond 1 year of age, 11 cases were healthy, without any laboratory or clinical abnormalities. However, hepatomegaly was found in 1 patient, and anemia in 2 cases. Dyslipidemia was revealed in 7 cases while failure to thrive (FTT) in 3, and in particular, 5 patients had FTT and dyslipidemia concurrently, constituting 5 additional patients with FTTDCD features. The remaining 9 patients were lost in contact.
Table 4. Molecular and clinical information of 60 new NICCD patients in China.
| Case | Patient | Gender | Mutations | Major presentations | Clinical outcomes |
| 57 | C0049 | Male | 851del4/851del4 | NICCD | Dyslipidemia |
| 58 | C0050 | Female | 851del4/851del4 | NICCD | FTTDCD |
| 59 | C0054 | Male | R467X/IVS4ins6kb | NICCD | Dyslipidemia |
| 60 | C0056 | Male | 851del4/851del4 | NICCD, ASD+VSD | α-Thalassemia |
| 61 | C0057 | Male | 851del4/1638-1660dup | NICCD | Normal |
| 62 | C0060 | Male | Q259X/c.72A>T | NICCD | Dyslipidemia |
| 63 | C0063 | Male | 851del4/851del4 | NICCD | FTTDCD |
| 64 | C0068 | Male | 851del4/851del4 | NICCD | Dyslipidemia |
| 65 | C0069 | Male | 851del4/IVS6+5G>A | NICCD | Lost contact |
| 66 | C0070 | Female | 851del4/851del4 | NICCD | Lost contact |
| 67 | C0071 | Female | 851del4/851del4 | NICCD | Dyslipidemia |
| 68 | C0073 | Female | 851del4/851del4 | NICCD | Lost contact |
| 69 | C0075 | Female | 851del4/851del4 | NICCD | Lost contact |
| 70 | C0076 | Female | 851del4/1638-1660dup | NICCD | FTTDCD |
| 71 | C0078 | Male | 851del4/851del4 | NICCD | Normal |
| 72 | C0079 | Male | IVS16ins3kb/c.1048G>A | NICCD | Lost contact |
| 73 | C0082 | Male | 1638-1660dup/1638-1660dup | NICCD | Lost contact |
| 74 | C0086 | Male | 851del4/851del4 | NICCD | Lost contact |
| 75 | C0087 | Male | 851del4/1638-1660dup | NICCD | Dyslipidemia |
| 76 | C0095 | Male | 851del4/IVS6+5G>A | NICCD | Normal |
| 77 | C0097 | Male | 851del4/851del4 | NICCD | Hepatomegaly |
| 78 | C0098 | Male | 1638-1660dup/c.1063C>G | NICCD | Lost contact |
| 79 | C0099 | Memale | 851del4/IVS6+5G>A | NICCD | Normal |
| 80 | C0108 | Female | 1638-1660dup/c.1364G>T | NICCD | Normal |
| 81 | C0109 | Male | 851del4/851del4 | NICCD | Normal |
| 82 | C0110 | Female | IVS6+5G>A/R319X | NICCD | Normal |
| 83 | C0116 | Male | IVS6+5G>A/IVS16ins3kb | NICCD | Normal |
| 84 | C0123 | Female | 851del4/IVS16ins3kb | NICCD, liver cirrhosis | FTTDCD, ascites, umbilical hernia, Hematochezia |
| 85 | C0124 | Male | IVS6+5G>A/? | NICCD, eczema | Normal |
| 86 | C0125 | Male | Q159X/R467X | NICCD | Dyslipidemia |
| 87 | C0129 | Male | 851del4/IVS6+5G>A | NICCD, liver cirrhosis | FTTDCD, Died of liver cirrhosis at his age of 2 years and 5 months |
| 88 | C0130 | Male | 851del4/851del4 | NICCD, Iron-deficient anemia | Iron-deficient anemia |
| 89 | C0134 | Male | 851del4/IVS6+5G>A | NICCD, cerebral palsy | FTT, improved cerebral palsy |
| 90 | C0136 | Female | 851del4/R467X | NICCD | Normal |
| 91 | C0139 | Female | 1092-1095delT/c.754G>A | NICCD | FTT |
| 92 | C0139B | Male | 1092-1095delT/c.754G>A | NICCD | Normal |
| 93 | C0140 | Female | IVS11+1G>A/IVS16ins3kb | NICCD | Improved cholestasis |
| 94 | C0142 | Female | 851del4/851del4 | NICCD | Improved cholestasis |
| 95 | C0143 | Male | 851del4/851del4 | NICCD | Improved cholestasis |
| 96 | C0144 | Female | 851del4/851del4 | NICCD | Normal |
| 97 | C0146 | Male | IVS16ins3kb/IVS16ins3kb | NICCD | FTT |
| 98 | C0147 | Male | 1638ins23/IVS6+5G>A | NICCD | Improved cholestasis |
| 99 | C0068S | Female | 851del4/851del4 | NICCD | Lost contact |
| 100 | C0148 | Female | 851del4/851del4 | NICCD | Improved cholestasis |
| 101 | C0149 | Female | 851del4/851del4 | NICCD | Improved cholestasis |
| 102 | C0150 | Male | IVS6+5G>A/R319X | NICCD | Improved cholestasis |
| 103 | C0151 | Female | 851del4/? | NICCD | Improved cholestasis |
| 104 | C0154 | Female | 851del4/851del4 | NICCD | Improved cholestasis |
| 105 | C0156 | Female | 851del4/IVS16ins3kb | NICCD | Improved cholestasis |
| 106 | C0157 | Male | 1638ins23/c.265delG | NICCD | Improved cholestasis, hydrocele |
| 107 | C0158 | Female | 851del4/IVS16ins3kb | NICCD | Improved cholestasis, congenital muscular torticollis, inguinal hernia |
| 108 | C0159 | Male | 851del4/851del4 | NICCD | Improved cholestasis |
| 109 | C0160 | Female | 851del4/IVS16ins3kb | NICCD | Improved cholestasis |
| 110 | C0161 | Female | 851del4/1092-1095delT | NICCD | Improved cholestasis |
| 111 | C0162 | Female | 851del4/851del4 | NICCD | Improved cholestasis |
| 112 | C0163 | Female | 851del4/851del4 | NICCD | Improved cholestasis |
| 113 | C0166 | Male | 1638ins23/1638ins23 | NICCD | Improved cholestasis |
| 114 | C0168 | Male | 851del4/c.1215G>T | NICCD | Improved cholestasis |
| 115 | C0169 | Female | 851del4/? | NICCD | Improved cholestasis |
| 116 | C0170 | Female | 851del4/c.1775A>C | NICCD | Improved cholestasis |
Demographic Features of the Chinese Pediatric Cohort
So far, 116 Chinese CD patients have been diagnosed by our group. This pediatric cohort consisted of 48 females and 68 males, and involved 113 families from 21 provinces, municipalities and autonomous regions in China, including Guangdong, Guangxi, Hainan, Hunan, Hubei, Yunnan, Guizhou, Sichuan, Chongqing, Jiangxi, Fujian, Zhejiang, Jiangsu, Anhui, Shanghai, Henan, Hebei, Shandong, Liaoning, Jilin and Inner Mongolia. As shown in Figure 4, 93 out of the 116 patients were from South China, especially from Guangdong province (51 patients).
Figure 4. Native places of the 116 Chinese patients with citrin deficiency.

By the end of February in 2013, 116 Chinese patients from 21 provinces, municipalities and autonomous regions in China were diagnosed by SLC25A13 gene analysis by our group. This figure indicated their native places, with the patient numbers in the parentheses behind.
The Distribution of SLC25A13 Mutations in the Chinese Pediatric Cohort
As shown in Table 5, the 4 mutations 851_854del4, 1638_1660dup, IVS6+5G>A and IVS16ins3kb were on the top of the list, and accounted for 83.19% of all mutated SLC25A13 alleles, constituting the high-frequency mutations in this Chinese CD cohort. The remaining 22 mutations contributed 14.11%, whilst the unknown ones, just 2.7%. In South China, the 4 high-frequency mutations contributed 87.9% of the total mutated alleles, while this proportion in the North was just 63.6%, and this difference in the mutation distribution between the two areas in China was proved to be statistically significant, with χ2 = 14.93 and P<0.01, as shown in Table S3.
Table 5. Frequency and proportion of the mutated SLC25A13 alleles in the large Chinese Pediatric cohort.
| No. | Mutations | Frequency | Percentage |
| 01 | 851_854del4 | 132 | 58.41 |
| 02 | 1638-1660dup | 20 | 8.85 |
| 03 | IVS6+5G>A | 19 | 8.41 |
| 04 | IVS16ins3kb | 17 | 7.52 |
| 05 | c.1399C>T(R467X) | 5 | 2.21 |
| 06 | c.955C>T(R319X) | 3 | 1.33 |
| 07 | IVS11+1G>A | 3 | 1.33 |
| 08 | c.754G>A | 2 | 0.88 |
| 09 | c.1092_1095delT | 2 | 0.88 |
| 10 | c.1078C>T(R360X) | 1 | 0.44 |
| 11 | c.1622C>A(A541D) | 1 | 0.44 |
| 12 | c.1231G>A(V411M) | 1 | 0.44 |
| 13 | c.847G>T(G283X) | 1 | 0.44 |
| 14 | c.998G>A(G333D) | 1 | 0.44 |
| 15 | c.475C>T(Q159X) | 1 | 0.44 |
| 16 | c.775C>T(Q259X) | 1 | 0.44 |
| 17 | g.2T>C | 1 | 0.44 |
| 18 | r.16-212dup | 1 | 0.44 |
| 19 | c.72T>A(Y24X) | 1 | 0.44 |
| 20 | c.1048G>A(D350N) | 1 | 0.44 |
| 21 | c.1063C>G(R355G) | 1 | 0.44 |
| 22 | c.1364G>T(R455L) | 1 | 0.44 |
| 23 | IVS4ins6kb | 1 | 0.44 |
| 24 | c.1215G>T(K405N) | 1 | 0.44 |
| 25 | c.265delG | 1 | 0.44 |
| 26 | c.1775A>C(Q592P) | 1 | 0.44 |
| 27 | Unknown | 6 | 2.7 |
| In total | 226* | 100 | |
Among the 116 patients, there were 3 siblings from 3 families. Therefore, the cohort comprised 226 mutated SLC25A13 alleles in total, including 6 unknown ones.
Discussion
Since SLC25A13 was cloned as the causative gene for CD [1], genetic analysis of this gene had been well-recognized as a reliable tool for the definitive diagnosis of CD patients. By direct DNA sequencing, 15 novel SLC25A13 variations were identified in this paper (Table 1 and Figure S1), and all of them proved to be CD-associated pathogenic mutations by laboratory and bioinformatic evidences (Table 1 and Figures 1 and 2). These novel mutations expanded the SLC25A13 mutation spectrum, and provided conclusive genetic evidences for the definitive diagnosis of the East Asian patients, along with the mutations revealed at screening analysis. However, routine DNA analytic approaches such as PCR-RFLP and sequencing could not identify all SLC25A13 mutations [13], [24], [44], and the patient C0054 in this paper was such a case, in whom a paternally-inherited c.1399C>T mutation was revealed by screening and direct sequencing, while the mutation of maternal origin once remained obscure on such routine DNA analytic tools.
Our recent cDNA cloning analysis of SLC25A13 ASVs in human PBLs uncovered the marked transcript diversity along with the abundant existence of the transcript r.213_328del that predicted the existence of a constructively novel isoform for citrin protein. Similar ASVs could be detected for mutated SLC25A13 alleles, but all of them carried the information of/from their corresponsive mutations [14]. SLC25A13 cDNA analysis in this study (Table 2 and Table S1) confirmed our previous findings, and particularly, the unique feature of exon 5 skipping in the ASVs of maternal origin in the patient C0054 (Table S2) provided a reliable evidence for the positioning of a large insertion, or deletion, within the SLC25A13 gene segment spanning from intron 4 to intron 5, and directly led to the final identification of the novel transposal insertion of IVS4ins6kb. These findings once again supported the concept that cDNA cloning analysis of SLC25A13 gene using human PBLs could be taken as a feasible tool for the molecular diagnosis of citrin deficiency, overcoming the technical limitation of the conventional DNA analysis.
The large IVS4ins6kb mutation in this study is the second transposal insertion besides the one reported in 2008 [22]. This large insertion was identical in sequence to a DNA segment of 6057bp at chromosome 16p11.2, with two 15bp repetitive sequences on the both sides as the target-site duplication for this DNA transposon (Figure 3). We have no direct evidences yet to clarify the pathogenic mechanism underlying this transposal insertion. However, since IVS4ins6kb occurred very closely to the branch point site within intron 4, a reasonable explanation might be that the large insertion interrupted the formation of the lariat structure and thus disrupted the excision of intron 4 during the splicing reaction. However, this transposal insertion did not affect the branch point site within intron 5, and the freed 5′ end of intron 4 thus had to joined to this site alternatively, forming a lariat and giving rise to exon 5 skipping in the SLC25A13 transcripts, as revealed by cDNA cloning analysis using PBLs in the patient C0054 (Table 2). The exon 5 skipping predictively resulted in a frameshift at codon 110, added 17 amino acids, and then introduced a stop codon at position 127, thus yielding a truncated citrin molecule p.E110fs127X.
To the best of our knowledge, Table 3 was the most comprehensive update of the reported SLC25A13 variations up to now, and the heterogenetic variations in this table constituted reliable molecular evidences for the definitive diagnosis of CD patients. However, it should be recognized that the list itself remained far from being perfect currently. One issue was the unclear mechanisms underlining specific mutations. For example, c.1311C>T had been reported as a synonymous mutation p.C437C [32], but this C>T substitution might cause abnormal splicing of pre-mRNA since it involved the last base of the exon 13 in SLC25A13 gene. Another issue lied in the limited experience on citrin protein analysis. Although the aberrant transcript r.16_212dup had been identified in a NICCD patient [14], its biochemical and structural effects on citrin protein remained obscure due to the technical limitation. The pathogenicity of some missense mutations was another issue. Actually, AGC2 function analysis had documented that c.1505C>T, c.1814G>A and c.1895C>T were all neutral, but not missense mutations [34]. The above issues once again necessitated the in-depth molecular analysis of CD by means of not only genetic, but also transcriptional, translational, and even functional tools.
Although the number of reported Chinese CD patients was relatively small, as a country with a population over 1.3 billion, China might be the largest victim of CD, currently having about 85 700 of such patients theoretically. We reported 60 new NICCD patients in this study (Table 4), and thus established a Pediatric cohort of 116 CD cases, providing a foundation for the in-depth investigation of this disease entity in China. In the SLC25A13 mutation spectrum, as shown in Table 5, 851_854del4, 1638_1660dup, IVS6+5G>A and IVS16ins3kb could be considered as high-frequency mutations in China while the remaining 22 mutations are mostly sporadic. This finding provided an important evidence for the screening of targeted SLC25A13 mutations in Chinese population. The native place analysis of the CD patients in Figure 4 uncovered that most patients are originated from south China, which could be explained partially by the higher carrier rate of SLC25A13 mutations in this area than in the north [44]. The geographic location of our hospital might be another likely reason. As the capital city of Guangdong province, Guangzhou was the largest city in South China.
The SLC25A13 mutations were distributed differently in the patients from different areas in China, and as shown in Table S3, the 4 high-frequency mutations were more common in southern Chinese than in northern. This distribution difference might be attributed to the founder effect and genetic drift. The 4 high-frequency mutations had occurred earlier in southern Chinese during the long history of human evolution, and then dispersed into northern Chinese along with the population migration. Actually, haplotype analysis has proved that the mutation 851_854 del arose in the southern mongoloid population [44], who originated in the Guangxi and Yunnan areas in the southwest China [50]. On the other hand, it has been proposed that the contemporary northern and southern Chinese have distinct origin site in the Yellow River valley and the Yangtze River valley, respectively, with the most likely boundary drawn at the latitude of 30°N [51], and anthropologic evidences have suggested that, in the early Middle Pleistocene, the early man in south of the Yangtze River dispersed to the Qinling Mountain and south of the Yellow River [49].
In conclusion, the 16 novel pathogenic mutations identified in this study enriched the variation spectrum of SLC25A13 gene, and the comprehensive update of SLC25A13 mutations provided a reliable molecular evidence for the definitive diagnosis of CD patients. And, the establishment of the large Chinese Pediatric cohort, the SLC25A13 mutations in this cohort, along with their distribution difference in the patients from different areas in China, formed a substantial contribution to the in-depth understanding of the genotypic features of Chinese CD patients.
Supporting Information
Direct DNA sequencing results of the point and deletion mutations in SLC25A13 gene. In this study, 1 deletion and 14 point mutations of SLC25A13 gene were identified in 15 patients with citrin deficiency. Arrows were used in this figure to indicate the mutated bases with description overhead, respectively.
(TIF)
The SLC25A13 ASVs detected by cDNA analysis in eight healthy volunteers.
(DOC)
Comparison of the SLC25A13 ASVs harboring r .329_468del in the patient C0054 and the 8 healthy volunteers.
(DOC)
Distribution of the mutated SLC25A13 alleles in the CD patients from South and North China.
(DOC)
Acknowledgments
The authors deeply appreciate Dr. Keiko Kobayashi, the Mother of Citrin Deficiency, for her substantial contribution and her long-term technical support to the molecular analysis of SLC25A13 gene. Also, we are grateful to all patients, their parents, and the control individuals for their kind cooperation. In addition, we thank Dr. Ituro Inoue for providing technical guidance for the application of the software PolyPhen2.
Funding Statement
This work was supported by the Fundamental Research Funds for the Central Universities (No. 21612430), the projects supported by the National Natural Science Foundation of China (NSFC, Nos. 81070279 and 81270957), and Grants-in-Aid for Asia-Africa Scientific Platform Program from the Japan Society for the Promotion of Science. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Kobayashi K, Sinasac DS, Iijima M, Boright AP, Begum L, et al. (1999) The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat Genet 22: 159–163. [DOI] [PubMed] [Google Scholar]
- 2.Kobayashi K, Saheki T, Song YZ. Citrin Deficiency. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2005 Sep 16 [Updated 2012 Jan 05].
- 3. Ohura T, Kobayashi K, Tazawa Y, Nishi I, Abukawa D, et al. (2001) Neonatal presentation of adult-onset type II citrullinemia. Hum Genet 108: 87–90. [DOI] [PubMed] [Google Scholar]
- 4. Tazawa Y, Kobayashi K, Ohura T, Abukawa D, Nishinomiya F, et al. (2001) Infantile cholestatic jaundice associated with adult-onset type II citrullinemia. J Pediatr 138: 735–740. [DOI] [PubMed] [Google Scholar]
- 5. Tomomasa T, Kobayashi K, Kaneko H, Shimura H, Fukusato T, et al. (2001) Possible clinical and histologic manifestations of adult-onset type II citrullinemia in early infancy. J Pediatr 138: 741–743. [DOI] [PubMed] [Google Scholar]
- 6. Saheki T, Kobayashi K (2002) Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD). J Hum Genet 47: 333–341. [DOI] [PubMed] [Google Scholar]
- 7. Saheki T, Kobayashi K, Iijima M, Nishi I, Yasuda T, et al. (2002) Pathogenesis and pathophysiology of citrin (a mitochondrial aspartate glutamate carrier) deficiency. Metab Brain Dis 17: 335–346. [DOI] [PubMed] [Google Scholar]
- 8. Yamaguchi N, Kobayashi K, Yasuda T, Nishi I, Iijima M, et al. (2002) Screening of SLC25A13 mutations in early and late onset patients with citrin deficiency and in the Japanese population: Identification of two novel mutations and establishment of multiple DNA diagnosis methods for nine mutations. Hum Mutat 19: 122–130. [DOI] [PubMed] [Google Scholar]
- 9. Nagasaka H, Okano Y, Tsukahara H, Shigematsu Y, Momoi T, et al. (2009) Sustaining hypercitrullinemia, hypercholesterolemia and augmented oxidative stress in Japanese children with aspartate/glutamate carrier isoform 2-citrin-deficiency even during the silent period. Mol Genet Metab 97: 21–26. [DOI] [PubMed] [Google Scholar]
- 10. Lee BH, Jin HY, Kim GH, Choi JH, Yoo HW (2010) Nonalcoholic fatty liver disease in 2 siblings with adult-onset type II citrullinemia. J Pediatr Gastroenterol Nutr 50: 682–685. [DOI] [PubMed] [Google Scholar]
- 11. Okano Y, Kobayashi K, Ihara K, Ito T, Yoshino M, et al. (2013) Fatigue and quality of life in citrin deficiency during adaptation and compensation stage. Mol Genet Metab 109: 9–13. [DOI] [PubMed] [Google Scholar]
- 12. Saheki T, Inoue K, Ono H, Tushima A, Katsura N, et al. (2011) Metabolomic analysis reveals hepatic metabolite perturbations in citrin/mitochondrial glycerol-3-phosphate dehydrogenase double-knockout mice, a model of human citrin deficiency. Mol Genet Metab 104: 492–500. [DOI] [PubMed] [Google Scholar]
- 13. Song YZ, Deng M, Chen FP, Wen F, Guo L, et al. (2011) Genotypic and phenotypic features of citrin deficiency: Five-year experience in a Chinese pediatric center. Int J Mol Med 28: 33–40. [DOI] [PubMed] [Google Scholar]
- 14. Zhang ZH, Lin WX, Deng M, Zhao XJ, Song YZ (2012) Molecular analysis of SLC25A13 gene in human peripheral blood lymphocytes: Marked transcript diversity, and the feasibility of cDNA cloning as a diagnostic tool for citrin deficiency. Gene 511: 227–234. [DOI] [PubMed] [Google Scholar]
- 15. Ohura T, Kobayashi K, Tazawa Y, Abukawa D, Sakamoto O, et al. (2007) Clinical pictures of 75 patients with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD). J Inherit Metab Dis 30: 139–144. [DOI] [PubMed] [Google Scholar]
- 16. Komatsu M, Yazaki M, Tanaka N, Sano K, Hashimoto E, et al. (2008) Citrin deficiency as a cause of chronic liver disorder mimicking non-alcoholic fatty liver disease. J Hepatol 49: 810–820. [DOI] [PubMed] [Google Scholar]
- 17. Song YZ, Li BX, Chen FP, Liu SR, Sheng JS, et al. (2009) Neonatal intrahepatic cholestasis caused by citrin deficiency: clinical and laboratory investigation of 13 subjects in mainland of China. Dig Liver Dis 41: 683–689. [DOI] [PubMed] [Google Scholar]
- 18. Kuhara T, Ohse M, Inoue Y, Cooper AJ (2011) A GC/MS-based metabolomic approach for diagnosing citrin deficiency. Anal Bioanal Chem 400: 1881–1894. [DOI] [PubMed] [Google Scholar]
- 19. Ben-Shalom E, Kobayashi K, Shaag A, Yasuda T, Gao HZ, et al. (2002) Infantile citrullinemia caused by citrin deficiency with increased dibasic amino acids. Mol Genet Metab 77: 202–208. [DOI] [PubMed] [Google Scholar]
- 20. Dimmock D, Kobayashi K, Iijima M, Tabata A, Wong LJ, et al. (2007) Citrin deficiency: a novel cause of failure to thrive that responds to a high-protein, low-carbohydrate diet. Pediatrics 119: e773–e777. [DOI] [PubMed] [Google Scholar]
- 21. Dimmock D, Maranda B, Dionisi-Vici C, Wang J, Kleppe S, et al. (2009) Citrin deficiency, a perplexing global disorder. Mol Genet Metab 96: 44–49. [DOI] [PubMed] [Google Scholar]
- 22. Tabata A, Sheng JS, Ushikai M, Song YZ, Gao HZ, et al. (2008) Identification of 13 novel mutations including a retrotransposal insertion in SLC25A13 gene and frequency of 30 mutations found in patients with citrin deficiency. J Hum Genet 53: 534–545. [DOI] [PubMed] [Google Scholar]
- 23. Fu HY, Zhang SR, Wang XH, Saheki T, Kobayashi K, et al. (2011) The mutation spectrum of the SLC25A13 gene in Chinese infants with intrahepatic cholestasis and aminoacidemia. J Gastroenterol 46: 510–518. [DOI] [PubMed] [Google Scholar]
- 24. Tokuhara D, Iijima M, Tamamori A, Ohura T, Takaya J, et al. (2007) Novel diagnostic approach to citrin deficiency: analysis of citrin protein in lymphocytes. Mol Genet Metab 90: 30–36. [DOI] [PubMed] [Google Scholar]
- 25. Luder AS, Tabata A, Iijima M, Kobayashi K, Mandel H (2006) Citrullinaemia type 2 outside East Asia: Israeli experience. J Inherit Metab Dis 29: S59. [Google Scholar]
- 26. Sheng JS, Ushikai M, Iijima M, Packman S, Weisiger K, et al. (2006) Identification of a novel mutation in a Taiwanese patient with citrin deficiency. J Inherit Metab Dis 29: S163. [Google Scholar]
- 27. Ko JM, Kim GH, Kim JH, Kim JY, Choi JH, et al. (2007) Six cases of citrin deficiency in Korea. Int J Mol Med 20: 809–815. [PubMed] [Google Scholar]
- 28.Song YZ, Sheng JS, Ushikai M, Hwu WL, Zhang CH, et al. (2008) Identification and diagnosis of three novel mutations in SLC25A13 gene of neonatal intrahepatic cholestasis caused by citrin deficiency. Zhonghua Er Ke Za Zhi 46: 411–415. [Article in Chinese]. [PubMed]
- 29. Thong MK, Boey CC, Sheng JS, Ushikai M, Kobayashi K (2010) Neonatal intrahepatic cholestasis caused by citrin deficiency in two Malaysian siblings: outcome at one year of life. Singapore Med J 51: e12–e14. [PubMed] [Google Scholar]
- 30.Xing YZ, Qiu WJ, Ye J, Han LS, Xu SS, et al. (2010) Studies on the clinical manifestation and SLC25A13 gene mutation of Chinese patients with neonatal intrahepatic cholestasis caused by citrin deficiency. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 27: 180–185. [Article in Chinese]. [DOI] [PubMed]
- 31. Lin JT, Hsiao KJ, Chen CY, Wu CC, Lin SJ, et al. (2011) High resolution melting analysis for the detection of SLC25A13 gene mutations in Taiwan. Clin Chim Acta 412: 460–465. [DOI] [PubMed] [Google Scholar]
- 32.Wen PQ, Wang GB, Chen ZL, Cui D, Yuan Q, et al. (2011) SLC25A13 gene analysis in neonates with intrahepatic cholestasis caused by citrin deficiency. Zhongguo Dang Dai Er Ke Za Zhi 13: 303–308.[Article in Chinese]. [PubMed]
- 33. Lin WX, Zhang ZH, Deng M, Cai XR, Song YZ (2012) Multiple ovarian antral follicles in a preterm infant with neonatal intrahepatic cholestasis caused by citrin deficiency: A clinical, genetic and transcriptional analysis. Gene 505: 269–275. [DOI] [PubMed] [Google Scholar]
- 34.Wongkittichote P, Tungpradabkul S, Wattanasirichaigoon D, Jensen LT (2012) Prediction of the functional effect of novel SLC25A13 variants using a S. cerevisiae model of AGC2 deficiency. J Inherit Metab Dis [Epub ahead of print]. [DOI] [PubMed]
- 35. Wong LJ, Dimmock D, Geraghty MT, Quan R, Lichter-Konecki U, et al. (2008) Utility of oligonucleotide array-based comparative genomic hybridization for detection of target gene deletions. Clin Chem 54: 1141–1148. [DOI] [PubMed] [Google Scholar]
- 36. Hutchin T, Preece MA, Hendriksz C, Chakrapani A, McClelland V, et al. (2009) Neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) as a cause of liver disease in infants in the UK. J Inherit Metab Dis 32: S151–S155. [DOI] [PubMed] [Google Scholar]
- 37. Hutchin T, Preece MA, Kobayashi K, Saheki T, Brown R, et al. (2006) Neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) in an European patient. J Inherit Metab Dis 29: S112. [DOI] [PubMed] [Google Scholar]
- 38. Fiermonte G, Parisi G, Martinelli D, De Leonardis F, Torre G, et al. (2011) A new Caucasian case of neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD): a clinical, molecular, and functional study. Mol Genet Metab 104: 501–506. [DOI] [PubMed] [Google Scholar]
- 39. Yasuda T, Yamaguchi N, Kobayashi K, Nishi I, Horinouchi H, et al. (2000) Identification of two novel mutations in the SLC25A13 gene and detection of seven mutations in 102 patients with adult-onset type II citrullinemia. Hum Genet 107: 537–545. [DOI] [PubMed] [Google Scholar]
- 40. Takaya J, Kobayashi K, Ohashi A, Ushikai M, Tabata A, et al. (2005) Variant clinical courses of 2 patients with neonatal intrahepatic cholestasis who have a novel mutation of SLC25A13. Metabolism 54: 1615–1619. [DOI] [PubMed] [Google Scholar]
- 41. Hayasaka K, Numakura C, Toyota K, Kimura T (2012) Treatment with lactose (galactose)-restricted and medium-chain triglyceride-supplemented formula for neonatal intrahepatic cholestasis caused by citrin deficiency. JIMD Rep 2: 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kikuchi A, Arai-Ichinoi N, Sakamoto O, Matsubara Y, Saheki T, et al. (2012) Simple and rapid genetic testing for citrin deficiency by screening 11 prevalent mutations in SLC25A13. Mol Genet Metab 105: 553–558. [DOI] [PubMed] [Google Scholar]
- 43. Takahashi Y, Koyama S, Tanaka H, Arawaka S, Wada M, et al. (2012) An Elderly Japanese Patient with Adult-onset Type II Citrullinemia with a Novel D493G Mutation in the SLC25A13 Gene. Intern Med 51: 2131–2134. [DOI] [PubMed] [Google Scholar]
- 44. Lu YB, Kobayashi K, Ushikai M, Tabata A, Iijima M, et al. (2005) Frequency and distribution in East Asia of 12 mutations identified in the SLC25A13 gene of Japanese patients with citrin deficiency. J Hum Genet 50: 338–346. [DOI] [PubMed] [Google Scholar]
- 45. Kobayashi K, Ushikai M, Song YZ, Gao HZ, Sheng JS, et al. (2008) Overview of citirn deficiency: SLC25A13 mutations and the frequency. J Appl Clin Pediatr 23: 1553–1557. [Google Scholar]
- 46. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, et al. (2010) A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. den Dunnen JT, Antonarakis SE (2001) Nomenclature for the description of human sequence variations. Hum Genet 109: 121–124. [DOI] [PubMed] [Google Scholar]
- 48. Zhao XJ, Tang XM, Zha QB, Shi SS, Song YZ, et al. (2011) Prenatal diagnosis of citrin deficiency in a Chinese family with a fatal proband. Tohoku J Exp Med 225: 273–276. [DOI] [PubMed] [Google Scholar]
- 49.Lin S (1987) Trends of distribution of early man in China. Acta Anthropologica Sinica 6: 190–195. [Article in Chinese].
- 50. Matsumoto H (1988) Characteristics of Mongoloid and neighboring populations based on the genetic markers of human immunoglobulins. Hum Genet 80: 207–218. [DOI] [PubMed] [Google Scholar]
- 51. Zhao TM, Lee TD (1989) Gm and Km allotypes in 74 Chinese populations: a hypothesis of the origin of the Chinese nation. Hum Genet 83: 101–110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Direct DNA sequencing results of the point and deletion mutations in SLC25A13 gene. In this study, 1 deletion and 14 point mutations of SLC25A13 gene were identified in 15 patients with citrin deficiency. Arrows were used in this figure to indicate the mutated bases with description overhead, respectively.
(TIF)
The SLC25A13 ASVs detected by cDNA analysis in eight healthy volunteers.
(DOC)
Comparison of the SLC25A13 ASVs harboring r .329_468del in the patient C0054 and the 8 healthy volunteers.
(DOC)
Distribution of the mutated SLC25A13 alleles in the CD patients from South and North China.
(DOC)