

Abstract
Animals have many ways of protecting themselves against stress; for example, they can induce animal-wide, stress-protective pathways and they can kill damaged cells via apoptosis. We have discovered an unexpected regulatory relationship between these two types of stress responses. We find that C. elegans mutations blocking the normal course of programmed cell death and clearance confer animal-wide resistance to a specific set of environmental stressors; namely, ER, heat and osmotic stress. Remarkably, this pattern of stress resistance is induced by mutations that affect cell death in different ways, including ced-3 (cell death defective) mutations, which block programmed cell death, ced-1 and ced-2 mutations, which prevent the engulfment of dying cells, and progranulin (pgrn-1) mutations, which accelerate the clearance of apoptotic cells. Stress resistance conferred by ced and pgrn-1 mutations is not additive and these mutants share altered patterns of gene expression, suggesting that they may act within the same pathway to achieve stress resistance. Together, our findings demonstrate that programmed cell death effectors influence the degree to which C. elegans tolerates environmental stress. While the mechanism is not entirely clear, it is intriguing that animals lacking the ability to efficiently and correctly remove dying cells should switch to a more global animal-wide system of stress resistance.
Author Summary
As an animal interacts with its environment, it invariably encounters stressful conditions such as extreme temperatures, drought, UV exposure and harmful xenobiotics. Since the ability to respond appropriately to stressful stimuli is paramount to survival, organisms have developed sophisticated stress response programs. Some stressful conditions cause damaged cells to commit suicide (undergo apoptosis), whereas others cause the entire organism to develop mechanisms to resist environmental stress. Studying the small roundworm C. elegans, we find that these two responses are somehow linked: perturbing the mechanisms that allow cells to undergo apoptosis changes the whole animal's response to environmental stress. In fact, perturbing the apoptosis machinery in any way—through mutations that prevent apoptosis altogether, or through mutations that either slow or accelerate the clearance of dying cells—causes the animal to become more stress resistant. Together our findings raise the possibility that the animal may have a way of detecting defects in the normal programmed cell death pathway, and that in response it induces a new program that protects itself from a harsh environment.
Introduction
In nature, animals are constantly exposed to changing environmental conditions. In order to survive, organisms must weather normal and oftentimes extreme variations in temperature, water availability, salt levels, xenobiotics and other environmental factors. To cope, animals have developed mechanisms for stress protection. At the cellular level, DNA damage and other forms of stress can induce apoptosis to remove damaged cells; at the organismal level, stressful conditions can sometimes induce responses that make the entire animal more stress resistant [1], [2].
The nematode C. elegans has evolved several stress-protective responses. These include aversive behaviors, such as avoidance of noxious stimuli, and the activation of alternative developmental programs, such as entry into the dauer state of diapause [3], [4]. The animal can also induce environmental stress resistance by turning on gene transcription to manage the stressor using, for example, the heat-shock transcription factor HSF-1 to combat heat stress [5], SKN-1/Nrf2 to combat xenobiotic stress [6] and the hypoxia-inducible factor HIF-1 to combat hypoxia [7].
In addition to coordinated stress responses at the organismal level, C. elegans, like other organisms, can protect itself against stress at the cellular level. For example, in C. elegans, germ cells undergo apoptosis in response to DNA damage from ionizing radiation [8], [9]. In general, these types of single-cell, live-or-die decisions may be made to sacrifice a part for the betterment of the whole. How these decisions are made and the mechanistic and molecular relationship, if any, between animal-wide stress responses and programmed cell death are, however, poorly understood.
As a fundamental process by which organisms remove unnecessary, abnormal or damaged cells, programmed cell death involves both cell killing via apoptosis and cell corpse removal via phagocytosis and degradation [10]. Although once considered a disinterested second party that simply removes the dead cell, the engulfing cell is now known to be an active participant in the cell death program. For example, in weak C. elegans caspase mutants, in which decisions about whether to complete the cell death program are made stochastically, a second mutation in an engulfment gene further reduces cell death [11], [12]. Likewise, in mammals, mutations affecting either the dying or engulfing cell can disrupt tissue homeostasis and produce developmental disorders, autoimmune disease, cancer and neurodegeneration [13].
Genes responsible for carrying out apoptosis and apoptotic-cell engulfment were first described in C. elegans (Figure S1) [14], [15]. In the apoptotic cell, the canonical programmed cell death pathway involves the Apaf-1 like protein CED-4, which is inhibited by the BCL-2-like protein, CED-9 [16]–[19]. When disinhibited by developmental cues, CED-4 activates the C. elegans executioner caspase CED-3 [20], [21]. In the engulfing cell, several partially redundant pathways govern the membrane and cytoskeletal rearrangements required for phagocytosis of the dying cell (Figure S1) [22]–[25].
C. elegans was instrumental in illuminating the core features of programmed cell death and clearance because it is highly amenable to genetic and experimental manipulation. Recently, we implicated the human disease gene progranulin in the regulation of programmed cell death using a C. elegans mutant [26]. The regulation and function of progranulin are particularly interesting because of its links to disease. Progranulin haploinsufficiency causes the human neurodegenerative disease frontotemporal lobar degeneration while homozygous null carriers develop neuronal ceroid lipofuscinosis [27]–[29]. Allelic variations in the gene have also been linked to Alzheimer Disease, Parkinson Disease and amyotrophic lateral sclerosis (ALS) [30]–[34], and altered progranulin levels have been implicated in autoimmune disease [35], [36], cancer [37]–[42] and ischemic injury [43], [44]. Thus, precise regulation of progranulin levels is important for maintaining health and homeostasis.
Previously, we showed that progranulin normally functions to regulate the rate of apoptotic cell engulfment during the process of programmed cell death [26]. In pgrn-1(-) mutants, apoptotic cells are cleared approximately twice as fast as normal. We also showed that macrophages from progranulin null-mutant mice are able to engulf apoptotic cells more rapidly than are wild-type macrophages. Thus, progranulin, like mtm-1, abl-1 and srgp-1, is a negative regulator of programmed cell death clearance [25], [26], [45]–[47] (Figure S1).
Given the close relationship between environmental stress and age-related disease, we asked whether pgrn-1(-) mutants exhibited an altered response to cellular stressors. We found that they did. However, unexpectedly, they demonstrated increased stress resistance. Even more surprisingly, we found that the same was true of mutations that perturb cell death in other ways, suggesting that a stress response pathway is activated when any part of the programmed cell death pathway does not proceed normally. Our findings reveal an unexpected link between mechanisms that control life-or-death decisions at the level of the individual cell and at the level of the entire animal.
Results
Loss of pgrn-1 Confers Stress Resistance
We tested the resistance of progranulin mutants to several environmental stressors. We found that compared to wild-type controls, pgrn-1(tm985) mutants were resistant to osmotic, heat and endoplasmic reticulum (ER) stress (the latter as measured by resistance to tunicamycin, an inhibitor of N-linked glycosylation) (Figure 1A–C, Tables S1A–C). In contrast, pgrn-1 mutants had normal responses to oxidative stress (paraquat), genotoxic stress (UV light) and pathogen exposure (P. aeruginosa and S. enterica; Figure S2A–C and data not shown). Reintroducing either the C. elegans or human progranulin gene into pgrn-1(-) mutants rescued or partially rescued the mutant stress resistance phenotypes (Figure 1C–D, Tables S1C–D). The partial rescue by human progranulin only at higher doses of tunicamycin could be due to species differences or differences in binding affinities to the progranulin receptor.
Figure 1. pgrn-1(-) mutants are resistant to osmotic, heat and ER stress.

N2 control and pgrn-1(tm985) animals were subjected to various stressors and then scored for survival or ability to develop from egg to L4 stage. (A) Day 1 adult animals were treated with 600 mM NaCl for 24 hours and scored for survival (Student's t test). (B) Day 1 adult animals were incubated at 35°C for 8 hours and scored for survival (Student's t test). (C) Embryos from WT, pgrn-1(-) or pgrn-1(-) expressing a C. elegans progranulin rescue construct were treated with 0, 1 or 5 µg/mL tunicamycin (TM) for 3 days to induce ER stress and scored for ability to develop to the L4 stage (Two-way ANOVA with Bonferroni post-tests). (D) Embryos from WT, pgrn-1(-) or pgrn-1(-) expressing a human progranulin rescue construct (two independently integrated and outcrossed strains) were treated with TM as in (C) (Two-way ANOVA with Bonferroni post-tests). Results shown are representative of at least 2 experiments. Error bars represent standard deviation. Statistical comparisons here are to N2 control. n.s. not significant, *p<0.05, **p<0.01, ***p<0.001. For additional statistical data, please see Table S1A–D.
What do heat, osmotic stress and tunicamycin have in common? One possibility is that they all beget unfolded proteins and induce the ER unfolded protein response. To address this idea, we tested the ability of each stressor to increase expression of hsp-4. HSP-4 is the nematode ortholog of mammalian grp78/BiP/HSP70, and is upregulated by heat and ER stress [48], [49]. We confirmed that heat stress and tunicamycin increased Phsp-4::gfp reporter levels (Figure S3A–B), and found that paraquat, UV irradiation and exposure to P. aeruginosa did not (Figure S4A–C). However, under our conditions, osmotic stress did not increase Phsp-4::gfp levels (Figure S3C). Thus, induction of the ER stress-resistance marker Phsp-4::gfp is not a feature that unifies heat, tunicamycin and osmotic stress.
Apoptosis-Defective Mutants Are Stress Resistant
Because loss of function of pgrn-1, a regulator of programmed cell death clearance, caused stress resistance, we asked whether other mutations affecting programmed cell death would also affect the stress response of the whole animal. In contrast to progranulin mutants, loss-of-function mutations in the gene encoding the executioner caspase ced-3 prevent apoptosis [15]. In ced-3 loss-of-function mutants, cells that normally die during development instead persist. Surprisingly, we found that ced-3(n717) mutant animals also exhibited increased resistance to ER stress (Figure 2A and Tables S2A). Moreover, pgrn-1(-); ced-3(n717) double mutants were no more stress-resistant than were either of the single mutants (Figure 2A and Table S2A), suggesting that pgrn-1 and ced-3 mutations may activate the same stress response pathway. ced-3(n717) mutants could also exhibit osmotic and heat stress resistance, albeit not as consistently as ER stress resistance (Figure 2B–C and Table S2B). Like pgrn-1 mutants, ced-3(n717) mutants did not exhibit resistance to paraquat or UV light (Figure S5).
Figure 2. Mutations in programmed cell death genes ced-3 and ced-9 confer stress resistance.

(A) ced-3(n717) animals were subjected to ER stress with indicated doses of tunicamycin (TM) and then scored for the ability to develop from egg to L4 stage. (B–C) Day 1 adult ced-3(n717) animals were exposed to 600 mM NaCl for 24 hours or thermal stress at 35°C for 8 hours and scored for survival. (D) Strong (n717), moderate (n1949, n2436) or weak (n2436) alleles of ced-3 were tested for response to ER stress. (E) A ced-9(n1950) gain-of-function allele was tested for ER stress resistance in wild-type, pgrn-1(-) or ced-3(-) backgrounds. (F) ced-4(n1162) and ced-9(n2812) loss-of-function mutants were treated with TM and scored for the ability to develop from egg to L4 stage. Results shown are representative of at least 2 experiments except for (C), which is an average of 3 experiments. Error bars represent standard deviation. Statistical comparisons are to N2 control (Student's t test or ANOVA with Bonferroni post-tests). n.s. not significant, *p<0.05, **p<0.01 ***p<0.001. For additional statistical data, see Table S2A–D.
Many alleles of ced-3 have been isolated, and they form an allelic series based on their ability to inhibit programmed cell death [50]. We found that ced-3 alleles exhibited graded levels of ER stress resistance that were correlated with their ability to block programmed cell death. The strong ced-3 allele n717 was more resistant to ER stress than were two intermediate strength (n1949 and n2436) alleles, and these, in turn, were more resistant than the weak (n2438) allele (Figure 2D and Table S2A). These results are consistent with a model in which the stress resistance conferred by ced-3 mutations is mechanistically related to the apoptotic killing conferred by ced-3.
In C. elegans, ced-9 and ced-4 regulate the ability of ced-3 to activate programmed cell death [17]. The Bcl-2-like protein CED-9 inhibits CED-4/Apaf1 activity, blocking cell death; whereas activated CED-4 cleaves CED-3 and activates its caspase function, leading to cell death [18], [51]. Therefore, ced-9 gain-of-function (gf) and ced-4 loss-of-function (lf or -) mutations are similar to ced-3(-) mutations in the sense that they impair programmed cell death, whereas ced-9(lf) mutations cause excessive cell death (and animal lethality) due to uncontrolled activity of CED-4 and CED-3. We tested ced-9(n1950gf) and ced-4(n1162lf) single mutants, as well as a ced-4(n1162lf) ced-9(n2812lf) double mutant, for their responses to ER stress. We found that ced-9(gf) mutants were resistant to ER stress (Figure 2E and Table S2C). Further, the stress resistance conferred by ced-9(gf) mutations was not additive with that conferred by either pgrn-1(-) or ced-3(-) mutations, once again suggesting that these genes affect the same stress-response pathway (Figure 2E, Table S2C). We also found that ced-4(lf) single mutants were resistant to ER stress at low doses of tunicamycin in one of two experiments (Table S2D), and that these mutants did not require intact ced-9 for this stress resistance (Figure 2F, Table S2D). These findings suggest that ced-4 (and likely ced-3) may be genetically downstream of ced-9 in the stress-resistance pathway, as it is in the cell death pathway. However, since ced-4(lf) animals displayed an incomplete degree of stress resistance compared to ced-3(lf) and ced-9(gf) mutants, it remains possible that ced-4 is dispensable or redundant in this stress response pathway.
Inhibition of Apoptotic Cell Engulfment Also Confers Stress Resistance
In addition to mutations that accelerate the clearance of apoptotic corpses, or prevent apoptosis altogether, we also asked whether mutations in apoptotic cell engulfment pathways affected stress resistance. We found that certain engulfment mutant alleles increased ER stress resistance, although the degree of resistance seen in engulfment mutants was generally less than that seen in animals carrying pgrn-1(-) or strong ced-3(-) mutant alleles. The engulfment mutants ced-1(e1735), ced-6(n1813), ced-7(n1892) and ced-2(e1752), ced-5(n1812), ced-10(n3246) exhibited resistance to ER stress at low doses of tunicamycin (1 µg/mL). However they exhibited variable responses to tunicamycin at higher doses (5 µg/mL), with ced-5, 7 and 10 mutants demonstrating ER stress resistance but not ced-1, 2 or 6 mutants (Figure 3A–B and Table S3A–B). Once again, double mutants containing pgrn-1 and engulfment mutations were not more resistant than pgrn-1(-) alone, suggesting that these mutations could potentially induce a common ER stress-resistance pathway (Figure 3A–B and Table S3A–B).
Figure 3. Engulfment mutations enhance ER stress resistance.

(A) Mutations in ced-1(e1735), ced-6(n1813) and ced-7(n1892) with or without pgrn-1(tm985) in the background were tested for ER stress resistance by tunicamycin (TM) treatment. (B) Mutations in ced-2(e1752), ced-5(n1812) and ced-10(n3246) with or without pgrn-1(tm985) in the background were tested for ER stress resistance by tunicamycin treatment. (C) Mutations in abl-1(ok171), abi-1(ok640) and abi-1(tm494) were tested for ER stress resistance by tunicamycin treatment. Results shown are representative of at least 2 experiments except in the case of (C) which was performed once. Error bars represent standard deviation. Statistical comparisons are to N2 control (ANOVA with Bonferroni post-tests). n.s. not significant, *p<0.05, **p<0.01, ***p<0.001. For additional statistical data, see Table S3A, B, and D.
We also tested the response of engulfment mutants to other stressors. We found that ced-1(e1735) and ced-2(e1752) mutants were resistant to osmotic stress (Figure S6A and Table S3C). The ced-2 mutant was also resistant to thermal stress in 1 of 2 trials (Figure S6 and Table S3C). As a group, the engulfment mutants were not as robustly resistant to environmental stressors as pgrn-1 and ced-3 mutants, which may be due to the partial functional redundancy of engulfment pathway genes (See Figure S1).
Like pgrn-1, the tyrosine kinase ABL-1 is a negative regulator of apoptotic corpse engulfment [45]. However, unlike pgrn-1, abl-1 does not act through the canonical engulfment pathways. Instead, abl-1 negatively regulates the engulfment gene abi-1 to inhibit cell death clearance (See Figure S1). We asked whether these two genes might influence ER stress resistance, and found that both abl-1 and abi-1 mutants exhibited resistance to low doses of tunicamycin (Figure 3C, Table S3D). At higher doses of TM, abl-1(-) and one mutant allele of abi-1, ok171, were resistant to tunicamycin stress compared to wild type. Curiously, in some situations, abl-1 mutations actually reduced ER stress resistance. For example, abl-1(n1963) mutations alone have no visible effect on engulfment of apoptotic corpses; however, abl-1 mutations reduce the severity of the engulfment phenotype of ced-1(n2091) and ced-6(n2095) mutants [45]. Likewise, we found that abl-1(n1963) mutations reduced the level of ER stress resistance conferred by ced-1(n2091) and ced-6(n2095) mutations (Figure S7A and Table S3E). We do not have a simple unifying explanation for these findings at this time, but they indicate that pgrn-1 is not the only negative regulator of cell engulfment that can affect ER stress resistance.
We also tested two additional genes that may modulate but are not directly involved in programmed cell death for stress response phenotypes. A mutation in unc-73 enhances the effect of other engulfment mutants but alone has no engulfment defect [52]. A mutation in unc-53 results in defective migration of cells and neuronal processes and the UNC-53 protein interacts with ABI-1 [53]. However, neither unc-73(e936) nor unc-53(e404) mutants exhibited ER stress resistance phenotypes (Figure S7B and Table S3F). Thus, not all genes involved in apoptotic cell engulfment are utilized for stress response.
Stress Resistance in a Non-Apoptotic Programmed Cell Death Pathway
Recently, pqn-41 was identified as a mediator of a type of non-apoptotic cell death [54]. This type of cell death, characterized by crenellation of the nuclear envelope and organelle swelling, occurs independently of the ced-3 caspase and engulfment genes [55], [56]. To determine if pqn-41 affects ER stress resistance, we tested a deletion mutant, ns924. We found that pqn-41(ns924) mutants were resistant to ER stress at a low dose of tunicamycin but not at a high dose, similar to some of the engulfment mutants we tested such as ced-1, ced-2 and ced-6 (Figure S7C and Table S3G). These data suggest that organismal stress resistance may be linked to both apoptotic and non-apoptotic programmed cell death.
Together, these findings indicate that perturbing C. elegans programmed cell death in a variety of ways, either by affecting the initiation of cell death or the engulfment of the dying cell, can confer whole-animal resistance to environmental stress.
The Unfolded Protein Response (UPR) Gene ire-1 Mediates ER Stress Resistance of pgrn-1 Mutants
Because of recent findings connecting the unfolded protein response with neurodegenerative diseases [57], we decided to investigate the mechanism by which a pgrn-1 mutation affected ER stress resistance. The UPR is the cellular program that responds to ER stress. The UPR is mediated by three ER resident proteins encoded by ire-1, pek-1 (mammalian Perk) and atf-6 [58]. Mutations in these genes impair the response to ER stress in C. elegans [59], [60]. Part of this stress response includes alternative splicing of xbp-1 mRNA by activated IRE-1 and the consequent upregulation of XBP-1 target genes, such as hsp-4, the nematode ortholog of mammalian grp78/BiP/HSP70 [48], [49]. To investigate the role of the UPR in the stress resistance of apoptosis mutants, we first tested whether pgrn-1 mutants required ire-1 for ER stress resistance. We tested a pgrn-1; ire-1 double mutant and found that resistance in pgrn-1 mutants was dependent on ire-1 (Figure 4A and Table S4A) suggesting that the mechanism of stress resistance of cell death mutants requires this branch of the UPR pathway. One possibility was that the IRE-1 pathway is constitutively activated in pgrn-1 mutants. To test this, we measured the levels of spliced xbp-1 mRNA. Interestingly, we found no changes in the levels of spliced xbp-1 mRNA in pgrn-1 mutants compared to wild type (Figure S8). We also investigated whether ced-3 or ced-1 mutants exhibited increased levels of spliced xbp-1 mRNA. Similar to pgrn-1 mutants, they did not (Figure S8).
Figure 4. pgrn-1(-) resistance to ER stress may be partially dependent on the UPR pathway, daf-16 and pmk-1.

(A) Embryos from the indicated strains were grown on plates with tunicamycin (TM) and assessed for their ability to develop to the L4 stage. (B) daf-2(e1370) and daf-16(mu86) were tested for ER stress resistance with and without pgrn-1(-) in the background. (C) daf-16(mu86); muIs113 and pgrn-1(tm985); muIs113 animals were tested for ER stress resistance. (D) pmk-1(km25) and pgrn-1(-); pmk-1(-) mutants were tested for ER stress resistance. Error bars represent standard deviation. Statistical comparisons are to N2 control (Two-way ANOVA with Bonferroni post-tests). *p<0.05, **p<0.01, ***p<0.001. For additional statistical data, see Table S4A–D.
Since the hsp-4 gene is a target of active XBP-1, we measured whether pgrn-1 mutants displayed increased Phsp-4::gfp reporter levels. We found that except for one time point at the L4 stage of larval development, Phsp-4::gfp levels in pgrn-1 mutants were largely unchanged compared to controls. Correspondingly, levels of Phsp-4::gfp in ced-3(n717) mutants were also generally unchanged compared to controls (Figure S9). These data suggest that although pgrn-1 mutations affect the ER stress response through the ire-1 gene, the downstream splicing of xbp-1 mRNA and expression of Phsp-4::gfp is not affected. pgrn-1 mutations may somehow make the IRE-1 branch of the UPR more effective without dramatically changing its activity.
The FOXO Transcription Factor DAF-16 and the MAPK PMK-1 Are Required for ER Stress Resistance of pgrn-1 Mutants
Mammalian progranulin has been demonstrated to activate the insulin/IGF-1 pathway and downstream MAP kinases [37], [61], [62]. Animals carrying mutations in the C. elegans insulin/IGF-1 receptor, daf-2, are long-lived [63], [64] and resistant to many stressors, including heat, osmotic stress and ER stress [63], [64]. The longevity and stress resistance of daf-2 mutants require the FOXO transcription factor daf-16 [60]. Upon inactivation of daf-2, DAF-16 accumulates in the nucleus [65] where it regulates transcription of stress response genes. We found that the degree of ER stress resistance of pgrn-1(tm985); daf-2(e1370) double mutants was similar to that of daf-2(-) single mutants (Figure 4B and Table S4B). pgrn-1 mutants also required intact daf-16 for stress resistance, as daf-16(mu86) pgrn-1(tm985) double mutants were no more stress resistant than were single daf-16(mu86) mutants (Figure 4B and Table S4B). These findings suggest that pgrn-1 may be part of the daf-2 pathway or act with daf-2 to confer stress resistance. However, unlike mutations in daf-2, pgrn-1(-) does not affect nuclear localization of DAF-16::GFP protein (Figure S10). DAF-16::GFP localization is also unaffected in ced-1 and ced-3 mutant animals (Figure S10). To determine if nuclear localization of DAF-16 would further increase stress resistance in pgrn-1 mutants, we crossed the daf-16aAM transgene (which causes DAF-16 nuclear accumulation due to mutation of its AKT-phosphorylation sites) into a pgrn-1(-) background [65]. We found that a pgrn-1(-); daf-16aAM strain was no more stress resistant than was the pgrn-1 mutant alone (Figure 4C and Table S4C).
Others have shown that adult-only ced-3 RNAi extends lifespan without altering DAF-16::GFP localization [66]. Given the correlation between lifespan extension and some forms of stress resistance [67], we tested the lifespan of ced-3, ced-1 and ced-2 mutants. In earlier work, we showed that pgrn-1(-) mutant lifespan is no different than wild type [26]. Whereas a ced-3(-) mutation significantly extended lifespan compared to wild type, ced-1 and ced-2 mutations did not (Figure S11), indicating that longevity and this type of organismal stress resistance can be dissociated.
Several MAP kinases are required for responses to cellular stressors in C. elegans. The PMK-1/p38 MAP kinase encoded by pmk-1 is required for resistance to oxidative stressors [68], pathogenic bacteria [69] and exogenously induced ER stress [70]. We confirmed that pmk-1 mutations increased sensitivity to ER stress and found that pgrn-1(-); pmk-1(km25) double mutants were no more resistant to ER stress than were pmk-1 single mutants. Thus, pmk-1 is required for the ER stress resistance induced by pgrn-1 mutations (Figure 4D and Table S4D).
Progranulin is a secreted protein. In mammals, two progranulin receptors have been identified, the tumor necrosis factor receptor (TNFR) and sortilin [71]. Thus, we tested a downstream TNF receptor associated factor (TRAF) mutant, trf-1(nr2014), for stress resistance and epistasis with pgrn-1(-). trf-1 mutants were not stress resistant compared to wild type and pgrn-1(-) did not require trf-1 for its stress resistance (Figure S12A). We also tested two mutant alleles of trk-1, a C. elegans neurotrophin receptor similar to a co-receptor for sortilin, the other mammalian progranulin receptor. Again, pgrn-1 mutants did not require trk-1 for stress resistance (Figure S12B). Thus, an as yet unidentified receptor(s) appears to be required for progranulin to influence ER stress resistance in C. elegans.
Apoptotic Cell Death Mutants Share Co-regulated Genes
If mutations that perturb cell death in different ways act in the same stress-resistance pathway, then they might share gene expression patterns that differ from wild type. To test this, we performed gene expression profiling by RNA sequencing (RNA-seq), comparing day 1 adult pgrn-1(tm985), ced-3(n717) and ced-1(e1735) mutants to wild-type animals. This allowed us to assess 1) whether these strains have altered gene expression, and 2) whether their differentially expressed genes are shared, suggesting the involvement of a common pathway. In spite of different cell death phenotypes of these mutants, RNA-seq revealed that all three mutants down-regulated the same 95 genes and up-regulated the same 9 genes, a highly significant portion of the total transcriptome (p<10−16) (Figure 5 and Table S5). Of the genes that share differential regulation in our mutants, a significant number are regulated by DAF-16 (Table S5). These findings are consistent with the possibility that the stress resistance phenotypes of these three mutants may be due to the involvement of shared pathways.
Figure 5. Programmed cell death mutants share differentially regulated genes.
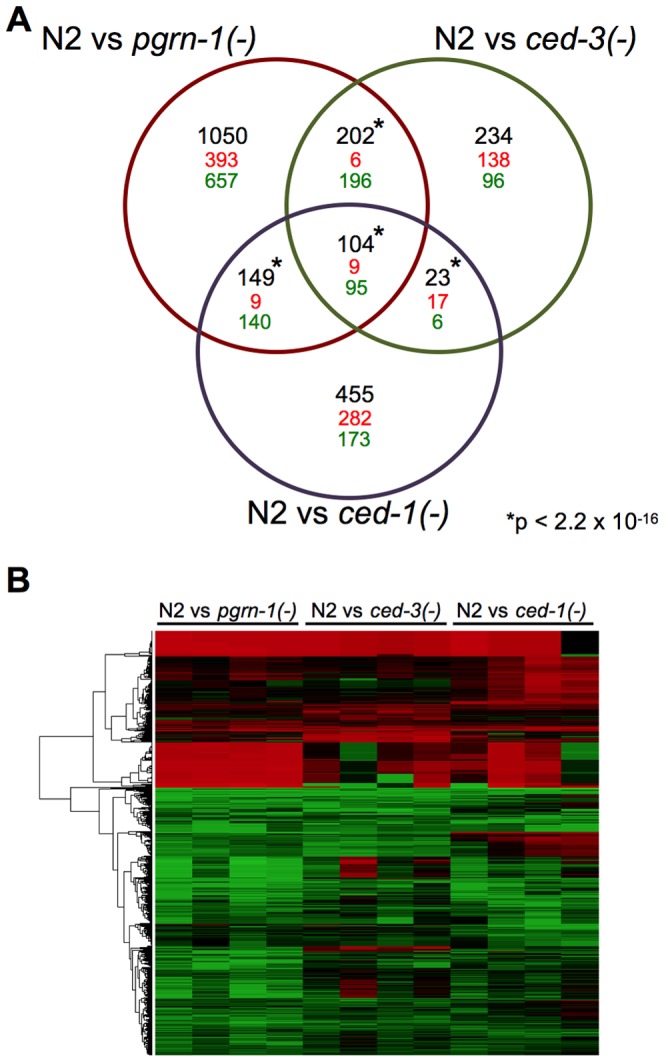
(A) Venn diagram of differentially regulated genes in day 1 adult wild-type (N2) animals versus pgrn-1(tm985), ced-3(n717) or ced-1(e1735) mutants. Significance cut-off was an FDR of <0.05. The numbers in black represent the total number of overlapping genes, with direction of change (up-regulated = red, down-regulated = green) indicated below. The significance of the overlaps between the mutant strains was calculated using Fisher's exact test. See the Excel file (Table S5) for gene list. *p value<2.2×10−16. (B) Heat map depicting the fold changes of gene expression in pgrn-1(tm985), ced-3(n717) and ced-1(e1735) mutants compared to N2 wild-type animals for each of four biological replicates.
Discussion
We have shown that mutations that impair C. elegans programmed cell death in any of three ways—by inhibiting apoptosis, by impairing corpse clearance or by accelerating corpse clearance—all enhance resistance to certain environmental stressors. These mutations do not confer resistance to all cellular stressors, as pgrn-1 and ced-3 mutants exhibit normal sensitivity to UV light and oxidative stress.
Several questions naturally follow from these findings. First, why are these mutants resistant to specific stressors? Tunicamycin is an N-linked glycosylation inhibitor that causes retention of translated proteins in the ER and induces the unfolded protein response. Similarly, heat and osmotic stress cause ER and/or cytosolic proteins to unfold and activate signaling programs that induce expression of heat shock proteins and other chaperones. Thus, it seems possible that cell death mutations all trigger a response specific for unfolded proteins, such as the ER unfolded protein response. Consistent with this, we found that the cell-death-related stressors tunicamycin and heat both activated the ER stress-response gene hsp-4/BIP, whereas the cell-death-unrelated stressors Pseudomonas, UV and paraquat did not. However, Phsp-4::gfp was not induced by osmotic stress and pgrn-1(-) mutation did not increase basal Phsp-4::gfp expression levels. Thus, defects in cell death can do more to protect the animal than simply to induce the canonical UPR. They must generate a more multifaceted response that can maintain proteostasis.
The mechanism by which cell death mutations induce animal-wide stress resistance is not known. If different cell-death mutations affected different pathways, or the same pathway to different extents, then one would expect double mutants to be even more stress resistant than the individual single mutants. As this was not the case, it is possible that all of these mutations trigger the same stress response, though other interpretations remain possible. We identified some of the genes required for pgrn-1 mutants to resist the ER stressor tunicamycin. We found that this stress resistance requires an intact ire-1 gene, the MAP kinase PMK-1 and the transcription factor DAF-16/FOXO. We also identified a number of genes that are differentially regulated by all three of our mutants (compared to wild type), suggesting that they may achieve stress resistance by recruiting shared genes and/or pathways. It will be very interesting to explore these shared genes in future studies.
In contrast to the selective stress resistance of pgrn-1 mutants, daf-2 mutants are resistant to most environmental stressors. Thus, decreased DAF-2/insulin/IGF-1 signaling activates a genetic program that more generally elevates organismal resilience. daf-2 mutants appear to increase their resistance to ER stress by making the ire-1/xbp-1 pathway more efficient, possibly by activating stress-response transcription factors like DAF-16 that collaborate with ire-1/xbp-1 to induce new protective genes [60]. In concordance with this, daf-2 mutants actually have reduced levels of expression of ire-1/xbp-1-regulated genes such as hsp-4/BIP. While this was not the case for pgrn-1 or ced-3 mutants, whose Phsp-4::gfp levels were not decreased, there are some unexpected similarities between the ER stress-resistance phenotypes of cell-death mutants and daf-2 mutants. First, the ER stress resistance phenotypes of pgrn-1 and daf-2 mutants require daf-16, either fully (pgrn-1 mutant) or partially (daf-2 mutant). Second, both ER stress responses are completely dependent on ire-1, yet in neither case is xbp-1 splicing increased. Additionally, the cell death mutants exhibited either no increase or only slight increases in Phsp-4::gfp expression, rather than the substantial increase one might expect if this branch of the ER pathway were constitutively active. Finally, the daf-2 and pgrn-1 mutant ER stress-resistance phenotypes are not additive. Thus the ER-stress resistance pathways activated by these mutations likely share at least some mechanistic features.
It was striking that mutations that perturb programmed cell death in such different ways all had similar effects on environmental stress resistance. Why should mutations that lead to undead cells (apoptosis mutations), lingering corpses (engulfment mutations), and prematurely-engulfed corpses (progranulin mutations) all activate what appears to be (from genetic tests) the same stress resistance pathway? From an evolutionary perspective, this linkage may make sense. Presumably apoptosis evolved not only to sculpt tissues during development, but also to remove cells that are damaged and unable to perform their normal functions, or perhaps that are overtly harmful to the animal. Viewed in this way, impairments in programmed cell death could be interpreted by the organism as an inability to respond normally to stress. Perhaps, under these conditions, the animal uses an alternative, back-up system to survive; namely, the system we have described in this study. Specifically, animals could have developed a sensitive surveillance system that can detect abnormalities in cell death, and respond to them by activating another pathway that enhances their overall level of stress resistance. The existence of this type of alternative system could have increased animal fitness during evolution in turbulent or adverse environments.
While this model makes sense from an evolutionary perspective, other models are possible as well. For example, perhaps cell-death proteins, which act in a multi-step pathway to remove unwanted cells, also act together in a different pathway that has the effect of sensitizing the animal to various forms of stress. Non-cell death functions have, in fact, been described for cell-death effectors. In C. elegans, ced-10(-) mutations impair not only cell engulfment but also cell migration [72]. Another engulfment gene, ced-1, has also been implicated in neuronal regulation of innate immunity [73], [74]. In mammals, a defect in the BCL2-family protein BID impairs cytokine production in response to immune activation independently of its cell death signaling function [75]. Further, certain mammalian executioner caspases can activate microglia in response to inflammogens without causing microglial death [76]. Finally, in a mouse model of Alzheimer Disease, caspase activation may be responsible for tau cleavage and aggregate formation, thereby serving a protective function [77]. Thus, programmed cell death effectors could hypothetically act together to sensitize animals to certain stressors or, alternatively, to inhibit a stress-response pathway. In either case, perturbing programmed cell death would increase organismal stress resistance.
A protein in the flowering plant Arabidopsis may support the model that programmed cell death effectors can sensitize an animal to environmental stress. Arabidopsis can express a protein called RD21 that, like CED-3, is a cysteine protease, and, like progranulin, contains a granulin domain. Osmotic stress induces RD21 and, possibly as a consequence, leaf senescence. Interestingly, in response to stress RD21 undergoes a process of maturation in which its caspase domain cleaves and releases the granulin domain [78]–[80]. In RD21, the caspase and granulin domains are contained within the same molecule. However, perhaps in C. elegans, the two domains reside in different proteins but nevertheless act together to influence organismal stress resistance.
In summary, our findings indicate that programmed cell death effectors not only kill and remove individual cells, but also influence environmental stress resistance at the level of the whole animal. To our knowledge this is the first time that cell-death effectors like ced-3, pgrn-1 and ced-1 have been implicated in organismal stress resistance, and these findings raise many interesting new questions about both mechanism and evolution.
Materials and Methods
Strains
Unless otherwise indicated, C. elegans were cultured at 20°C using standard procedures [81]. Strains were kindly provided by the Mitani Laboratory (National Bioresource Project) at the Tokyo Women's Medical University and the Caenorhabditis Genetics Center (CGC) at the University of Minnesota. Strains were outcrossed four times to the laboratory N2 control strain (N2 Bristol). Descriptions of strains can be found at www.wormbase.org. The following strains were used:
AWK2 ced-9(n1950gf) III; ced-3(n717) IV
AWK74 daf-16(mu86) I; ced-3(n717) IV
AWK76 daf-16(mu86) pgrn-1(tm985) I; muIs109[Pdaf-16::daf-16::gfp+Podr-1::RFP]
AWK77 daf-16(mu86) I; ced-3(n717) IV; muIs109[Pdaf-16::daf-16::gfp+Podr-1::RFP]
AWK78 ced-1(e1735) daf-16(mu86) I
AWK80 ced-1(e1735) daf-16(mu86) I; muIs109[Pdaf-16::daf-16::gfp+Podr-1::RFP]
AWK109 pgrn-1(tm985) I; pqn-41(ns294) III
AWK111 pgrn-1(tm985) I; muIs113[Pdaf-16::daf-16AM::gfp+rol-6]
CB404 unc-53(e404) II
CB936 unc-73(e936) I
CF1037 daf-16(mu86) I
CF1041 daf-2(e1370) III
CF1934 daf-16(mu86) I; muIs109[Pdaf-16::daf-16::gfp+Podr-1::RFP]
CF2260 N2; zcIs4[Phsp-4::gfp] V
CF2473 ire-1(ok799) II
CF3050 pgrn-1(tm985) I
CF3165 pgrn-1(tm985) I; zcIs4[Phsp-4-4::gfp] V
CF3170 pgrn-1(tm985) I; ire-1(ok799) II
CF3196 daf-16(mu86) pgrn-1(tm985) I
CF3206 pgrn-1(tm985) I; daf-2(e1370) III
CF3324 ced-3(n717) IV
CF3419 pgrn-1(tm985) I; ced-3(n717) IV
CF3447 pgrn-1(tm985) I; muIs189[Ppgrn-1:: pgrn-1::polycistronic mCherry+Podr-1::CFP]
CF3762 ced-3(n2436) IV
CF3656 ced-2(e1752) IV
CF3660 ced-10(n3246) IV
CF3662 pgrn-1(tm985) I; ced-2(e1752) IV
CF3667 ced-1(e1735) I
CF3672 pgrn-1(tm985) ced-1(e1735) I
CF3675 pgrn-1(tm985) I; ced-10(n3246) IV
CF3680 ced-5(n1812) IV
CF3683 pgrn-1(tm985) I; ced-7(n1892) III
CF3684 pgrn-1(tm985) I; ced-5(n1812) IV
CF3685 pgrn-1(tm985) I; ced-6(n1813) III
CF3687 pgrn-1(tm986) I; muIs211[Pegl-3::huPGRN::polycistronic mCherry+Podr-1::CFP] Line 1
CF3688 pgrn-1(tm986) I; muIs211[Pegl-3::huPGRN::polycistronic mCherry+Podr-1::CFP] Line 2
CF3762 ced-3(n1949) IV
CF3802 ced-3(n717) IV; zcIs4[Phsp-4::gfp] V
CF3808 trk-1(tm3985) X
CF3809 trk-1(tm4054) X
CF3817 pgrn-1(tm985) I; trk-1(tm3985) X
CF3818 pgrn-1(tm985) I; trk-1(tm4054) X
CF3821 trf-1(nr2014) III
CF3833 pgrn-1(tm985) I; trf-1(nr2014) III
CF3879 pgrn-1(tm985) I; pmk-1(km25) IV
CF3881 pgrn-1(tm985) I; ced-9(n1950gf) III
FX494 abi-1(tm494) III
KU25 pmk-1(km25) IV
MT2547 ced-4(n1162) III
MT4433 ced-6(n1813) III
MT4982 ced-7(n1892) III
MT4770 ced-9(n1950gf) III
MT7384 ced-4(n1162) ced-9(n2812lf) III
MT16077 ced-1(n2091) I; abl-1(n1963) X
MT19956 ced-6(n2095) III; abl-1(ok171) X
OS4023 pqn-41(ns294) III
RB829 abi-1(ok640) III
XR1 abl-1(ok171) X
Generation of Transgenic C. elegans Strains
To generate a C. elegans pgrn-1 rescue construct, full-length pgrn-1a+TAA stop codon and its endogenous 0.5 kB upstream promoter were cloned into a Gateway polycistronic mCherry vector (courtesy K. Ashrafi lab, UCSF). The resulting plasmid (Ppgrn-1::pgrn-1+TAA::polycistronic mCherry) expresses both progranulin and mCherry and functions as a full length rescuing construct when expressed in the pgrn-1 mutant. To generate a human progranulin rescue strain, the pan-neuronal egl-3 promoter (courtesy of K. Ashrafi lab) and the human progranulin cDNA sequence were cloned into a Gateway polycistronic mCherry vector (Pegl-1::human PGRN::polycistronic mCherry).
The constructs were microinjected separately into the gonads of day 1 adult C. elegans. Stable monogenic lines were isolated and analyzed using Leica fluorescent, Zeiss Axioplan 2 or Nikon Spectral Confocal microscopes. Extrachromosomal arrays were integrated by UV irradiation by the method of C. Frank et al. [82] and outcrossed at least 5 times to our lab's wild-type N2 control strain.
Stress Assays
For thermal and osmotic stress assays on Day 1 worms, L4-stage animals were picked and grown at 20°C overnight. For thermal stress assays, worms were moved to a 35°C incubator for 12 hours and then scored for survival. Osmotic stress assays were performed by the method of Lamitina et al. [83] with the following modifications: worms were fed OP50 bacteria, worms were cultured at 20°C prior to the assay, and assays were performed on NG-based plates at 20°C with increasing amounts of NaCl added as indicated. For paraquat stress assays, individual animals were placed in 96-well plates with 100 µL of 250 µM methyl viologen (paraquat, Sigma-Aldrich) dissolved in M9 and scored for movement every 1 hour at 25°C. For genotoxic stress assays, day 1 adult animals were transferred to unseeded plates and treated with 1200 J/m2 UV light in a Stratalinker 1800 (Stratagene). Animals were then scored for survival every 24 hours. Pathogen stress was performed by transferring worms to plates seeded with P. aeruginosa or S. enterica starting at day 1 and scoring each subsequent day for survival. In all assays, animals that failed to move in response to a gentle touch with a metal pick were scored as dead.
For ER stress assays, synchronized eggs were transferred to plates containing 0, 1, 2 or 5 µg/mL of tunicamycin (EMD Chemicals). After 3 days, animals that developed to the L4 stage were quantified. Figures show fraction of animals that develop to L4 stage normalized to percent hatching on 0 µg/mL tunicamycin for each strain.
Statistical analyses were performed in GraphPad Prism statistical package with tests as indicated in figure legends.
Lifespan Analysis
Wild-type, ced-1(e1735), ced-2(e1752) and ced-3(n717) strains were grown at 20 degrees Celsius (C), then picked to fresh OP50 at the L4 stage and shifted to 25 degrees C. Subsequent lifespan analysis was done at this temperature. Animals were transferred every day to fresh plates until progeny production ceased. Animals that crawled off the plate, exploded, bagged, or became contaminated were censored. GraphPad Prism was used to calculate mean life spans and perform statistical analyses. P values were determined using log-rank (Mantel-Cox) statistics.
xbp-1 RT-PCR
C. elegans eggs were obtained by bleaching, then plated onto E. coli OP50 and allowed to develop at 20°C to day 1 of adulthood. At this point, positive controls were exposed to 5 mg/ml tunicamycin for 5 hours while all other worms were left untreated. After washing animals off plates, Trizol was added and samples were frozen in liquid nitrogen. Animals were lysed in a Mini-Beadbeater (Biospec products) for 10 minutes at the maximal setting. Total RNA was isolated using a phenol/chloroform extraction and DNA contamination was removed with DNA-free treatment (Ambion). cDNA was synthesized (iScript) using oligo (dT) primers and RT-PCR was performed using primers that amplify an ∼200 bp unspliced transcript and an ∼180 bp spliced transcript. Forward primer sequence: 5′ ctacgaagaagaagtcgtcgg 3′ and reverse primer sequence: 5′ ttcttgttgcgatccatgtg 3′. RT-PCR products were analyzed by running them out on a 3% agarose gel stained with ethidium bromide.
Quantification of Fluorescence
Animals expressing the Phsp-4::gfp transgene were anaesthetized on agarose pads containing 2.5 mM levamisole. Whole worm images were taken using a Retiga EXi Fast1394 CCD digital camera (QImaging, Burnaby, BC, Canada) using the 5× objective on a Zeiss Axioplan 2 compound microscope (Zeiss Corporation, Germany). Each image was taken so that the intestine was in focus and exposure time was calibrated to minimize number of saturated pixels for the set of animals. Images within each experiment were acquired using identical settings and exposure times to allow direct comparisons. Fluorescence intensity was measured by outlining the entire worm. Openlab 4.0.2 software (Improvision, Coventry, UK) was then used to quantify total intensity of each pixel in the selected area. Measurements were obtained by subtracting the minimum intensity from the mean intensity and taking the average of these calculations for 8–10 animals per time-point.
RNA-Seq Analysis
Total RNA was isolated from each of the strains pgrn-1(tm985), ced-3(n717), ced-1(e1735) and wild-type (N2E) using a phenol/chloroform extraction, and DNA contamination was removed with DNA-free treatment (Ambion). Samples were extracted in quadruplicates (four biological replicates for each strain), for a total of 16 samples. Total RNA was quantified using the RiboGreen assay and RNA quality was checked using an Agilent Bioanalyzer (Agilent). RNA Integrity Numbers (RINs) were >8 in all the samples. Libraries for RNA-seq were prepared using the Illumina TruSeq library preparation protocol (Illumina Inc), multiplexed into a single pool and sequenced using an Illumina HiSeq 2500 sequencer across 4 lanes of 2 Rapid Run SR 1×50 flow cells. After demultiplexing, we obtained between 13 and 32 million reads per sample, each one 50 bases long. Quality control was performed on base qualities and nucleotide composition of sequences. Alignment to the C. elegans genome (ce10) was performed using the STAR spliced read aligner (PMID 23104886) with default parameters. Additional QC was performed after the alignment to examine the following: level of mismatch rate, mapping rate to the whole genome, repeats, chromosomes, and key transcriptomic regions (exons, introns, UTRs, genes). Between 92 and 93% of the reads mapped uniquely to the worm genome. Total counts of read-fragments aligned to candidate gene regions within the C. elegans reference gene annotation were derived using HTS-seq program and used as a basis for the quantification of gene expression. Only uniquely mapped reads were used for subsequent analyses. Following alignment and read quantification, we performed quality control using a variety of indices, including sample clustering, consistency of replicates, and average gene coverage. Differential expression analysis was performed using the EdgeR Bioconductor package (19910308), and differentially expressed genes were selected based on False Discovery Rate (FDR Benjamini Hochberg adjusted p- values) estimated at ≤5%. Clustering and overlap analyses were performed using Bioconductor packages within the statistical environment R (www.r-project.org/). Gene Ontology annotation was performed using DAVID (david.abcc.ncifcrf.gov/). DAF-16 dependent genes were curated from published reports [84], [85] and Wormmart annotation (http://caprica.caltech.edu:9002/biomart/martview).
Supporting Information
Genetic pathways that regulate programmed cell death in apoptotic and engulfing cells in C. elegans. Genes that normally promote cell death and engulfment are in green while those that normally inhibit them are shown in red. Genes tested in this study are in bold.
(TIFF)
Related to Figure 1. A mutation in pgrn-1 does not confer oxidative, UV or pathogen stress resistance. Day 1 adult N2 or pgrn-1(tm985) animals were subjected to oxidative stress with 250 µM paraquat (A), genotoxic stress with 1200 J/m2 UV light (B) or pathogen stress by feeding P. aeruginosa (C). Worms were scored for survival at indicated times and analyses by log-rank Mantel-Cox test were performed. (A) p = 0.1, N>30 animals/strain. (B) p = 0.054, n>50 animals/strain. (C) p = 0.078, N>50 animals/strain. Results shown are representative of at least 2 experiments except in A, which was performed 3 times with p values of 0.0034, 0.01 and 0.125.
(TIFF)
Related to Figure 1. Heat and tunicamycin stress increase HSP-4::GFP levels while osmotic stress does not. (A) Phsp-4::hsp-4::gfp animals were incubated at 20°C (Control treated) or 35°C (Heat stress treated) for 4 hours then imaged on a Zeiss AxioImager. (B) Phsp-4::hsp-4::gfp animals were incubated at 20°C on agarose plates containing 0 µg/mL (Control treated) or 5 µg/mL (ER stress treated) tunicamycin. Twenty-four hours later, they were imaged on a Zeiss AxioImager. (C) Phsp-4::hsp-4::gfp animals were incubated at 20°C on agarose plates containing 51 mM NaCl (Control treated) or 400 mM NaCl (Osmotic stress treated). After 24 hours, they were imaged on a Zeiss AxioImager.
(TIFF)
Related to Figure 1. UV light, paraquat and P. aeruginosa do not increase HSP-4::GFP levels. (A) Phsp-4::hsp-4::gfp animals were mock-treated (Control treated) or exposed to 1200 J/m2 of ultraviolet light (UV stress treated). After 5 hours at 20°C, the animals were imaged on a Zeiss AxioImager. (B) Phsp-4::hsp-4::gfp animals were placed onto agarose plates with either OP50 bacteria (Control treated) or P. aeruginosa PA14 (Pathogen treated) bacteria. After incubation for 24 hours at 20°C, animals were imaged on a Zeiss AxioImager. (C) Phsp-4::hsp-4::gfp animals were placed in M9 solution containing either vehicle alone (Control treated) or 200 mM paraquat (Oxidative stress treated) for 3 hours. The animals that were still alive at that time were imaged on a Zeiss AxioImager.
(TIFF)
Related to Figure 2. A mutation in ced-3 does not confer oxidative or UV stress resistance. Day 1 adult N2 or ced-3(n717) animals were subjected to oxidative stress with 250 µM paraquat (A) or genotoxic stress with 1200 J/m2 UV light (B). Worms were scored for survival at indicated times and analyses by log-rank Mantel-Cox test were performed. (A) p>0.05, N = 20 animals/strain. (B) p>0.05, N>35 animals/strain.
(TIFF)
Related to Figure 3. Mutations in engulfment genes confer resistance to osmotic and heat stresses. Day 1 adult ced-1(e1735) and ced-2(e1752) animals were exposed to 600 mM NaCl for 24 hours (A) or thermal stress at 35°C for 8 hours (B) and scored for survival. Error bars represent standard deviation. Statistical comparisons here are to N2 control (Student's t test or ANOVA with Bonferroni post-tests). *p<0.05, ***p<0.001. For additional statistical data, see Supplemental Table S3C.
(TIFF)
Related to Figure 3. ER stress resistance in related cell death mutants. A) Mutations in ced-1(e2091) and ced-6(n2095) with or without abi-1(n2091 or ok171) in the background were tested for ER stress resistance by tunicamycin (TM) treatment. (B) Mutations in unc-53(e404) and unc-73(e936) were tested for ER stress resistance by tunicamycin treatment. (C) Mutations in pqn-41(ns924) with and without pgrn-1(tm985) in the background were tested for ER stress resistance by tunicamycin treatment. Results shown are representative of at least 2 experiments except in the case of (C) which was performed once. Error bars represent standard deviation. Statistical comparisons are to N2 control (ANOVA with Bonferroni post-tests). *p<0.05, **p<0.01, ***p<0.001. For additional statistical data, see Supplemental Tables S3E–G.
(TIFF)
Related to Figure 4. Levels of spliced xbp-1 mRNA is unchanged in pgrn-1(-), ced-1(-) or ced-3(-) mutants compared to wild type. Representative RT-PCR of xbp-1 from RNA isolated from day 1 animals. Arrows point to the unspliced and spliced xbp-1 bands and the loading control actin band. + TM refers to treatment with 5 mg/mL tunicamycin.
(TIFF)
Related to Figure 4. HSP-4 does not mediate stress resistance of pgrn-1(-) or ced-3(-) mutants. Baseline levels of HSP-4::GFP in wild-type, pgrn-1(-) and ced-3(-) animals. Animals expressing Phsp-4::hsp-4::gfp in an otherwise wild-type, pgrn-1(tm985) or ced-3(n717) background were synchronized by washing adults and larvae away from eggs. L1 animals were collected from hatched embryos 1 hour later while L2, L3 and L4 larval stages were identified by size and anatomical landmarks. All animals were imaged and fluorescence quantified as in Experimental Methods.
(TIFF)
Related to Figure 4. DAF-16 nuclear localization is unchanged in pgrn-1(-), ced-1(-) or ced-3(-) mutants compared to wild type. Day 1 daf-16(-) adult animals expressing Pdaf-16::daf-16::GFP in an otherwise wild-type, daf-2(e1370), pgrn-1(tm985), ced-1(e1735) or ced-3(n717) background were imaged on a Zeiss AxioImager and DAF-16::GFP was scored as either nuclear or diffuse. Images shown are representative of 10 to 15 animals observed for each genotype except for ced-3(-) for which three animals were observed. Top row shows two representative animals and bottom row displays close-ups of these animals.
(TIFF)
Lifespan analysis of ced mutants. Survival of N2 Control, ced-3(n717), ced-1(e1735) and ced-2(e1752) animals was plotted across time. Compared to control, ced-3(-) animals lived significantly longer (p = 0.0007, Mantel-Cox Test). N = 100 animals per strain.
(TIFF)
TNFR/neurotrophin receptor-related genes are not resistant to ER stress. (A) A C. elegans TRAF mutant, trf-1(nr2010), was tested for stress resistance. (B) Two alleles of the neurotrophin receptor trk-1 (tm3985 and tm4054) were tested for ER stress resistance. Error bars represent standard deviation. N = 50 animals in triplicate per strain per condition. *p<0.05, **p<0.01, ***p<0.001.
(TIFF)
Related to Figure 1. pgrn-1 mutants are resistant to osmotic, thermal and unfolded protein stress. (A) Day-1 adult wild-type control animals and pgrn-1(-) mutants were exposed to osmotic stress with 600 mM NaCl for 24 hours and then scored for survival. Shown are mean survival ± SD and p value versus control (Student's t test). (B) Day-1 adult wild-type control animals and pgrn-1(-) mutants were exposed to thermal stress at 35°C for 8 hours and then scored for survival. Shown are mean survival ± SD and p value versus control (Student's t test). (C) Newly-laid wild-type control, pgrn-1(tm985) and pgrn-1(-); pgrn-1-rescue embryos were collected and placed onto plates with varying doses of tunicamycin. Three days later, those animals that had developed to L4 stage were counted. The fraction of animals that developed to L4 stage ± SD are shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests). (D) Newly laid embryos from wild-type control, pgrn-1(tm985) and pgrn-1(-); human PGRN-rescue (2 independent lines) were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD are shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests).
(DOCX)
Related to Figure 2. ced-3(n717lf), ced-4(n162lf) and ced-9(n1950gf) mutations inhibit programmed cell death and confer stress resistance. (A) ER stress resistance is correlated with ced-3 allele strength. Newly laid embryos from wild-type control and ced-3 mutant alleles with graded abilities to inhibit programmed cell death (n717—strong, n1949—intermediate, n2436—intermediate and n2438—weak) were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD is shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests). (B) Day-1 adult wild-type control animals and ced-3(-) mutants were exposed to osmotic stress with 600 mM NaCl for 24 hours or thermal stress at 35°C for 8 hours and scored for survival. Shown are mean survival ± SD and p value versus control (Student's t test). (C) A gain-of-function mutation in ced-9 increases stress resistance. Newly laid wild-type control, pgrn-1(tm985), ced-3(n717), ced-9(n1950gf), pgrn-1(-); ced-9(gf) and ced-3(n717); ced-9(gf) embryos were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD are shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests). (D) Newly laid wild-type control, pgrn-1(tm985), ced-4(n1162lf) and ced-4(n1162lf); ced-9(n2812lf) embryos were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD are shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests).
(DOCX)
Related to Figure 3. Engulfment mutants are resistant to unfolded protein stress. (A) Newly laid wild-type control, pgrn-1(tm985), ced-1(e1735), ced-6(n1813) and ced-7(n1892) embryos were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD are shown. (B) Newly laid wild-type control, pgrn-1(tm985), ced-2(e1752), ced-5(n1812) and ced-10(n3246) embryos were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD are shown. (C) Day 1 adult wild-type control, ced-1(e1735), ced-6(n1813), ced-7(n1892), ced-2(e1752) and ced-5(n1812) mutants were exposed to osmotic stress with 600 mM NaCl for 24 hours or thermal stress at 35°C for 10 hours and then scored for survival. Shown are mean survival ± SD and p value versus control (ANOVA with Tukey post-test). (D) Newly laid wild-type control, pgrn-1(tm985), abl-1(ok171), abi-1(ok640) and abi-1(tm494) embryos were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD are shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests). (E) Newly laid wild-type control, pgrn-1(tm985), ced-1(n2091), ced-1(n2091); abi-1(n1963), ced-6(n2095), and ced-6(n2095); abi-1(n1963) embryos were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD are shown. P value versus control and ced-1 or ced-6 mutants are shown (ANOVA with Bonferroni post-tests). (F) Newly laid wild-type control, pgrn-1(tm985), unc-53(e404), and unc-73(e936) embryos were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD are shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests). (G) Newly laid wild-type control, pgrn-1(tm985), pqn-41(ns294), and pgrn-1(tm985); pqn-41(ns294) embryos were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD are shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests).
(DOCX)
Related to Figure 4. pgrn-1(-) resistance to ER stress may be partially dependent on the UPR pathway, daf-16 and pmk-1. (A) Newly laid embryos from wild-type control, pgrn-1(tm985), ire-1(ok799) and pgrn-1(-); ire-1(-) mutants were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD is shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests). (B) Newly laid embryos from wild-type control, pgrn-1(tm985), daf-2(e1370), pgrn-1(-);daf-2(-), daf-16(mu86) and pgrn-1(-); daf-16(-) mutants were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD is shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests). (C) Newly laid embryos from wild-type control, pgrn-1(tm985), daf-16(mu86); muIs113 and pgrn-1(tm985); muIs113 mutants were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD is shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests). (D) Newly laid embryos from wild-type control, pgrn-1(tm985), pmk-1(km25) and pgrn-1(-); pmk-1(-) mutants were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD is shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests).
(DOCX)
Supplementary dataset of differentially expressed genes. Each worksheet contains lists of genes differentially expressed with FDR<0.05. Lists include log2 fold-changes, p-values, and FDR for each gene. Fold-changes are color coded to denote up- or down-regulation (up-regulated = red, down-regulated = green). Please see first tab for contents of other tabs. Tab E shows genes differentially expressed in pgrn-1(tm985), ced-3(n717), and ced-1(e1735) compared to N2E that are also regulated by daf-16. Hypergeometric distribution, p value<2×10−9.
(XLSX)
Acknowledgments
Some strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440). Additional strains were provided by the labs of Kaveh Ashrafi, Shai Shaham, Robert Horvitz and Shohei Mitani with the National Bioresource Project (Tokyo, Japan). We thank Kaveh Ashrafi, Sivan Korenblit, Arjumand Ghazi, and Michael Cary for helpful discussions regarding the manuscript and Sergio Baranzini and Iryna Lobach for statistical advice.
Funding Statement
This work was supported by an NIH RO1 AG11816 to CK and grants from the Consortium for Frontotemporal Dementia Research (CFR), Hellman Family Foundation, Alzheimer's Disease Research Center, and NIH (K08 NS59604) to AWK. CK is an American Cancer Society Professor and director of the UCSF Hillblom Foundation for the Biology of Aging. MEJ was supported by a post-doctoral fellowship from the Consortium for Frontotemporal Dementia Research (CFR). GC and FG are supported by the NINDS Informatics Center for Neurogenetics and Neurogenomics (P30 NS062691). This study was initiated by AWK while a postdoctoral fellow with CK and continued in her own lab. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Cypser JR, Tedesco P, Johnson TE (2006) Hormesis and aging in Caenorhabditis elegans . Exp Gerontol 41: 935–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gartner A, Boag PR, Blackwell TK (2008) Germline survival and apoptosis. WormBook 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bargmann CI (1993) Genetic and cellular analysis of behavior in C. elegans . Annu Rev Neurosci 16: 47–71. [DOI] [PubMed] [Google Scholar]
- 4. Golden JW, Riddle DL (1982) A pheromone influences larval development in the nematode Caenorhabditis elegans . Science 218: 578–580. [DOI] [PubMed] [Google Scholar]
- 5. Hajdu-Cronin YM, Chen WJ, Sternberg PW (2004) The L-type cyclin CYL-1 and the heat-shock-factor HSF-1 are required for heat-shock-induced protein expression in Caenorhabditis elegans . Genetics 168: 1937–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. An JH, Blackwell TK (2003) SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev 17: 1882–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jiang H, Guo R, Powell-Coffman JA (2001) The Caenorhabditis elegans hif-1 gene encodes a bHLH-PAS protein that is required for adaptation to hypoxia. Proc Natl Acad Sci U S A 98: 7916–7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gartner A, Milstein S, Ahmed S, Hodgkin J, Hengartner MO (2000) A conserved checkpoint pathway mediates DNA damage–induced apoptosis and cell cycle arrest in C. elegans . Mol Cell 5: 435–443. [DOI] [PubMed] [Google Scholar]
- 9. Gumienny TL, Lambie E, Hartwieg E, Horvitz HR, Hengartner MO (1999) Genetic control of programmed cell death in the Caenorhabditis elegans hermaphrodite germline. Development 126: 1011–1022. [DOI] [PubMed] [Google Scholar]
- 10. Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26: 239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoeppner DJ, Hengartner MO, Schnabel R (2001) Engulfment genes cooperate with ced-3 to promote cell death in Caenorhabditis elegans . Nature 412: 202–206. [DOI] [PubMed] [Google Scholar]
- 12. Reddien PW, Cameron S, Horvitz HR (2001) Phagocytosis promotes programmed cell death in C. elegans . Nature 412: 198–202. [DOI] [PubMed] [Google Scholar]
- 13. Elliott MR, Ravichandran KS (2010) Clearance of apoptotic cells: implications in health and disease. J Cell Biol 189: 1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sulston JE, Schierenberg E, White JG, Thomson JN (1983) The embryonic cell lineage of the nematode Caenorhabditis elegans . Dev Biol 100: 64–119. [DOI] [PubMed] [Google Scholar]
- 15. Ellis HM, Horvitz HR (1986) Genetic control of programmed cell death in the nematode C. elegans . Cell 44: 817–829. [DOI] [PubMed] [Google Scholar]
- 16. Hengartner MO, Ellis RE, Horvitz HR (1992) Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature 356: 494–499. [DOI] [PubMed] [Google Scholar]
- 17. Spector MS, Desnoyers S, Hoeppner DJ, Hengartner MO (1997) Interaction between the C. elegans cell-death regulators CED-9 and CED-4. Nature 385: 653–656. [DOI] [PubMed] [Google Scholar]
- 18. Wu D, Wallen HD, Nunez G (1997) Interaction and regulation of subcellular localization of CED-4 by CED-9. Science 275: 1126–1129. [DOI] [PubMed] [Google Scholar]
- 19. Conradt B, Horvitz HR (1998) The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell 93: 519–529. [DOI] [PubMed] [Google Scholar]
- 20. Yuan JY, Horvitz HR (1990) The Caenorhabditis elegans genes ced-3 and ced-4 act cell autonomously to cause programmed cell death. Dev Biol 138: 33–41. [DOI] [PubMed] [Google Scholar]
- 21. Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR (1993) The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell 75: 641–652. [DOI] [PubMed] [Google Scholar]
- 22. Hedgecock EM, Sulston JE, Thomson JN (1983) Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans . Science 220: 1277–1279. [DOI] [PubMed] [Google Scholar]
- 23. Ellis RE, Jacobson DM, Horvitz HR (1991) Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditis elegans . Genetics 129: 79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Conradt B, Xue D (2005) Programmed cell death. WormBook 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zou W, Lu Q, Zhao D, Li W, Mapes J, et al. (2009) Caenorhabditis elegans myotubularin MTM-1 negatively regulates the engulfment of apoptotic cells. PLoS Genet 5: e1000679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kao AW, Eisenhut RJ, Herl Martens L, Nakamura A, Huang A, et al. (2011) A neurodegenerative disease mutation that accelerates the clearance of apoptotic cells. Proc Natl Acad Sci U S A 108: 4441–4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, et al. (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442: 916–919. [DOI] [PubMed] [Google Scholar]
- 28. Cruts M, Kumar-Singh S, Van Broeckhoven C (2006) Progranulin mutations in ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Curr Alzheimer Res 3: 485–491. [DOI] [PubMed] [Google Scholar]
- 29. Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, et al. (2012) Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet 90: 1102–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brouwers N, Nuytemans K, van der Zee J, Gijselinck I, Engelborghs S, et al. (2007) Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch Neurol 64: 1436–1446. [DOI] [PubMed] [Google Scholar]
- 31. Brouwers N, Sleegers K, Engelborghs S, Maurer-Stroh S, Gijselinck I, et al. (2008) Genetic variability in progranulin contributes to risk for clinically diagnosed Alzheimer disease. Neurology 71: 656–664. [DOI] [PubMed] [Google Scholar]
- 32. Sleegers K, Brouwers N, Maurer-Stroh S, van Es MA, Van Damme P, et al. (2008) Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology 71: 253–259. [DOI] [PubMed] [Google Scholar]
- 33. Viswanathan J, Makinen P, Helisalmi S, Haapasalo A, Soininen H, et al. (2009) An association study between granulin gene polymorphisms and Alzheimer's disease in Finnish population. Am J Med Genet B Neuropsychiatr Genet 150B: 747–750. [DOI] [PubMed] [Google Scholar]
- 34. Lee MJ, Chen TF, Cheng TW, Chiu MJ (2011) rs5848 variant of progranulin gene is a risk of Alzheimer's disease in the Taiwanese population. Neurodegener Dis 8: 216–220. [DOI] [PubMed] [Google Scholar]
- 35. Tang W, Lu Y, Tian QY, Zhang Y, Guo FJ, et al. (2011) The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science 332: 478–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matsubara T, Mita A, Minami K, Hosooka T, Kitazawa S, et al. (2012) PGRN is a key adipokine mediating high fat diet-induced insulin resistance and obesity through IL-6 in adipose tissue. Cell Metab 15: 38–50. [DOI] [PubMed] [Google Scholar]
- 37. He Z, Ismail A, Kriazhev L, Sadvakassova G, Bateman A (2002) Progranulin (PC-cell-derived growth factor/acrogranin) regulates invasion and cell survival. Cancer Res 62: 5590–5596. [PubMed] [Google Scholar]
- 38. Ho JC, Ip YC, Cheung ST, Lee YT, Chan KF, et al. (2008) Granulin-epithelin precursor as a therapeutic target for hepatocellular carcinoma. Hepatology 47: 1524–1532. [DOI] [PubMed] [Google Scholar]
- 39. Kamrava M, Simpkins F, Alejandro E, Michener C, Meltzer E, et al. (2005) Lysophosphatidic acid and endothelin-induced proliferation of ovarian cancer cell lines is mitigated by neutralization of granulin-epithelin precursor (GEP), a prosurvival factor for ovarian cancer. Oncogene 24: 7084–7093. [DOI] [PubMed] [Google Scholar]
- 40. Matsumura N, Mandai M, Miyanishi M, Fukuhara K, Baba T, et al. (2006) Oncogenic property of acrogranin in human uterine leiomyosarcoma: direct evidence of genetic contribution in in vivo tumorigenesis. Clin Cancer Res 12: 1402–1411. [DOI] [PubMed] [Google Scholar]
- 41. Pan CX, Kinch MS, Kiener PA, Langermann S, Serrero G, et al. (2004) PC cell-derived growth factor expression in prostatic intraepithelial neoplasia and prostatic adenocarcinoma. Clin Cancer Res 10: 1333–1337. [DOI] [PubMed] [Google Scholar]
- 42. Wang M, Li G, Yin J, Lin T, Zhang J (2011) Progranulin overexpression predicts overall survival in patients with glioblastoma. Med Oncol 29 ((4)): 2423–31. [DOI] [PubMed] [Google Scholar]
- 43. Tao J, Ji F, Wang F, Liu B, Zhu Y (2011) Neuroprotective effects of progranulin in ischemic mice. Brain Res 1436: 130–136. [DOI] [PubMed] [Google Scholar]
- 44. Xu J, Xilouri M, Bruban J, Shioi J, Shao Z, et al. (2011) Extracellular progranulin protects cortical neurons from toxic insults by activating survival signaling. Neurobiol Aging 32: 2326 e2325–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hurwitz ME, Vanderzalm PJ, Bloom L, Goldman J, Garriga G, et al. (2009) Abl kinase inhibits the engulfment of apoptotic cells in Caenorhabditis elegans . PLoS Biol 7: e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Neukomm LJ, Frei AP, Cabello J, Kinchen JM, Zaidel-Bar R, et al. (2011) Loss of the RhoGAP SRGP-1 promotes the clearance of dead and injured cells in Caenorhabditis elegans . Nat Cell Biol 13: 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Neukomm LJ, Nicot AS, Kinchen JM, Almendinger J, Pinto SM, et al. (2011) The phosphoinositide phosphatase MTM-1 regulates apoptotic cell corpse clearance through CED-5-CED-12 in C. elegans . Development 138: 2003–2014. [DOI] [PubMed] [Google Scholar]
- 48. Olsen A, Vantipalli MC, Lithgow GJ (2006) Lifespan extension of Caenorhabditis elegans following repeated mild hormetic heat treatments. Biogerontology 7: 221–230. [DOI] [PubMed] [Google Scholar]
- 49. Urano F, Calfon M, Yoneda T, Yun C, Kiraly M, et al. (2002) A survival pathway for Caenorhabditis elegans with a blocked unfolded protein response. J Cell Biol 158: 639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shaham S, Reddien PW, Davies B, Horvitz HR (1999) Mutational analysis of the Caenorhabditis elegans cell-death gene ced-3 . Genetics 153: 1655–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang X, Chang HY, Baltimore D (1998) Essential role of CED-4 oligomerization in CED-3 activation and apoptosis. Science 281: 1355–1357. [DOI] [PubMed] [Google Scholar]
- 52. deBakker CD, Haney LB, Kinchen JM, Grimsley C, Lu M, et al. (2004) Phagocytosis of apoptotic cells is regulated by a UNC-73/TRIO-MIG-2/RhoG signaling module and armadillo repeats of CED-12/ELMO. Curr Biol 14: 2208–2216. [DOI] [PubMed] [Google Scholar]
- 53. Schmidt KL, Marcus-Gueret N, Adeleye A, Webber J, Baillie D, et al. (2009) The cell migration molecule UNC-53/NAV2 is linked to the ARP2/3 complex by ABI-1. Development 136: 563–574. [DOI] [PubMed] [Google Scholar]
- 54. Blum ES, Abraham MC, Yoshimura S, Lu Y, Shaham S (2012) Control of nonapoptotic developmental cell death in Caenorhabditis elegans by a polyglutamine-repeat protein. Science 335: 970–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Abraham MC, Lu Y, Shaham S (2007) A morphologically conserved nonapoptotic program promotes linker cell death in Caenorhabditis elegans . Dev Cell 12: 73–86. [DOI] [PubMed] [Google Scholar]
- 56. Blum ES, Driscoll M, Shaham S (2008) Noncanonical cell death programs in the nematode Caenorhabditis elegans . Cell Death Differ 15: 1124–1131. [DOI] [PubMed] [Google Scholar]
- 57. Ozcan L, Tabas I (2012) Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu Rev Med 63: 317–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8: 519–529. [DOI] [PubMed] [Google Scholar]
- 59. Shen X, Ellis RE, Lee K, Liu CY, Yang K, et al. (2001) Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development . Cell 107: 893–903. [DOI] [PubMed] [Google Scholar]
- 60. Henis-Korenblit S, Zhang P, Hansen M, McCormick M, Lee SJ, et al. (2010) Insulin/IGF-1 signaling mutants reprogram ER stress response regulators to promote longevity. Proc Natl Acad Sci U S A 107: 9730–9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xu SQ, Tang D, Chamberlain S, Pronk G, Masiarz FR, et al. (1998) The granulin/epithelin precursor abrogates the requirement for the insulin-like growth factor 1 receptor for growth in vitro. J Biol Chem 273: 20078–20083. [DOI] [PubMed] [Google Scholar]
- 62. Zanocco-Marani T, Bateman A, Romano G, Valentinis B, He ZH, et al. (1999) Biological activities and signaling pathways of the granulin/epithelin precursor. Cancer Res 59: 5331–5340. [PubMed] [Google Scholar]
- 63. Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R (1993) A C. elegans mutant that lives twice as long as wild type. Nature 366: 461–464. [DOI] [PubMed] [Google Scholar]
- 64. Lithgow GJ, White TM, Melov S, Johnson TE (1995) Thermotolerance and extended life-span conferred by single-gene mutations and induced by thermal stress. Proc Natl Acad Sci U S A 92: 7540–7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lin K, Hsin H, Libina N, Kenyon C (2001) Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet 28: 139–145. [DOI] [PubMed] [Google Scholar]
- 66. Curran SP, Ruvkun G (2007) Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet 3: e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shore DE, Carr CE, Ruvkun G (2012) Induction of cytoprotective pathways is central to the extension of lifespan conferred by multiple longevity pathways. PLoS Genet 8: e1002792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Inoue H, Hisamoto N, An JH, Oliveira RP, Nishida E, et al. (2005) The C. elegans p38 MAPK pathway regulates nuclear localization of the transcription factor SKN-1 in oxidative stress response. Genes Dev 19: 2278–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Aballay A, Drenkard E, Hilbun LR, Ausubel FM (2003) Caenorhabditis elegans innate immune response triggered by Salmonella enterica requires intact LPS and is mediated by a MAPK signaling pathway. Curr Biol 13: 47–52. [DOI] [PubMed] [Google Scholar]
- 70. Richardson CE, Kinkel S, Kim DH (2011) Physiological IRE-1-XBP-1 and PEK-1 signaling in Caenorhabditis elegans larval development and immunity. PLoS Genet 7: e1002391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hu F, Padukkavidana T, Vægter CB, Brady OA, Zheng Y, et al. (2010) Sortilin-Mediated Endocytosis Determines Levels of the Frontotemporal Dementia Protein, Progranulin. Neuron 68: 654–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Reddien PW, Horvitz HR (2000) CED-2/CrkII and CED-10/Rac control phagocytosis and cell migration in Caenorhabditis elegans . Nat Cell Biol 2: 131–136. [DOI] [PubMed] [Google Scholar]
- 73. Haskins KA, Russell JF, Gaddis N, Dressman HK, Aballay A (2008) Unfolded protein response genes regulated by CED-1 are required for Caenorhabditis elegans innate immunity. Dev Cell 15: 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sun J, Singh V, Kajino-Sakamoto R, Aballay A (2011) Neuronal GPCR controls innate immunity by regulating noncanonical unfolded protein response genes. Science 332: 729–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yeretssian G, Correa RG, Doiron K, Fitzgerald P, Dillon CP, et al. (2011) Non-apoptotic role of BID in inflammation and innate immunity. Nature 474: 96–99. [DOI] [PubMed] [Google Scholar]
- 76. Burguillos MA, Deierborg T, Kavanagh E, Persson A, Hajji N, et al. (2010) Caspase signalling controls microglia activation and neurotoxicity. Nature 472: 319–324. [DOI] [PubMed] [Google Scholar]
- 77. de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, et al. (2011) Caspase activation precedes and leads to tangles. Nature 464: 1201–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gu C, Shabab M, Strasser R, Wolters PJ, Shindo T, et al. (2012) Post-translational regulation and trafficking of the granulin-containing protease RD21 of Arabidopsis thaliana . PLoS One 7: e32422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yamada K, Matsushima R, Nishimura M, Hara-Nishimura I (2001) A slow maturation of a cysteine protease with a granulin domain in the vacuoles of senescing Arabidopsis leaves. Plant Physiol 127: 1626–1634. [PMC free article] [PubMed] [Google Scholar]
- 80. Koizumi M, Yamaguchi-Shinozaki K, Tsuji H, Shinozaki K (1993) Structure and expression of two genes that encode distinct drought-inducible cysteine proteinases in Arabidopsis thaliana . Gene 129: 175–182. [DOI] [PubMed] [Google Scholar]
- 81. Brenner S (1974) The genetics of Caenorhabditis elegans. Genetics 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Frank CA, Baum PD, Garriga G (2003) HLH-14 is a C. elegans achaete-scute protein that promotes neurogenesis through asymmetric cell division. Development 130: 6507–6518. [DOI] [PubMed] [Google Scholar]
- 83. Lamitina ST, Morrison R, Moeckel GW, Strange K (2004) Adaptation of the nematode Caenorhabditis elegans to extreme osmotic stress. Am J Physiol Cell Physiol 286: C785–791. [DOI] [PubMed] [Google Scholar]
- 84. Schuster E, McElwee JJ, Tullet JM, Doonan R, Matthijssens F, et al. (2010) DamID in C. elegans reveals longevity-associated targets of DAF-16/FoxO. Mol Syst Biol 6: 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, et al. (2003) Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans . Nature 424: 277–283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genetic pathways that regulate programmed cell death in apoptotic and engulfing cells in C. elegans. Genes that normally promote cell death and engulfment are in green while those that normally inhibit them are shown in red. Genes tested in this study are in bold.
(TIFF)
Related to Figure 1. A mutation in pgrn-1 does not confer oxidative, UV or pathogen stress resistance. Day 1 adult N2 or pgrn-1(tm985) animals were subjected to oxidative stress with 250 µM paraquat (A), genotoxic stress with 1200 J/m2 UV light (B) or pathogen stress by feeding P. aeruginosa (C). Worms were scored for survival at indicated times and analyses by log-rank Mantel-Cox test were performed. (A) p = 0.1, N>30 animals/strain. (B) p = 0.054, n>50 animals/strain. (C) p = 0.078, N>50 animals/strain. Results shown are representative of at least 2 experiments except in A, which was performed 3 times with p values of 0.0034, 0.01 and 0.125.
(TIFF)
Related to Figure 1. Heat and tunicamycin stress increase HSP-4::GFP levels while osmotic stress does not. (A) Phsp-4::hsp-4::gfp animals were incubated at 20°C (Control treated) or 35°C (Heat stress treated) for 4 hours then imaged on a Zeiss AxioImager. (B) Phsp-4::hsp-4::gfp animals were incubated at 20°C on agarose plates containing 0 µg/mL (Control treated) or 5 µg/mL (ER stress treated) tunicamycin. Twenty-four hours later, they were imaged on a Zeiss AxioImager. (C) Phsp-4::hsp-4::gfp animals were incubated at 20°C on agarose plates containing 51 mM NaCl (Control treated) or 400 mM NaCl (Osmotic stress treated). After 24 hours, they were imaged on a Zeiss AxioImager.
(TIFF)
Related to Figure 1. UV light, paraquat and P. aeruginosa do not increase HSP-4::GFP levels. (A) Phsp-4::hsp-4::gfp animals were mock-treated (Control treated) or exposed to 1200 J/m2 of ultraviolet light (UV stress treated). After 5 hours at 20°C, the animals were imaged on a Zeiss AxioImager. (B) Phsp-4::hsp-4::gfp animals were placed onto agarose plates with either OP50 bacteria (Control treated) or P. aeruginosa PA14 (Pathogen treated) bacteria. After incubation for 24 hours at 20°C, animals were imaged on a Zeiss AxioImager. (C) Phsp-4::hsp-4::gfp animals were placed in M9 solution containing either vehicle alone (Control treated) or 200 mM paraquat (Oxidative stress treated) for 3 hours. The animals that were still alive at that time were imaged on a Zeiss AxioImager.
(TIFF)
Related to Figure 2. A mutation in ced-3 does not confer oxidative or UV stress resistance. Day 1 adult N2 or ced-3(n717) animals were subjected to oxidative stress with 250 µM paraquat (A) or genotoxic stress with 1200 J/m2 UV light (B). Worms were scored for survival at indicated times and analyses by log-rank Mantel-Cox test were performed. (A) p>0.05, N = 20 animals/strain. (B) p>0.05, N>35 animals/strain.
(TIFF)
Related to Figure 3. Mutations in engulfment genes confer resistance to osmotic and heat stresses. Day 1 adult ced-1(e1735) and ced-2(e1752) animals were exposed to 600 mM NaCl for 24 hours (A) or thermal stress at 35°C for 8 hours (B) and scored for survival. Error bars represent standard deviation. Statistical comparisons here are to N2 control (Student's t test or ANOVA with Bonferroni post-tests). *p<0.05, ***p<0.001. For additional statistical data, see Supplemental Table S3C.
(TIFF)
Related to Figure 3. ER stress resistance in related cell death mutants. A) Mutations in ced-1(e2091) and ced-6(n2095) with or without abi-1(n2091 or ok171) in the background were tested for ER stress resistance by tunicamycin (TM) treatment. (B) Mutations in unc-53(e404) and unc-73(e936) were tested for ER stress resistance by tunicamycin treatment. (C) Mutations in pqn-41(ns924) with and without pgrn-1(tm985) in the background were tested for ER stress resistance by tunicamycin treatment. Results shown are representative of at least 2 experiments except in the case of (C) which was performed once. Error bars represent standard deviation. Statistical comparisons are to N2 control (ANOVA with Bonferroni post-tests). *p<0.05, **p<0.01, ***p<0.001. For additional statistical data, see Supplemental Tables S3E–G.
(TIFF)
Related to Figure 4. Levels of spliced xbp-1 mRNA is unchanged in pgrn-1(-), ced-1(-) or ced-3(-) mutants compared to wild type. Representative RT-PCR of xbp-1 from RNA isolated from day 1 animals. Arrows point to the unspliced and spliced xbp-1 bands and the loading control actin band. + TM refers to treatment with 5 mg/mL tunicamycin.
(TIFF)
Related to Figure 4. HSP-4 does not mediate stress resistance of pgrn-1(-) or ced-3(-) mutants. Baseline levels of HSP-4::GFP in wild-type, pgrn-1(-) and ced-3(-) animals. Animals expressing Phsp-4::hsp-4::gfp in an otherwise wild-type, pgrn-1(tm985) or ced-3(n717) background were synchronized by washing adults and larvae away from eggs. L1 animals were collected from hatched embryos 1 hour later while L2, L3 and L4 larval stages were identified by size and anatomical landmarks. All animals were imaged and fluorescence quantified as in Experimental Methods.
(TIFF)
Related to Figure 4. DAF-16 nuclear localization is unchanged in pgrn-1(-), ced-1(-) or ced-3(-) mutants compared to wild type. Day 1 daf-16(-) adult animals expressing Pdaf-16::daf-16::GFP in an otherwise wild-type, daf-2(e1370), pgrn-1(tm985), ced-1(e1735) or ced-3(n717) background were imaged on a Zeiss AxioImager and DAF-16::GFP was scored as either nuclear or diffuse. Images shown are representative of 10 to 15 animals observed for each genotype except for ced-3(-) for which three animals were observed. Top row shows two representative animals and bottom row displays close-ups of these animals.
(TIFF)
Lifespan analysis of ced mutants. Survival of N2 Control, ced-3(n717), ced-1(e1735) and ced-2(e1752) animals was plotted across time. Compared to control, ced-3(-) animals lived significantly longer (p = 0.0007, Mantel-Cox Test). N = 100 animals per strain.
(TIFF)
TNFR/neurotrophin receptor-related genes are not resistant to ER stress. (A) A C. elegans TRAF mutant, trf-1(nr2010), was tested for stress resistance. (B) Two alleles of the neurotrophin receptor trk-1 (tm3985 and tm4054) were tested for ER stress resistance. Error bars represent standard deviation. N = 50 animals in triplicate per strain per condition. *p<0.05, **p<0.01, ***p<0.001.
(TIFF)
Related to Figure 1. pgrn-1 mutants are resistant to osmotic, thermal and unfolded protein stress. (A) Day-1 adult wild-type control animals and pgrn-1(-) mutants were exposed to osmotic stress with 600 mM NaCl for 24 hours and then scored for survival. Shown are mean survival ± SD and p value versus control (Student's t test). (B) Day-1 adult wild-type control animals and pgrn-1(-) mutants were exposed to thermal stress at 35°C for 8 hours and then scored for survival. Shown are mean survival ± SD and p value versus control (Student's t test). (C) Newly-laid wild-type control, pgrn-1(tm985) and pgrn-1(-); pgrn-1-rescue embryos were collected and placed onto plates with varying doses of tunicamycin. Three days later, those animals that had developed to L4 stage were counted. The fraction of animals that developed to L4 stage ± SD are shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests). (D) Newly laid embryos from wild-type control, pgrn-1(tm985) and pgrn-1(-); human PGRN-rescue (2 independent lines) were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD are shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests).
(DOCX)
Related to Figure 2. ced-3(n717lf), ced-4(n162lf) and ced-9(n1950gf) mutations inhibit programmed cell death and confer stress resistance. (A) ER stress resistance is correlated with ced-3 allele strength. Newly laid embryos from wild-type control and ced-3 mutant alleles with graded abilities to inhibit programmed cell death (n717—strong, n1949—intermediate, n2436—intermediate and n2438—weak) were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD is shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests). (B) Day-1 adult wild-type control animals and ced-3(-) mutants were exposed to osmotic stress with 600 mM NaCl for 24 hours or thermal stress at 35°C for 8 hours and scored for survival. Shown are mean survival ± SD and p value versus control (Student's t test). (C) A gain-of-function mutation in ced-9 increases stress resistance. Newly laid wild-type control, pgrn-1(tm985), ced-3(n717), ced-9(n1950gf), pgrn-1(-); ced-9(gf) and ced-3(n717); ced-9(gf) embryos were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD are shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests). (D) Newly laid wild-type control, pgrn-1(tm985), ced-4(n1162lf) and ced-4(n1162lf); ced-9(n2812lf) embryos were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD are shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests).
(DOCX)
Related to Figure 3. Engulfment mutants are resistant to unfolded protein stress. (A) Newly laid wild-type control, pgrn-1(tm985), ced-1(e1735), ced-6(n1813) and ced-7(n1892) embryos were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD are shown. (B) Newly laid wild-type control, pgrn-1(tm985), ced-2(e1752), ced-5(n1812) and ced-10(n3246) embryos were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD are shown. (C) Day 1 adult wild-type control, ced-1(e1735), ced-6(n1813), ced-7(n1892), ced-2(e1752) and ced-5(n1812) mutants were exposed to osmotic stress with 600 mM NaCl for 24 hours or thermal stress at 35°C for 10 hours and then scored for survival. Shown are mean survival ± SD and p value versus control (ANOVA with Tukey post-test). (D) Newly laid wild-type control, pgrn-1(tm985), abl-1(ok171), abi-1(ok640) and abi-1(tm494) embryos were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD are shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests). (E) Newly laid wild-type control, pgrn-1(tm985), ced-1(n2091), ced-1(n2091); abi-1(n1963), ced-6(n2095), and ced-6(n2095); abi-1(n1963) embryos were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD are shown. P value versus control and ced-1 or ced-6 mutants are shown (ANOVA with Bonferroni post-tests). (F) Newly laid wild-type control, pgrn-1(tm985), unc-53(e404), and unc-73(e936) embryos were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD are shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests). (G) Newly laid wild-type control, pgrn-1(tm985), pqn-41(ns294), and pgrn-1(tm985); pqn-41(ns294) embryos were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD are shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests).
(DOCX)
Related to Figure 4. pgrn-1(-) resistance to ER stress may be partially dependent on the UPR pathway, daf-16 and pmk-1. (A) Newly laid embryos from wild-type control, pgrn-1(tm985), ire-1(ok799) and pgrn-1(-); ire-1(-) mutants were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD is shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests). (B) Newly laid embryos from wild-type control, pgrn-1(tm985), daf-2(e1370), pgrn-1(-);daf-2(-), daf-16(mu86) and pgrn-1(-); daf-16(-) mutants were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD is shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests). (C) Newly laid embryos from wild-type control, pgrn-1(tm985), daf-16(mu86); muIs113 and pgrn-1(tm985); muIs113 mutants were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD is shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests). (D) Newly laid embryos from wild-type control, pgrn-1(tm985), pmk-1(km25) and pgrn-1(-); pmk-1(-) mutants were collected and placed onto plates with varying doses of tunicamycin. Three days later, the number of animals that had developed to L4 stage was determined. The fraction of animals that developed to L4 stage ± SD is shown. P value versus control and pgrn-1 mutant are shown (ANOVA with Bonferroni post-tests).
(DOCX)
Supplementary dataset of differentially expressed genes. Each worksheet contains lists of genes differentially expressed with FDR<0.05. Lists include log2 fold-changes, p-values, and FDR for each gene. Fold-changes are color coded to denote up- or down-regulation (up-regulated = red, down-regulated = green). Please see first tab for contents of other tabs. Tab E shows genes differentially expressed in pgrn-1(tm985), ced-3(n717), and ced-1(e1735) compared to N2E that are also regulated by daf-16. Hypergeometric distribution, p value<2×10−9.
(XLSX)