

Abstract
L-dopa-induced dyskinesias (LIDs) are a side effect of Parkinson’s disease therapy that is thought to arise, at least in part, because of excessive dopaminergic activity. Thus, drugs that regulate dopaminergic tone may provide an approach to manage LIDs. Our previous studies showed that nicotine treatment reduced LIDs in parkinsonian animal models. The present work investigates whether nicotine may exert its beneficial effects by modulating presynaptic dopaminergic function. Rats were unilaterally lesioned by injection of 6-OHDA (2 × 3 ug per site) into the medial forebrain bundle to yield moderate parkinsonism. They were then implanted with minipumps containing vehicle or nicotine (2.0 mg/kg/d) and rendered dyskinetic with L-dopa (8 mg/kg plus 15 mg/kg benserazide). Lesioning alone decreased the striatal dopamine transporter, nicotinic receptor (nAChR) levels and nAChR-mediated 3H-dopamine release, consistent with previous results. Nicotine administration reduced L-dopa induced abnormal involuntary movements throughout the course of the study (4 months). Nicotine treatment led to declines in the striatal dopamine transporter, α6β2* nAChRs and various components of α6β2* and α4β2* nAChR-mediated release. L-dopa treatment had no effect. These data suggest that nicotine may improve LIDs in parkinsonian animal models by dampening striatal dopaminergic activity.
Keywords: LIDs, dopamine, nicotine, nicotinic receptors, nigrostriatal lesion
Introduction
Parkinson’s disease symptoms are greatly improved by dopamine replacement therapy with L-dopa, but within a few years of treatment unwanted side effects such as abnormal involuntary movements or dyskinesias develop, which may be mild to severely incapacitating (Rascol et al. 2011, Schapira & Jenner 2011). Current treatments to reduce L-dopa-induced dyskinesias (LIDs) are of limited success and include reductions in L-dopa dose, adjunct therapy with amantadine or surgical intervention (Rascol et al. 2011, Schapira & Jenner 2011). Further studies to identify the mechanisms that contribute to LIDs are thus important as they may yield better therapeutic strategies.
Extensive evidence indicates that both pre- and postsynaptic dopaminergic factors contribute to the development of LIDs. Presynaptic mechanisms of particular relevance include the nigrostriatal dopaminergic neuronal loss that results in a decreased dopamine buffering capacity and the increased extracellular dopamine levels that arise with L-dopa treatment (Carta & Bezard 2011, Cenci 2007, Lindgren et al. 2009, Fisone & Bezard 2011). Typical oral L-dopa doses produce large transient increases in the levels of dopamine in striatum of patients affected by LIDs (Pavese et al. 2006). Animal models also reveal excessive extracellular striatal dopamine concentrations after L-dopa treatment (Meissner et al. 2006, Lundblad et al. 2009, Carta & Bezard 2011). The loss of presynaptic dopamine transporters responsible for the clearance of dopamine from the synaptic cleft, coupled with increased dopamine synthesis, is proposed to contribute to the enhanced dopaminergic activity with L-dopa use (Murer & Moratalla 2011, Lundblad et al. 2009, Carta & Bezard 2011). Thus, drugs that normalize striatal dopaminergic tone may reduce LIDs. Since nicotinic acetylcholine receptors (nAChRs) are significant modulators of striatal dopamine release, balancing dopaminergic tone via nAChR regulation may provide an approach to attenuate LIDs.
Evidence for a role for nAChRs is based on studies showing that both nicotine and nAChR agonists reduce LIDs (Quik et al. 2007, Quik et al. 2012a, Quik et al. 2012b, Bordia et al. 2008, Bordia et al. 2010, Huang et al. 2011a, Huang et al. 2011b). Notably, a nAChR antagonist mecamylamine also reduced LIDs (Bordia et al. 2010). A similar effect of both nAChR agonists and antagonists suggests that agonists may induce their effects by nAChR desensitization, which would effectively result in a functional blockade. This interpretation is consistent with recent studies suggesting that nAChR desensitization may represent a mechanism whereby nicotine and nAChR drugs modulate alcoholism, addiction and anxiety (Dopico & Lovinger 2009, Anderson & Brunzell 2012).
Numerous nAChRs are expressed on different neuronal elements in the nigrostriatal pathway. Within the large family of striatal nAChRs, the majority contain the β2 subunit. The α6β2* nAChRs are exclusively expressed on dopaminergic neurons while α4β2* receptors are located on both dopaminergic and non-dopaminergic neurons (Zoli et al. 2002, Grady et al. 2010b, Gotti et al. 2010, Quik & Wonnacott 2011). The asterisk indicates the possible presence of other subunits in the receptor complex. Both α6β2* and α4β2* receptors are decreased with nigrostriatal degeneration (Quik et al. 2003, Perez et al. 2009) and play an important role in nicotine-mediated reduction in L-dopa induced dyskinesias (Quik et al. 2012b, Huang et al. 2011b).
The objective of the present study was to understand the mechanisms whereby nicotine treatment may reduce LIDs. To approach this, 6-OHDA lesioned rats, treated with or without nicotine, were rendered dyskinetic using L-dopa. They were then killed and striatal dopaminergic function, specifically striatal 3H-dopamine release, was assessed. Long-term nicotine treatment led to significant decreases in various dopaminergic measures, nAChRs and nAChR-mediated dopamine release in striatum. Overall, these data suggest that the antidyskinetic effect of nicotine in moderate Parkinson’s disease may be due to its ability to reduce dopaminergic tone via an interaction at nAChRs.
Materials and methods
Lesioning and parkinsonian rating
Adult male Sprague-Dawley rats (200–250 g) were purchased from Charles River (Livermore, CA). They were housed 3–4 per cage under standard conditions, with free access to food and water. Three days after arrival, they were unilaterally lesioned with 6-hydroxydopamine (6-OHDA, Sigma-Aldrich, St. Louis, MO) under isofluorane anesthesia. 6-OHDA was dissolved in 0.02% ascorbic acid/saline (1.5 μg free base/μl) with a 2 μl aliquot stereotaxically injected at two sites in the medial forebrain bundle (Cenci & Lundblad 2007, Bordia et al. 2008, Bordia et al. 2010). All procedures conformed to the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee. Three to 4 wk following lesioning, parkinsonism was evaluated using the forelimb use asymmetry test (Schallert et al. 2000, Bordia et al. 2008, Bordia et al. 2010).
Nicotine treatment
Four weeks following 6-OHDA lesioning, the rats were randomly divided into two groups and surgically implanted with osmotic minipumps (Fig. 1) (Alzet model 2006; Durect Corporation, Cupertino, CA) releasing vehicle or 2 mg/kg/d nicotine (free base; Sigma-Aldrich Co., St. Louis, MO). Our previous work had shown that minipumps releasing 2 mg/kg/d nicotine resulted in plasma cotinine values comparable to those in smokers (Hukkanen et al. 2005, Bordia et al. 2008, Bordia et al. 2010). Cotinine levels were not measured in the current study. The minipumps were replaced every two months.
Fig. 1.
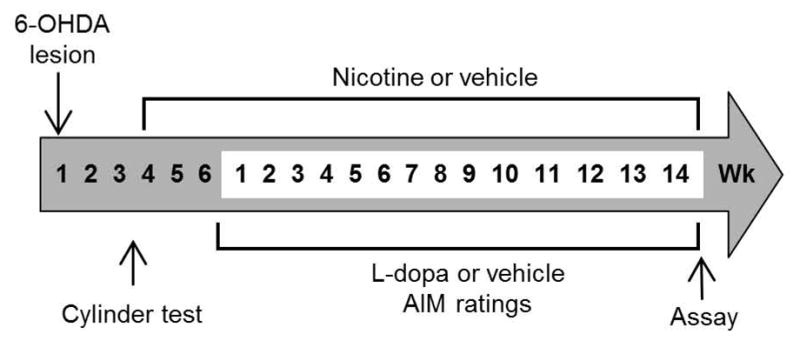
Treatment timeline depicting 6-OHDA lesioning, drug treatment and behavioral testing.
L-dopa treatment and AIM ratings
Two week after minipump implantation, rats were administered L-dopa methyl ester (8 mg/kg, sc, once daily until the end of the study) plus benserazide (15 mg/kg, sc) (Sigma-Aldrich Co., St. Louis, MO) or vehicle (Fig 1). L-dopa-induced abnormal involuntary movements (AIMs) were then rated 3 wk after the start of L-dopa treatment, at which time they have maximally developed, as described (Cenci & Lundblad 2007, Bordia et al. 2008, Bordia et al. 2010). The total maximum AIM score for each rat was 108.
Tissue preparation
Rats were killed by decapitation, and the brains rapidly removed. They were then bisected coronally at a mid-striatal level, with the striatum from the anterior portion used for 3H-dopamine release and the striatum from the caudal portion frozen in isopentane on dry ice and stored at −80°C. The caudal portion was later cut into 8 μm sections at −15°C in a cryostat (Leica Microsystems Inc., Deerfield, IL). The sections were thaw-mounted onto poly-L-lysine coated slides, dried, and stored at −80°C.
Nicotine-evoked 3H-dopamine release
Nicotine-evoked 3H-dopamine release from the rat striatal synaptosomes was performed as described (Bordia et al. 2012, Quik et al. 2003). About 15 mg of striatal tissue was homogenized in 2 ml of ice-cold 0.32 M sucrose buffered with 5.0 mM HEPES (pH 7.5) and then centrifuged. The pellets were re-suspended and processed for measurement of 3H-dopamine release. Data were analyzed using the curve-fitting algorithm of SigmaPlot (Systat Software Inc, San Jose, CA). CPMs were calculated as counts over basal release from samples prior to and following nicotine stimulation, with results expressed as cpm/mg tissue.
Autoradiography
125I-RTI-121 (specific activity, 2200 Ci/mmol; PerkinElmer Life and Analytical Sciences, Waltham, MA) autoradiography was performed to determine the dopamine transporter levels (Quik et al. 2003, Bordia et al. 2010). Nonspecific binding was determined in the presence of the uptake inhibitor nomifensine (100 μM).
125I-α-conotoxinMII (α-CtxMII; specific activity, 2200 Ci/mmol) autoradiography was done to measure α6β2* nAChRs (Quik et al. 2003, Bordia et al. 2010). Striatal α4β2* nAChRs levels were determined using 125I-epibatidine (specific activity, 2200 Ci/mmol; PerkinElmer Life and Analytical Sciences, Waltham, MA) autoradiography in the presence of 100 nM α-CtxMII (Quik et al. 2003, Bordia et al. 2010). Nicotine (100 μM) was used to assess nonspecific binding in both studies.
Data analyses
Autoradiographic images were quantitated using the ImageQuant system (GE Healthcare (Chalfront, St. Giles, Buckinghamshire, UK). Standard curves, generated using 3H-microscale standards, were calibrated for 125I autoradiography as described (Artymyshyn et al. 1990). A statistical comparison for data involving 3 study groups (lesion x L-DOPA x nicotine) was done using 3-way ANOVA to identify significance, using SPSS. Lower order ANOVAs were then used to identify specific effects using GraphPad Prism (San Diego, CA) as indicated in the figure legends. A value of p = 0.05 was considered statistically significant. Values are expressed as the mean ± S.E.M. of the indicated number of rats.
Results
Characterization of the treatment groups
Rats were administered two intracranial injections of 6-OHDA (3 μg per site) into the right medial forebrain bundle as this paradigm generated moderate nigrostriatal damage with only 50–60% loss of striatal dopamine terminals (Bordia et al. 2008, Huang et al. 2011b). The maintenance of a residual population of dopamine terminals is important as our earlier work had shown that nicotine best reduces L-dopa-induced dyskinesias in animals with moderate nigrostriatal damage (Huang et al. 2011a, Quik et al. 2012a, Quik et al. 2012b). As well, a partial lesion was essential as this would allow for measureable dopamine release from the remaining terminals.
Parkinsonism was measured 3 to 4 wk after 6-OHDA administration when the degenerative effects of the toxin are complete. The forepaw placement or cylinder test was used because it allows for a functional assessment without the use of dopaminergic stimulant drugs (Schallert et al. 2000, Bordia et al. 2008, Bordia et al. 2010). The overall % forelimb use on the side affected by the lesion was 23 ± 1.6% (n = 45), indicative of a partial lesion. The rats were then divided into 4 groups such that the % forelimb use in each group was similar, as follows: no nicotine and no L-dopa group, 26 ±3.1% (n = 9); no nicotine and L-dopa group, 21 ± 3.6% (n = 13); nicotine and no L-dopa group, 22 ± 3.7% (n = 11); and nicotine and L-dopa group, 23 ± 3.0% (n = 12).
Two wk after implantation of minipumps containing nicotine or vehicle, the rats were treated with 8 mg/kg L-dopa methyl ester plus benserazide once daily sc (Fig. 1). We used this L-dopa dose as it is within the standard dose range (6 to 10 mg/kg per injection) for rat dyskinesia studies (Cenci & Lindgren 2007). AIMs were tested 3 wk after the start of L-dopa treatment, when they have maximally developed (Cenci & Lundblad 2007). Although the lesion yielded only moderate nigrostriatal damage, we consistently observed the development of L-dopa-induced AIMs as in our previous studies (Bordia et al. 2008, Huang et al. 2011b).
Nicotine treatment significantly decreased total AIMs at 3 wk (p < 0.05), an effect that persisted throughout the study with 40–50% decreases in total AIMs at wk 8 (p < 0.01) and wk 12 (p < 0.05) (Fig. 2, top panels). The daily time course of total L-dopa-induced AIMs at wk 12 (bottom panel left) shows that AIMs were significantly reduced at most time points after L-dopa administration, with a significant main effect of treatment (p < 0.001) and time (p < 0.001), but no interaction (two-way ANOVA). Analyses of the individual AIM components showed that nicotine significantly lowered oral (p < 0.01) and forelimb AIMs (p < 0.05) (Fig. 2 lower right panels), with no significant effect on axial AIMs (data not shown). These data are consistent with our previous studies which showed that the development of axial AIMs is variable in rats with moderate nigrostriatal damage (Bordia et al. 2008, Bordia et al. 2010, Huang et al. 2011b). When axial AIMs did arise, the effect of nicotine and nicotinic agonists to reduce this type of AIM was also variable (Bordia et al. 2008, Bordia et al. 2010, Huang et al. 2011b).
Fig. 2.

Nicotine treatment reduced L-dopa-induced AIMs in rats. 6-OHDA lesioned rats were implanted with minipumps containing nicotine (2 mg/kg) or vehicle. They were then injected with 8 mg/kg L-dopa methyl ester plus benserazide and AIMs rated as depicted in Fig. 1. AIMs were rated 3 wk after the start of L-dopa treatment with significant declines at 3, 8 and 12 wk after L-dopa treatment (top panels). The daily time course of total AIMs at wk 12 (bottom panel left) shows a reduction in AIMs at all time points after L-dopa administration using two-way ANOVA followed by a Bonferroni post hoc test. The nicotine-mediated decline in total AIMs was due to a reduction in oral and forelimb AIMs (bottom panels right) using Student’s t-test. Values are the mean ± S.E.M. of 9–13 rats. Significance of difference from control group, *p < 0.05, **p < 0.01, ***p < 0.001.
Long-term nicotine treatment decreases total nicotine-evoked dopamine release from rat striatal synaptosomes
Nicotine is well known to regulate striatal dopamine release (Bordia et al. 2012, Grady et al. 2010a, Marks et al. 2009). Because dopamine has a major role in the control of locomotor activity and because aberrant dopaminergic activity underlies the development of LIDs, we investigated whether alterations in dopaminergic function may contribute to nicotine’s improvements in LIDs.
To approach this, we tested the long-term effect of nicotine treatment on nAChR-evoked 3H-dopamine release from striatal synaptosomes. Release is provided as total counts (cpms) of stimulated 3H-dopamine release minus baseline or unstimulated release. Since the lesion itself decreases release, release was not normalized relative to baseline as this may obscure the actual results. 3H-dopamine release was measured in response to both 1.0 and 10.0 μM nicotine to examine responsiveness with submaximal and maximal stimulation, respectively. Table 1 provides the striatal 3H-dopamine release values, expressed as cpm/mg tissue, in response to 1.0 μM and 10.0 μM nicotine for control unlesioned rats. Release was about half-maximal at 1.0 μM nicotine and maximal at 10.0 μM nicotine.
Table 1.
| nAChRs |
3H-dopamine release (cpm/mg tissue)
|
|||
|---|---|---|---|---|
| 1.0 μM nicotine | 10 μM nicotine | |||
|
| ||||
| Intact side | Lesioned side | Intact side | Lesioned side | |
| Total | 5,148 ± 908 | 1923 ± 688* | 12,491 ± 1,690 | 6,558 ± 2012* |
| α4β2* | 2,857 ± 494 | 1429 ± 489* | 7,125 ± 940 | 4,025 ± 1164* |
| α6β2* | 2,291 ± 532 | 746 ± 299* | 5,366 ± 884 | 2421 ± 972* |
Synaptosomal 3H-dopamine release was measured from intact and lesioned striatum. Release was stimulated by either 1.0 or 10 μM nicotine to obtain a measure of submaximal and maximal 3H-dopamine release, respectively. Values represent the mean ± SEM of 9 rats. Significance of difference from the intact side using Student’s t-test,
p < 0.05.
6-OHDA lesioning alone resulted in significant declines (p < 0.05) in total nAChR-mediated 3H-dopamine release (Table 1) on the lesioned compared to the intact side, in agreement with previous findings (Quik et al. 2003). Total dopamine release was reduced by 63% at 1 μM nicotine stimulation and 48% using 10 μM nicotine.
Long-term nicotine treatment reduced 1.0 μM nicotine-evoked 3H-dopamine release by ~40% in the lesioned striatum of rats treated with and without L-dopa, with a significant main effect of nicotine (p < 0.05) using two-way ANOVA (Fig. 3). There was also a trend for a decline in release in the intact striatum with nicotine treatment (Fig. 3). In addition, there was a significant main effect of nicotine (p < 0.05) using 10.0 μM nicotine to evoke 3H-dopamine release in both the intact and lesioned striatum of rats treated with and without L-dopa (Fig. 3). L-dopa administration had no effect on nicotine-evoked 3H-dopamine release. This finding was unexpected, but most likely reflects the fact that we are measuring release of exogenously added 3H-dopamine and not endogenous dopamine stores that may be increased with L-dopa treatment.
Fig. 3.

Nicotine treatment decreases total nicotine-evoked dopamine release from rat striatal synaptosomes. 6-OHDA-lesioned rats, implanted with minipumps containing nicotine (Nic, 2 mg/kg/d) or vehicle, were injected with 8 mg/kg L-dopa methyl ester (LD) plus benserazide until the end of the study. They were then killed 60 min after L-dopa administration, when its effects are maximal. NAChR-mediated 3H-dopamine release was determined in response to 1 and 10 μM nicotine. 6-OHDA lesioning resulted in ~50% decline in dopamine release. Nicotine treatment significantly reduced 3H-dopamine release in all cases, except with 1 μM nicotine stimulation in intact striatum. Data are expressed as % intact control, that is, the intact side of animals not treated with nicotine and L-dopa. Values represent the mean ± S.E.M. of 9–13 rats. Significant main effect of nicotine (p < 0.05) using two-way ANOVA.
Long-term nicotine treatment decreases high affinity α6β2* nAChR-mediated release from striatal synaptosomes
The striatum contains two major nAChRs populations, the α4β2* and α6β2* subtypes (Quik & Wonnacott 2011). To determine how nicotine treatment modified α4β2* and α6β2* nAChR release, 3H-dopamine release was measured from striatal synaptosomes in the absence and presence of α-CtxMII, which blocks α6β2* nAChRs. Release measured in the presence of α-CtxMII represents that occurring via α4β2* nAChRs, while the difference between total andα4β2* nAChR release is defined as α6β2* nAChR-mediated release. 6-OHDA lesioning alone resulted in significant declines in α6β2* nAChR-mediated release (Fig. 4, Table 1). Dopamine release was reduced by 67% at 1 μM nicotine stimulation (p < 0.05) and 55% using 10 μM nicotine (p < 0.05). L-dopa administration had no effect on 3H-dopamine release.
Fig. 4.

Nicotine treatment decreases α6β2* nAChR-mediated dopamine release from rat striatal synaptosomes. Striatal synaptosomes were prepared from 6-OHDA-lesioned rats, treated with and without nicotine (Nic) and/or L-dopa (LD). At the end of the treatment phase, the rats were killed and synaptosomal 3H-dopamine release determined in response to 1 and 10 μM nicotine. 6-OHDA lesioning resulted in ~50% decline in α6β2* nAChR-mediated release. Nicotine treatment significantly reduced 3H-dopamine release with submaximal (1 μM) nicotine stimulation. Values represent the mean ± S.E.M. of 9–13 rats. Significant main effect of nicotine (*p < 0.05) using two-way ANOVA.
Long-term nicotine treatment reduced α6β2* nAChR-mediated 3H-dopamine release by ~50% with submaximal (1 μM) nicotine stimulation, with a significant main effect of nicotine (p < 0.05) on both the intact and lesioned side (Fig. 4). By contrast, long-term nicotine dosing did not reduce release when evoked with 10 μM nicotine. This suggests that a specific α6β2* nAChR subtype is involved.
Long-term nicotine treatment decreases lower affinity α4β2* nAChR-mediated release from striatal synaptosomes
Lesioning alone also led to significant declines in α4β2* nAChR-evoked 3H-dopamine release from striatal synaptosomes (Table 1). Dopamine release was reduced by 50% at 1 μM nicotine stimulation (p < 0.05) and 44% using 10 μM nicotine (p < 0.05) (Table 1). L-dopa administration again did not modulate 3H-dopamine release under any condition (Fig. 5).
Fig. 5.

Nicotine treatment decreases α4β2* nAChR-mediated dopamine release from rat striatal synaptosomes. Striatal synaptosomes were prepared from 6-OHDA-lesioned rats, treated with and without nicotine (Nic) and/or L-dopa (LD). At the end of the treatment phase, the rats were killed and synaptosomal 3H-dopamine release determined in response to 1 and 10 μM nicotine. 6-OHDA lesioning resulted in ~50% decline in α4β2* nAChR-mediated release. Nicotine treatment significantly reduced release on both the intact and lesioned side at maximal nicotine stimulation (10 μM). Values represent the mean ± S.E.M. of 9–13 rats. Significant main effect of nicotine (*p < 0.05, **p < 0.01) using two-way ANOVA.
Long-term nicotine treatment reduced release on both the intact and lesioned side at maximal nicotine stimulation (10 μM), with significant main effects of nicotine of p < 0.05 and p < 0.01, respectively on the two sides. However, long-term nicotine treatment did not modify release on either the intact or lesioned side at the 1.0 μM nicotine concentration. These observations again suggests that long-term nicotine treatment selectively affects function mediated via a specific nAChR subtype(s). The two primary α4β2* nAChR subtypes in striatum are the α4β2 andα4α5β2 receptors, with the former thought to be of lower affinity (Grady et al. 2010b). These data thus suggest that the declines in nAChR-mediated dopamine release may be mediated via the lower affinity α4β2 nAChR population, as further clarified in the Discussion.
Basal 3H-dopamine release with nicotine and L-dopa treatment in intact and lesioned striatum
In the above studies, we show the effect of chronic nicotine treatment on nAChR-evoked 3H-dopamine release, which provides a measure of release in response to drug stimulation. We also assessed basal activity that reflects tonic release in the absence of stimulation. The data in Table 2 show that lesioning alone significantly decreased basal 3H-dopamine release, as previously shown (Quik et al., 2003). There was no further decrease with either L-dopa and/or nicotine treatment. The lack of effect of chronic nicotine treatment on basal release suggests that nicotine selectively modifies mechanisms linked to presynaptic nAChRs to reduce dopamine release.
Table 2.
6-OHDA lesioning decreased basal 3H-dopamine release
| Group | Treatment | Basal 3H-dopamine release (cpm/mg tissue) | ||
|---|---|---|---|---|
|
| ||||
| L-dopa | Nicotine | Intact side | Lesioned side | |
| 1 | No | No | 2,016 ± 164 | 1,396 ± 199* |
| 2 | Yes | No | 1,990 ± 78 | 1,298 ± 153*** |
| 3 | No | Yes | 2,041 ± 136 | 1,013 ± 188*** |
| 4 | Yes | Yes | 1,842 ± 122 | 1,134 ± 191** |
Basal 3H-dopamine release from striatum was defined as release occurring in the absence of a stimulus. 6-OHDA lesioning decreased basal release, with no effect of either L-dopa and/or nicotine treatments. Values are the mean ± S.E.M. of 9–13 rats. Significance of difference from the intact side using Student’s t-test,
p < 0.05,
p < 0.01,
p < 0.001.
Effect of lesioning and nicotine treatment on striatal dopamine transport
Measurement of the dopamine transporter, a molecular marker of dopamine nerve terminal integrity (Quik et al. 2003) showed that lesioning resulted in ~60% decline, consistent with the 6-OHDA dosing paradigm used in the present experiments (Fig. 6 top panels). Previous studies had suggested that L-dopa-induced (AIMs) develop more readily in rats in which the striatal dopamine depletion >80%. We therefore investigated whether there might be a differential pattern of striatal dopamine transporter loss to explain the consistent occurrence of AIMs in our studies by quantitating the density in different striatal areas. However, no differential losses were observed. Long-term nicotine treatment had no effect on the transporter on the intact side; however, it led to small, but significant reductions on the lesioned side (p < 0.05 and p < 0.01). L-dopa treatment did not alter the dopamine transporter.
Fig. 6.

Effect of lesioning and drug treatment on dopamine transport. The dopamine transporter was measured using 125I-RTI-121 autoradiography, with representative autoradiographic images from the intact and lesioned striatum of nicotine (Nic)-treated rats in the top panels. Quantitative analyses of the data from all treatment groups are provided in the middle panel. Lesioning significantly reduced the transporter by about 60%. Nicotine treatment also led to a small reduction in the transporter on the lesioned side. As an indirect measure of dopamine transport, we also measured synaptosomal 3H-dopamine content when release was complete (bottom panel). Results were similar to those observed with dopamine transporter autoradiography. Values are the mean ± S.E.M. of 9–13 rats. Significance of difference from own intact side: **p < 0.01, ***p < 0.001; significance of difference from own no nicotine-treated group, #p < 0.05, ##p < 0.01 using two-way ANOVA followed by a Bonferroni post hoc test.
As an alternate approach to evaluate dopamine transporter function, we also measured 3H-dopamine content in the synaptosomes when the release studies were completed. The results paralleled those obtained with dopamine transporter autoradiography. 6-OHDA lesioning resulted in ~40% decline in 3H-dopamine content (Fig. 6 bottom panels). Long-term nicotine treatment had no effect on the transporter on the intact side, but resulted in a small, significant decrease on the lesioned side in the rats not treated with L-dopa (p < 0.05). L-dopa treatment had no effect on synaptosomal 3H-dopamine levels.
Effect of lesioning and drug treatment on striatal α6β2* and α4β2* nAChRs
α6β2* nAChRs were measured using the α6β2* nAChR selective radioligand 125I-α-CtxMII (Fig. 7). As previously shown, 6-OHDA lesioning decreased α6β2* nAChRs in parallel with the dopamine transporter (Lai et al. 2005, Perry et al. 2007, Mugnaini et al. 2006). Nicotine treatment also significantly decreased 125I-α-CtxMII binding on the intact and lesioned side, consistent with previous findings (Quik et al. 2003). L-dopa treatment had no effect on α6β2* receptor levels.
Fig. 7.

Effect of lesioning and drug treatment on striatal α6β2* nAChRs. α6β2* nAChR were then measured using 125I-α-CtxMII autoradiography. Representative autoradiograms from the intact and lesioned striatum of nicotine (Nic)-treated rats are depicted in the upper panels. Quantitative analyses of the data from all treatment groups are provided in the lower panel. Lesioning significantly reduced binding. Nicotine treatment also decreased binding on the lesioned and intact side. Values are the mean ± S.E.M. of 9–13 rats. Significance of difference from own intact side: ***p < 0.001; significance of difference from own no nicotine-treated group, #p < 0.05, ##p < 0.01, ###p < 0.001 using two-way ANOVA followed by a Bonferroni post hoc test.
α4β2* nAChRs were quantified using 125I-epibatidine in the presence of α-CtxMII to block binding of the radioligand to α6β2* nAChRs (Fig. 8). Since only 20–30% of the α4β2* nAChRs are present on striatal dopamine terminals, nigrostriatal damage resulted in only a small (~15%) decline (p < 0.05) in this subtype, in agreement with previous findings (Quik et al. 2003). L-dopa treatment did not affect striatal 125I-epibatidine binding compared to vehicle under any treatment condition. Consistent with earlier studies (Marks et al. 1992), long-term nicotine treatment enhanced α4β2* nAChR levels (p < 0.001) in striatum.
Fig. 8.

Effect of lesioning and drug treatment on striatal α4β2* nAChRs. α4β2* nAChRs were measured using 125I-epibatidine autoradiography in the presence of α-CtxMII. Representative autoradiograms from the intact and lesioned striatum of nicotine (Nic)-treated rats are depicted in the upper panels. Quantitative analyses of the data from all treatment groups are provided in the lower panel. Lesioning led to a small decline in binding. Nicotine treatment increased binding on both the lesioned and intact side. Values are the mean ± S.E.M. of 9–13 rats. Significance of difference from own intact side: *p < 0.05; significance of difference from own no nicotine-treated group, ###p < 0.001 using two-way ANOVA followed by a Bonferroni post hoc test.
Discussion
Aberrant presynaptic dopaminergic activity is thought to have a major impact in the development of LIDs (Brotchie & Jenner 2011, Carta & Bezard 2011, Fisone et al. 2007, Bezard et al. 2001, Jenner 2008, Cenci & Konradi 2010). The nigrostriatal dopamine terminal loss that occurs in Parkinson’s disease and the excessive dopaminergic stimulation in response to intermittent L-dopa dosing both appear to play a critical role. These findings suggest that treatments that modulate striatal dopaminergic tone may improve LIDs.
Our work showed that nicotine and nAChR agonists reduce LIDs in parkinsonian rats, mice and monkeys by acting, in part, at nAChRs on nigrostriatal dopamine terminals (Quik et al. 2007, Quik et al. 2012a, Quik et al. 2012b, Bordia et al. 2008, Bordia et al. 2010, Huang et al. 2011a, Huang et al. 2011b). This finding was initially unexpected since acute nAChR stimulation enhances dopamine release (Gotti et al. 2010, Quik & Wonnacott 2011) and thus might be expected to worsen LIDs. However, long-term nicotine administration has been shown to elicit a very different response, with nAChR desensitization and/or downregulation and corresponding declines in dopaminergic function (Picciotto et al. 2008, Buccafusco et al. 2009). The goal of the present experiments was to investigate whether a long-term decrease in striatal nAChR-mediated dopaminergic function may represent a mechanism that underlies the nicotine-mediated decline in LIDs.
To approach this, we measured nAChR-evoked 3H-dopamine release from synaptosomes, a well-established technique used to assess striatal dopamine function in vitro (Rapier et al. 1990, Grady et al. 1992, Wonnacott et al. 2000, Grady et al. 2002, Quik et al. 2003). Our results show that chronic nicotine administration decreased nAChR-mediated dopamine release from striatum, providing support for the idea that nicotine may reduce L-dopa-induced AIMs by dampening dopaminergic tone, as depicted schematically in Fig. 9.
Fig. 9.

Schematic representation of the effect of long-term nicotine treatment on dopamine (DA) release from nigrostriatal terminals. The nicotine-induced decline in striatal dopamine release may counterbalance the effect of excess dopamine production (due to L-dopa) that is not cleared because of the dopamine nerve terminal loss.
Since nicotine elicits striatal dopamine release by acting at α4β2* and/or α6β2* nAChRs (Quik & Wonnacott 2011), we next evaluated the contribution of these two subtypes to the nicotine-mediated decline in striatal 3H-dopamine release. To study this, we used the α6β2* nAChR-directed neurotoxin α-CtxMII, which selectively blocks α6β2* but not α4β2* nAChR-mediated function (McIntosh et al. 2004). The results showed that long-term nicotine treatment decreased α6β2* nAChR-mediated 3H-dopamine release in response to 1 μM, but not 10.0 μM, application of nicotine to the synaptosomal preparation from either the intact or lesioned striatum. These two concentrations were selected as the former leads to submaximal and the later to maximal dopamine release (Salminen et al. 2004). The differential response at these two nicotine concentrations can be explained by the presence of least two α6β2* nAChRs in striatum, the α6α4β2β3 and α6β2β3 subtypes, with the former being more sensitive to nicotine (Bordia et al. 2007, Gotti et al. 2010, Salminen et al. 2004). Thus, 1 μM nicotine primarily elicits dopamine release mediated by the α6α4β2β3 subtype, while 10 μM nicotine predominantly evokes release at the α6β2β3 receptor subtype. A decline in release only at the 1.0 μM nicotine concentration may relate to the fact that α6α4β2β3, but not α6β2β3 receptors are downregulated with long-term nicotine treatment (Perez et al. 2008). Low nicotine (1.0 μM) application would elicit less release because the remaining α6β2β3 require a higher nicotine concentration to be activated. These data are consistent with the 125I-α-CtxMII binding studies which show that α6β2* nAChRs are deceased with long-term nicotine treatment although it is not known whether these represent α6α4β2β3 or α6β2β3 receptor subtypes. Interestingly, accumulating studies suggest that the α6α4β2β3 nAChR may be the one critical for a variety of different nAChR-mediated behaviors including locomotor activity and addiction (Exley et al. 2011, Zhao-Shea et al. 2011). The present studies suggest that a decline in α6α4β2β3 nAChRs is linked to the nicotine-mediated decline in L-dopa-induced AIMs.
We also investigated the effect of long-term nicotine treatment on α4β2* nAChR-mediated dopamine release from striatal synaptosomes. In this case, long-term nicotine administration significantly decreased 3H-dopamine release elicited by the 10 μM, but not the 1.0 μM nicotine concentration. This varying responsiveness at the two concentrations may be due to differential effects at striatal α4β2* nAChR subtypes, with the two main populations being the lower affinity α4β2 and higher affinity α4α5β2 receptor (Gotti et al. 2010, Grady et al. 2010b). If chronic nicotine treatment selectively downregulates the lower affinity α4β2 subtype, one might expect a decrease in 3H-dopamine release only at the 10 μM nicotine concentration. Release at the 1.0 μM dose of nicotine would be unchanged since α4β2 receptors are not reduced by long-term nicotine treatment.
α4β2* nAChR binding studies were done to evaluate the effect of chronic nicotine treatment on receptor levels. However, correlation of these results with α4β2* nAChR-mediated dopamine release is complicated by the fact that only 20% of the α4β2* nAChRs in the striatum are present on the dopamine terminals and the remaining 80% on other neurons in the striatum (Zoli et al. 2002, Quik & Wonnacott 2011). This finding is supported by the current data which show that a ~50% dopaminergic nerve terminal loss with 6-OHDA lesioning leads to only a ~10% decline in α4β2* nAChR binding. This small population (20%) of α4β2* nAChRs on the dopamine terminals may be controlled in a similar manner as those on striatal neurons, that is, upregulated by nicotine. The release studies, however, demonstrate a decline in α4β2* nAChR function. This is most likely due to α4β2* nAChR desensitization (Buccafusco et al. 2009, Picciotto et al. 2008). Long-term α4β2* receptor desensitization or inactivation would dampen dopaminergic tone. It is also possible that α4β2* nAChR desensitization leads to changes in downstream signaling mechanisms that improve LIDs. The molecular and cellular mechanisms that underlie the nicotine-mediated reduction are long-term events since several weeks of nicotine treatment are required to decrease LIDs (Bordia et al. 2010, Quik et al. 2007, Huang et al. 2011b, Huang et al. 2011a).
The current data show that nicotine treatment decreased dopamine release in both intact and lesioned striatum. This observation raises questions about the relevance of these findings to the nicotine-mediated decline in LIDs, which is generally associated with nigrostriatal damage. One possibility is that the decrease in release on the intact side does not lead to behavioral consequences under non-pathological conditions. On the other hand, the reduction in dopamine release on the lesioned side may contribute to the decline in LIDs because there are fewer terminals to clear the excess dopamine that arises with L-dopa administration. Another point of note is that the level of nAChR-evoked 3H-dopamine release in nicotine-treated rats is greater on the intact as compared to the lesioned striatum. Thus, there may be a reserve of striatal dopamine on the intact side such that overall behavior is not affected under control conditions.
Although long-term nicotine treatment decreased nicotine-evoked 3H-dopamine release, L-dopa administration did affect the release of radioactive dopamine from striatal synaptosomes. This finding was not unexpected since the current studies evaluate release of exogenously added 3H-dopamine loaded into a synaptosomal preparation. By contrast, release of endogenous dopamine from synaptosomes is most likely increased in striatum of L-dopa-treated rats, especially since striata are harvested an hour after L-dopa administration. Evidence for such an assumption is based on the studies showing that striatal dopamine levels are elevated after L-dopa treatment. Interestingly, the increase was most prominent in striatum of dyskinetic compared to nondyskinetic rats, suggesting that enhanced dopamine levels may underlie the development of dyskinesias (Carta et al. 2006).
The present studies in rats show that chronic nicotine treatment reduces various components of both striatal α6β2* and α4β2* nAChR-mediated dopamine release. However, we found no decrease in striatal 3H-dopamine release in a recent study in parkinsonian monkeys treated with nicotine (Quik et al. 2012a). These discrepant results may be due to differences in the treatment regimen since the rats were implanted with nicotine minipumps, which provides a constant level of nicotine exposure while the monkeys were given nicotine in the drinking water which yields intermittent nicotine exposure. Minipump nicotine administration has been reported to yield more pronounced changes in striatal nAChRs (Moretti et al. 2010). Differential effects may also be due to species differences in nAChR regulation. Long-term nicotine treatment resulted in 30–50% declines in α6β2* nAChR levels in rodents, but only a small or no change in α6β2* nAChR levels in monkeys (Lai et al. 2005, McCallum et al. 2006). The increased number of animals in the current study (9–13 rats/group) also improves the ability to discriminate differences between treatment groups.
In addition to presynaptic dopaminergic mechanisms, L-dopa treatment also results in a host of postsynaptic changes that contribute to LIDs. These may arise as a result of enhanced stimulation of dopamine D1, and possibly D2, receptors, and the consequent activation of downstream signaling mechanisms by dopamine derived from exogenous L-dopa (Cenci & Konradi 2010, Berthet & Bezard 2009). An accumulating literature also indicates that dysregulated release of dopamine from serotonergic neurons may exacerbate LIDs that arise with therapeutic doses of L-dopa (Munoz et al. 2008, Nahimi et al. 2012, Carta et al. 2007, Cenci 2007). Studies to determine whether nicotine modulates postsynaptic mechanisms remain to be done.
In summary, our previous work had shown that chronic nicotine treatment improved L-dopa-induced AIMs in several parkinsonian animal models. The present results provide a mechanistic basis for these findings by showing that long-term nicotine treatment decreased both α6β2* and α4β2* nAChR-mediated 3H-dopamine release from striatal synaptosomes. Long-term nicotine treatment also reduced 125I-α-CtxMII binding sites, suggesting that receptor downregulation may underlie the α6β2* nAChR-mediated decrease in release. By contrast, α4β2* nAChR desensitization may contribute to the α4β2* nAChR-mediated decline in striatal 3H-dopamine release. Overall, these data suggest that nicotine may improve LIDs in moderate Parkinson’s disease by decreasing α6β2* and α4β2* nAChR striatal dopaminergic function.
Acknowledgments
This work was supported by NIH grants NS59910 (MQ), NS65851 (MQ), GM103801 (JMM) and GM48677 (JMM). The authors thank Matthew Chin for technical assistance and help with the manuscript.
Abbreviations used
- ANOVA
analysis of variance
- nAChRs
nicotinic acetylcholine receptors
- [125I]RTI-121
[125I]-3β-(4-iodophenyl)tropane-2β-carboxylic acid isopropyl ester
- *
the asterisk indicates the possible presence of other nicotinic subunits in the receptor complex
Footnotes
The authors have no conflicts of interest to declare.
References
- Anderson SM, Brunzell DH. Low Dose Nicotine and Antagonism of beta2 Subunit Containing Nicotinic Acetylcholine Receptors Have Similar Effects on Affective Behavior in Mice. PLoS One. 2012;7:e48665. doi: 10.1371/journal.pone.0048665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artymyshyn R, Smith A, Wolfe BB. The use of 3H standards in 125I autoradiography. J Neurosci Methods. 1990;32:185–192. doi: 10.1016/0165-0270(90)90139-7. [DOI] [PubMed] [Google Scholar]
- Berthet A, Bezard E. Dopamine receptors and L-dopa-induced dyskinesia. Parkinsonism Relat Disord. 2009;15(Suppl 4):S8–12. doi: 10.1016/S1353-8020(09)70827-2. [DOI] [PubMed] [Google Scholar]
- Bezard E, Brotchie JM, Gross CE. Pathophysiology of levodopa-induced dyskinesia: potential for new therapies. Nat Rev Neurosci. 2001;2:577–588. doi: 10.1038/35086062. [DOI] [PubMed] [Google Scholar]
- Bordia T, Campos C, Huang L, Quik M. Continuous and intermittent nicotine treatment reduces L-3,4-dihydroxyphenylalanine (L-DOPA)-induced dyskinesias in a rat model of Parkinson’s disease. J Pharmacol Exp Ther. 2008;327:239–247. doi: 10.1124/jpet.108.140897. [DOI] [PubMed] [Google Scholar]
- Bordia T, Campos C, McIntosh JM, Quik M. Nicotinic receptor-mediated reduction in L-dopa-induced dyskinesias may occur via desensitization. J Pharmacol Exp Ther. 2010;333:929–938. doi: 10.1124/jpet.109.162396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordia T, Grady SR, McIntosh JM, Quik M. Nigrostriatal damage preferentially decreases a subpopulation of {alpha}6{beta}2* nAChRs in mouse, monkey and Parkinson’s disease striatum. Mol Pharmacol. 2007;72:52–61. doi: 10.1124/mol.107.035998. [DOI] [PubMed] [Google Scholar]
- Bordia T, Hrachova M, Chin M, McIntosh JM, Quik M. Varenicline is a potent partial agonist at alpha6beta2* nAChRs in rat and monkey striatum. J Pharmacol Exp Ther. 2012;342:327–334. doi: 10.1124/jpet.112.194852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brotchie J, Jenner P. New approaches to therapy. Int Rev Neurobiol. 2011;98:123–150. doi: 10.1016/B978-0-12-381328-2.00005-5. [DOI] [PubMed] [Google Scholar]
- Buccafusco JJ, Beach JW, Terry AV. Desensitization of nicotinic acetylcholine receptors as a strategy for drug development. J Pharmacol Exp Ther. 2009;328:364–370. doi: 10.1124/jpet.108.145292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta M, Bezard E. Contribution of pre-synaptic mechanisms to l-DOPA-induced dyskinesia. Neuroscience. 2011;198:245–251. doi: 10.1016/j.neuroscience.2011.07.070. [DOI] [PubMed] [Google Scholar]
- Carta M, Carlsson T, Kirik D, Bjorklund A. Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in parkinsonian rats. Brain. 2007;130:1819–1833. doi: 10.1093/brain/awm082. [DOI] [PubMed] [Google Scholar]
- Carta M, Lindgren HS, Lundblad M, Stancampiano R, Fadda F, Cenci MA. Role of striatal L-DOPA in the production of dyskinesia in 6-hydroxydopamine lesioned rats. J Neurochem. 2006;96:1718–1727. doi: 10.1111/j.1471-4159.2006.03696.x. [DOI] [PubMed] [Google Scholar]
- Cenci M, Lindgren H. Advances in understanding l-DOPA-induced dyskinesia. Curr Opin Neurobiol. 2007;17:665–671. doi: 10.1016/j.conb.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Cenci MA. Dopamine dysregulation of movement control in L-DOPA-induced dyskinesia. Trends Neurosci. 2007;30:236–243. doi: 10.1016/j.tins.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Konradi C. Maladaptive striatal plasticity in l-DOPA-induced dyskinesia. Progress in brain research. 2010;183C:209–233. doi: 10.1016/S0079-6123(10)83011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci MA, Lundblad M. Ratings of L-DOPA-induced dyskinesia in the unilateral 6-OHDA lesion model of Parkinson’s disease in rats and mice. Curr Protoc Neurosci. 2007;Chapter 9(Unit 9):25. doi: 10.1002/0471142301.ns0925s41. [DOI] [PubMed] [Google Scholar]
- Dopico AM, Lovinger DM. Acute alcohol action and desensitization of ligand-gated ion channels. Pharmacol Rev. 2009;61:98–114. doi: 10.1124/pr.108.000430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exley R, Maubourguet N, David V, et al. Distinct contributions of nicotinic acetylcholine receptor subunit alpha4 and subunit alpha6 to the reinforcing effects of nicotine. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7577–7582. doi: 10.1073/pnas.1103000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisone G, Bezard E. Molecular mechanisms of l-DOPA-induced dyskinesia. Int Rev Neurobiol. 2011;98:95–122. doi: 10.1016/B978-0-12-381328-2.00004-3. [DOI] [PubMed] [Google Scholar]
- Fisone G, Hakansson K, Borgkvist A, Santini E. Signaling in the basal ganglia: postsynaptic and presynaptic mechanisms. Physiol Behav. 2007;92:8–14. doi: 10.1016/j.physbeh.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Gotti C, Guiducci S, Tedesco V, et al. Nicotinic acetylcholine receptors in the mesolimbic pathway: primary role of ventral tegmental area alpha6beta2* receptors in mediating systemic nicotine effects on dopamine release, locomotion, and reinforcement. J Neurosci. 2010;30:5311–5325. doi: 10.1523/JNEUROSCI.5095-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady S, Marks MJ, Wonnacott S, Collins AC. Characterization of nicotinic receptor-mediated [3H]dopamine release from synaptosomes prepared from mouse striatum. J Neurochem. 1992;59:848–856. doi: 10.1111/j.1471-4159.1992.tb08322.x. [DOI] [PubMed] [Google Scholar]
- Grady SR, Drenan RM, Breining SR, et al. Structural differences determine the relative selectivity of nicotinic compounds for native alpha 4 beta 2*-, alpha 6 beta 2*-, alpha 3 beta 4*- and alpha 7-nicotine acetylcholine receptors. Neuropharmacology. 2010a;58:1054–1066. doi: 10.1016/j.neuropharm.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady SR, Murphy KL, Cao J, Marks MJ, McIntosh JM, Collins AC. Characterization of nicotinic agonist-induced [(3)H]dopamine release from synaptosomes prepared from four mouse brain regions. J Pharmacol Exp Ther. 2002;301:651–660. doi: 10.1124/jpet.301.2.651. [DOI] [PubMed] [Google Scholar]
- Grady SR, Salminen O, McIntosh JM, Marks MJ, Collins AC. Mouse striatal dopamine nerve terminals express alpha4alpha5beta2 and two stoichiometric forms of alpha4beta2*-nicotinic acetylcholine receptors. J Mol Neurosci. 2010b;40:91–95. doi: 10.1007/s12031-009-9263-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Grady SR, Quik M. Nicotine Reduces L-Dopa-Induced Dyskinesias by Acting at {beta}2 Nicotinic Receptors. J Pharmacol Exp Ther. 2011a;338:932–941. doi: 10.1124/jpet.111.182949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LZ, Campos C, Ly J, Carroll FI, Quik M. Nicotinic receptor agonists decrease L-dopa-induced dyskinesias most effectively in moderately lesioned parkinsonian rats. Neuropharmacology. 2011b;60:861–868. doi: 10.1016/j.neuropharm.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hukkanen J, Jacob P, 3rd, Benowitz NL. Metabolism and disposition kinetics of nicotine. Pharmacol Rev. 2005;57:79–115. doi: 10.1124/pr.57.1.3. [DOI] [PubMed] [Google Scholar]
- Jenner P. Molecular mechanisms of L-DOPA-induced dyskinesia. Nat Rev Neurosci. 2008;9:665–677. doi: 10.1038/nrn2471. [DOI] [PubMed] [Google Scholar]
- Lai A, Parameswaran N, Khwaja M, Whiteaker P, Lindstrom JM, Fan H, McIntosh JM, Grady SR, Quik M. Long-term nicotine treatment decreases striatal alpha6* nicotinic acetylcholine receptor sites and function in mice. Mol Pharmacol. 2005;67:1639–1647. doi: 10.1124/mol.104.006429. [DOI] [PubMed] [Google Scholar]
- Lindgren HS, Andersson DR, Lagerkvist S, Nissbrandt H, Cenci MA. L-DOPA-induced dopamine efflux in the striatum and the substantia nigra in a rat model of Parkinson’s disease: temporal and quantitative relationship to the expression of dyskinesia. J Neurochem. 2009 doi: 10.1111/j.1471-4159.2009.06556.x. [DOI] [PubMed] [Google Scholar]
- Lundblad M, af Bjerken S, Cenci MA, Pomerleau F, Gerhardt GA, Stromberg I. Chronic intermittent L-DOPA treatment induces changes in dopamine release. J Neurochem. 2009;108:998–1008. doi: 10.1111/j.1471-4159.2008.05848.x. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF, Collins AC. Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J Neurosci. 1992;12:2765–2784. doi: 10.1523/JNEUROSCI.12-07-02765.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MJ, Wageman CR, Grady SR, Gopalakrishnan M, Briggs CA. Selectivity of ABT-089 for alpha4beta2* and alpha6beta2* nicotinic acetylcholine receptors in brain. Biochem Pharmacol. 2009;78:795–802. doi: 10.1016/j.bcp.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum S, Parameswaran N, Bordia T, Fan H, McIntosh M, Quik M. Differential regulation of mesolimbic {alpha}3*/{alpha}6{beta}2* and {alpha}4{beta}2* nAChR sites and function after long-term oral nicotine to monkeys. J Pharmacol Exp Ther. 2006;318:381–388. doi: 10.1124/jpet.106.104414. [DOI] [PubMed] [Google Scholar]
- McIntosh JM, Azam L, Staheli S, Dowell C, Lindstrom JM, Kuryatov A, Garrett JE, Marks MJ, Whiteaker P. Analogs of alpha-conotoxin MII are selective for alpha6-containing nicotinic acetylcholine receptors. Mol Pharmacol. 2004;65:944–952. doi: 10.1124/mol.65.4.944. [DOI] [PubMed] [Google Scholar]
- Meissner W, Ravenscroft P, Reese R, et al. Increased slow oscillatory activity in substantia nigra pars reticulata triggers abnormal involuntary movements in the 6-OHDA-lesioned rat in the presence of excessive extracellular striatal dopamine. Neurobiol Dis. 2006;22:586–598. doi: 10.1016/j.nbd.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Moretti M, Mugnaini M, Tessari M, Zoli M, Gaimarri A, Manfredi I, Pistillo F, Clementi F, Gotti C. A comparative study of the effects of the intravenous self-administration or subcutaneous minipump infusion of nicotine on the expression of brain neuronal nicotinic receptor subtypes. Mol Pharmacol. 2010;78:287–296. doi: 10.1124/mol.110.064071. [DOI] [PubMed] [Google Scholar]
- Mugnaini M, Garzotti M, Sartori I, Pilla M, Repeto P, Heidbreder CA, Tessari M. Selective down-regulation of [(125)I]Y(0)-alpha-conotoxin MII binding in rat mesostriatal dopamine pathway following continuous infusion of nicotine. Neuroscience. 2006;137:565–572. doi: 10.1016/j.neuroscience.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Munoz A, Li Q, Gardoni F, et al. Combined 5-HT1A and 5-HT1B receptor agonists for the treatment of L-DOPA-induced dyskinesia. Brain. 2008;131:3380–3394. doi: 10.1093/brain/awn235. [DOI] [PubMed] [Google Scholar]
- Murer MG, Moratalla R. Striatal Signaling in L-DOPA-Induced Dyskinesia: Common Mechanisms with Drug Abuse and Long Term Memory Involving D1 Dopamine Receptor Stimulation. Front Neuroanat. 2011;5:51. doi: 10.3389/fnana.2011.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahimi A, Holtzermann M, Landau AM, et al. Serotonergic modulation of receptor occupancy in rats treated with L-DOPA after unilateral 6-OHDA lesioning. J Neurochem. 2012;120:806–817. doi: 10.1111/j.1471-4159.2011.07598.x. [DOI] [PubMed] [Google Scholar]
- Pavese N, Evans AH, Tai YF, Hotton G, Brooks DJ, Lees AJ, Piccini P. Clinical correlates of levodopa-induced dopamine release in Parkinson disease: a PET study. Neurology. 2006;67:1612–1617. doi: 10.1212/01.wnl.0000242888.30755.5d. [DOI] [PubMed] [Google Scholar]
- Perez X, O’Leary K, Parameswaran N, McIntosh JM, Quik M. Prominent role of {alpha}3/{alpha}6{beta}2* nAChRs in regulating evoked dopamine release in primate putamen; effect of long-term nicotine treatment. Mol Pharmacol. 2009;75:938–946. doi: 10.1124/mol.108.053801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez XA, Bordia T, McIntosh JM, Grady SR, Quik M. Long-term nicotine treatment differentially regulates striatal alpha6alpha4beta2* and alpha6(nonalpha4)beta2* nAChR expression and function. Mol Pharmacol. 2008;74:844–853. doi: 10.1124/mol.108.048843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry DC, Mao D, Gold AB, McIntosh JM, Pezzullo JC, Kellar KJ. Chronic nicotine differentially regulates alpha6- and beta3-containing nicotinic cholinergic receptors in rat brain. J Pharmacol Exp Ther. 2007;322:306–315. doi: 10.1124/jpet.107.121228. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Addy NA, Mineur YS, Brunzell DH. It is not “either/or”: Activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog Neurobiol. 2008;84:329–342. doi: 10.1016/j.pneurobio.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quik M, Cox H, Parameswaran N, O’Leary K, Langston JW, Di Monte D. Nicotine reduces levodopa-induced dyskinesias in lesioned monkeys. Annals of neurology. 2007;62:588–596. doi: 10.1002/ana.21203. [DOI] [PubMed] [Google Scholar]
- Quik M, Mallela A, Chin M, McIntosh JM, Perez XA, Bordia T. Nicotine-mediated improvement in L-dopa-induced dyskinesias in MPTP-lesioned monkeys is dependent on dopamine nerve terminal function. Neurobiol Dis. 2012a doi: 10.1016/j.nbd.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quik M, Park KM, Hrachova M, Mallela A, Huang LZ, McIntosh JM, Grady SR. Role for alpha6 nicotinic receptors in l-dopa-induced dyskinesias in parkinsonian mice. Neuropharmacology. 2012b;63:450–459. doi: 10.1016/j.neuropharm.2012.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quik M, Sum JD, Whiteaker P, McCallum SE, Marks MJ, Musachio J, McIntosh JM, Collins AC, Grady SR. Differential declines in striatal nicotinic receptor subtype function after nigrostriatal damage in mice. Mol Pharmacol. 2003;63:1169–1179. doi: 10.1124/mol.63.5.1169. [DOI] [PubMed] [Google Scholar]
- Quik M, Wonnacott S. {alpha}6{beta}2* and {alpha}4{beta}2* Nicotinic Acetylcholine Receptors As Drug Targets for Parkinson’s Disease. Pharmacol Rev. 2011;63:938–966. doi: 10.1124/pr.110.003269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapier C, Lunt GG, Wonnacott S. Nicotinic modulation of [3H]dopamine release from striatal synaptosomes: pharmacological characterisation. J Neurochem. 1990;54:937–945. doi: 10.1111/j.1471-4159.1990.tb02341.x. [DOI] [PubMed] [Google Scholar]
- Rascol O, Fitzer-Attas CJ, Hauser R, et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease (the ADAGIO study): prespecified and post-hoc analyses of the need for additional therapies, changes in UPDRS scores, and non-motor outcomes. Lancet Neurol. 2011;10:415–423. doi: 10.1016/S1474-4422(11)70073-4. [DOI] [PubMed] [Google Scholar]
- Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, Grady SR. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol. 2004;65:1526–1535. doi: 10.1124/mol.65.6.1526. [DOI] [PubMed] [Google Scholar]
- Schallert T, Fleming SM, Leasure JL, Tillerson JL, Bland ST. CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology. 2000;39:777–787. doi: 10.1016/s0028-3908(00)00005-8. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Jenner P. Etiology and pathogenesis of Parkinson’s disease. Mov Disord. 2011;26:1049–1055. doi: 10.1002/mds.23732. [DOI] [PubMed] [Google Scholar]
- Wonnacott S, Kaiser S, Mogg A, Soliakov L, Jones IW. Presynaptic nicotinic receptors modulating dopamine release in the rat striatum. European journal of pharmacology. 2000;393:51–58. doi: 10.1016/s0014-2999(00)00005-4. [DOI] [PubMed] [Google Scholar]
- Zhao-Shea R, Liu L, Soll LG, et al. Nicotine-Mediated Activation of Dopaminergic Neurons in Distinct Regions of the Ventral Tegmental Area. Neuropsychopharmacology. 2011;36:1021–1032. doi: 10.1038/npp.2010.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoli M, Moretti M, Zanardi A, McIntosh JM, Clementi F, Gotti C. Identification of the nicotinic receptor subtypes expressed on dopaminergic terminals in the rat striatum. J Neurosci. 2002;22:8785–8789. doi: 10.1523/JNEUROSCI.22-20-08785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]