

Abstract
Cure rates in pediatric acute myeloid leukemia (AML) remain suboptimal. Overexpression of the surface receptor CXCR4 is associated with poor outcome in acute lymphoblastic leukemia (ALL) and AML. Certain non-chemotherapeutic agents have been shown to modulate CXCR4 expression and alter leukemia interactions with stromal cells in the bone marrow microenvironment. Because chemotherapy is the mainstay of AML treatment, we hypothesized that standard cytotoxic chemotherapeutic agents induce dynamic changes in leukemia surface CXCR4 expression, and that chemotherapy-induced upregulation of CXCR4 represents a mechanism of acquired chemotherapy resistance. Here, we show that cell lines variably upregulate CXCR4 with chemotherapy treatment. Those that showed upregulation were differentially protected from chemotherapy-induced apoptosis when co-cultured with stroma. We further explored the functional effects of chemotherapy-induced CXCR4 upregulation in an AML cell line (MOLM-14, which consistently upregulated CXCR4) and primary samples. We found enhanced stromal-cell derived factor-1α (SDF-1α)-mediated chemotaxis and stromal protection from additional chemotherapy-induced apoptosis. Further, treatment with the CXCR4 inhibitor plerixafor preferentially decreased stromal protection in cells with higher chemotherapy-induced upregulation of surface CXCR4. Upregulation of surface CXCR4 by standard chemotherapy may represent a mechanism of chemotherapy resistance in pediatric AML and may be a biomarker that can identify optimal patients for CXCR4 inhibition.
Keywords: CXCR4, AML, microenvironment, stroma, plerixafor
Introduction
Stepwise advances in the treatment of children with acute leukemia have led to remarkable improvements in cure rates and survival.(1) The greatest improvements have been achieved in pediatric ALL and cure rates are approaching 90 percent.(1) However, children with high-risk acute leukemias continue to have suboptimal outcomes. Further, a disproportionate number of children with AML are not cured of their disease. Research in AML continues to be focused primarily on factors intrinsic to the leukemic blast that can predict outcome and response to therapy. For example, studies have established that the core binding factor mutations inv(16) and t(8;21) portend a favorable outcome in AML, while monosomy 7, del(5q), MLL gene rearrangements (MLL-R), and FLT3/ITD are associated with poor prognosis.(1) As a result, recent clinical trials have focused on intensifying therapy for children with high-risk features, as well as identifying other potentially prognostic mutations, including those in nucleophosmin (NPM1)(2) and CCAAT/enhancer binding protein-alpha (CEBPA).(3) However, 5-year event-free survival rates continue to be between 50 and 60 percent with contemporary therapies.(1)
Recently, research has broadened to include extrinsic mechanisms of leukemia cell survival and proliferation, namely the bone marrow microenvironment. The bone marrow microenvironment consists of stromal cells, endothelial cells, osteoblasts, and other cells that support the development of hematopoietic stem cells (HSCs).(4, 5) Many studies have also demonstrated that the bone marrow microenvironment has a similar supportive effect on leukemias.(6, 7) For example, co-culture with bone marrow stromal cells clearly contributes to the ability of leukemia cells to survive in vitro.(8, 9) We also previously demonstrated that bone marrow stroma selectively provides more protection to MLL-R ALL compared to non-MLL-R ALL. We also found that co-culture of primary samples of infant MLL-R ALL with bone marrow stroma attenuated cytotoxicity induced by the FLT3 inhibitor lestaurtinib and promoted the survival of leukemic stem cells (LSCs).(10)
Multiple receptor-ligand interactions regulate the migration, adhesion, quiescence, and proliferation of leukemia cells within the bone marrow microenvironment, such as the interactions between very late adhesion molecule-4 (VLA-4) and its ligands fibronectin(11) and vascular cell adhesion molecule-1 (VCAM-1).(12) Children’s Oncology Group reported that VLA-4 surface expression is predictive of outcome and relapse risk, suggesting that leukemia-stromal interactions are important in AML.(13) Another important mediator of leukemia-microenvironment interactions is CXCR4, a transmembrane G-protein-coupled receptor expressed by HSCs, various white blood cells, and leukemia cells.(14-16) CXCR4 is activated by stromal cell-derived factor 1 (SDF-1 or CXCL12), a chemokine constitutively secreted by stromal and endothelial cells in the bone marrow microenvironment.(17-19) Interactions between CXCR4 and SDF-1 are paramount in the migration and survival of leukemia cells in vitro and in vivo.(16, 20, 21) High CXCR4 expression has also been implicated as a marker of poor prognosis in both ALL(22, 23) and AML.(24-26) In addition, preclinical models of ALL and AML demonstrate that inhibition of CXCR4 enhances the efficacy of chemotherapy and tyrosine kinase inhibitors (TKIs).(10, 27-31) These findings suggest that CXCR4 expression is a factor that helps mediate response to therapy through increased leukemia-microenvironment interactions.
Therefore, we sought to better elucidate the role of CXCR4 in chemotherapy resistance. Previous studies have shown that CXCR4 expression in leukemias is dynamic and is modulated by non-chemotherapeutic agents such as TKIs and histone deacetylase (HDAC) inhibitors.(32-35) Because chemotherapy is the foundation of pediatric leukemia treatment, we hypothesized that surface CXCR4 expression by acute leukemias changes dynamically in response to standard cytotoxic chemotherapeutic agents. We also hypothesized that chemotherapy-mediated upregulation of surface CXCR4 would lead to increased protection by normal human bone marrow stroma due to enhanced SDF-1α/CXCR4 signaling. Our data demonstrate for the first time that surface CXCR4 expression is modulated by standard chemotherapeutic agents and that the direction of CXCR4 modulation (ie, up or down) is predictive of the degree of stromal protection. We also show for the first time that chemotherapy-induced upregulation of surface CXCR4 results in increased SDF-1α-mediated chemotaxis and a stromal-mediated survival advantage in an AML cell line and AML primary samples. Finally, we show that treatment with plerixafor decreases stromal protection preferentially in leukemias that upregulate surface CXCR4 in response to chemotherapy. These novel findings suggest that standard therapies for AML may increase treatment resistance in surviving leukemic blasts and that this resistance may be overcome by CXCR4 inhibition.
Materials and Methods
Cell lines and reagents
Cell culture reagents included RPMI 1640 medium, 100x penicillin-streptomycin solution (PS) and 200 mmol/L L-glutamine solution (LG) (all purchased from Invitrogen, Carlsbad, CA), fetal bovine serum (FBS, Gemini Bio-Products, West Sacramento, CA), and 50 μmol/L hydrocortisone solution (Sigma-Aldrich, St. Louis, MO). ALL (697, HB-1119, Nalm-6, SEM-K2) and AML (MOLM-14, MV4-11) cell lines were purchased from ATCC (Manassas, VA) and DSMZ (Braunschweig, Germany). All cell line cultures were maintained in RPMI 1640 supplemented with 10% FBS, 1% PS, and 1% LG and incubated at 37°C in 5% CO2. Stock solutions of all chemotherapeutic agents (cytarabine, daunorubicin, etoposide, methotrexate, and vincristine; Sigma-Aldrich) were prepared according to the manufacturer’s instructions and stored at -80°C until ready for use. Plerixafor was kindly provided by Genzyme (Cambridge, MA).
Primary leukemia samples
Diagnostic bone marrow and peripheral blood samples were collected from newly diagnosed children with AML enrolled on a Johns Hopkins institutional review board-approved protocol. Following collection, primary leukemia cells were isolated using density centrifugation (Ficoll-Paque PLUS, GE Healthcare, Piscataway, NJ). Samples were viably cryopreserved by suspension in 90% FBS/10% DMSO and storage in liquid nitrogen. To perform the experiments, primary AML cells were maintained in RPMI 1640 supplemented with 20% FBS, 1% PS, and 1% LG.
Normal human bone marrow stromal cell cultures
Normal human bone marrow was collected from healthy adult bone marrow transplant donors according to a Johns Hopkins institutional review board-approved protocol. Establishment and maintenance of stromal cell cultures were performed as previously described.(10) All stromal cell cultures were maintained at 33°C in 5% CO2. For stromal co-culture experiments, 96 well plate stroma cultures were washed 3 times with phosphate-buffered saline prior to the addition of leukemia cells. All co-culture plates were incubated at 37°C in 5% CO2.
Measurement of CXCR4 expression by flow cytometry
Cells were stained with APC-conjugated anti-human CD184 (12G5, eBioscience, San Diego, CA) and read on a FACSCalibur (BD Biosciences, San Diego, CA). To evaluate intracellular CXCR4 expression, surface CXCR4 was blocked with nonconjugated anti-human CXCR4 antibody (4G10, Santa Cruz Biotechnology, Santa Cruz, CA), fixed with 4% paraformaldehyde (Sigma-Aldrich), permeabilized with 0.2% Triton X-100 (Sigma-Aldrich), stained with APC-conjugated anti-human CD184, and read on a FACSCalibur. Results were analyzed using FlowJo (Tree Star, Inc., Ashland, OR). Gates were drawn around the live populations, as determined by forward scatter and side scatter properties, and the mean fluorescence index (MFI) of CXCR4 of live cells was calculated.
Measurement of CXCR4 expression by quantitative real-time PCR (qRT-PCR)
To evaluate gene expression of CXCR4 and its positively-regulating transcription factor NRF-1, RNA was isolated from leukemic cells using the RNEasy Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. RNA was stored at -80°C until ready for use. cDNA was made using SuperScript III Reverse Transcriptase and associated reagents (Invitrogen) and run on a Mastercycler (Eppendorf, Hamburg, Germany). qRT-PCR was performed using TaqMan primers for CXCR4 and NRF-1, with GAPDH as a control (Invitrogen), according to the manufacturer’s instructions. qRT-PCR was performed and results were analyzed using the CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA).
Apoptosis assays
Cell lines and primary AML samples were treated with dose ranges of chemotherapy for 48 to 72 hours in 96 well plates. Cells were harvested and stained with PE-conjugated Annexin V and 7-AAD (BD Pharmingen, San Diego, CA), read on a FACSCalibur, and analyzed with FlowJo. In co-culture experiments, leukemia cells were distinguished from stromal cells by their forward scatter/side scatter properties.
Chemotaxis
Culture medium with and without human recombinant SDF-1α (Peprotech, Rocky Hill, NJ) was placed in the bottom of 24 well plates. Millicell hanging cell culture inserts (8 μm membrane pore size, Millipore, Billerica, MA) were placed into each well. Leukemic cells were seeded into each insert at a concentration of 1×106/mL. Plates were incubated at 37°C. After incubation, trypsin at a 1:10 dilution was added to the bottom of each well and plates were incubated for an additional 5 minutes. Migrated cells were then quantified by harvesting cells from the bottom of each well and counting in triplicate using a hemacytometer and Trypan Blue exclusion. Results are expressed as the number of migrated cells in an SDF-1α-containing well divided by the number of migrated cells in a media only-containing well.
Flow cytometric measurement of phosphorylated Akt and ERK1/2
Cells were treated with vehicle control or cytarabine for 72 hours and then serum-starved overnight. Cells were then prepared according to the BD Phosflow Protocol for Human PBMCs (BD Biosciences). Briefly, cells were fixed using Cytofix Buffer, permeabilized using Perm Buffer III, stained with PE-conjugated anti-human Akt (pS473) and Alexa Fluor 488-conjugated anti-human ERK1/2 (pT202/pY204) (all purchased from BD Biosciences), and read on a FACSCalibur. The MFI of live cells was calculated using FlowJo.
Statistical analysis
Paired student’s t-tests and independent two-sample t-tests were used to calculate p values. Alpha was set to 0.05. Error bars in all figures represent the standard error of the mean (SEM).
Results
Baseline surface CXCR4 levels affect the degree of chemotaxis toward an SDF-1α gradient
We measured baseline levels of surface CXCR4 of a representative panel of ALL (697, HB1119, Nalm-6, SEM-K2) and AML (MOLM-14, MV4-11) cell lines by flow cytometry (Fig. 1A for cell line characteristics(36-41)). Surface CXCR4 levels varied at baseline, with Nalm-6 and MOLM-14 having the highest and lowest values of the cell lines tested, respectively (Fig. 1A). Previous work has suggested that SDF-1α-induced chemotaxis is dependent on surface CXCR4 expression in CD34+ HSCs.(42) Therefore, we measured chemotaxis of MOLM-14 and Nalm-6 toward a gradient of SDF-1α to determine if this phenomenon was also true in leukemia. Cells were seeded in hanging cell culture inserts placed into wells containing SDF-1α or media alone. Migrated cells were measured after 4 and 24 hours of incubation. Nalm-6, which had the highest expression of surface CXCR4 among the cell lines tested, demonstrated striking SDF-1α-induced chemotaxis that was both time- and SDF-1α-concentration-dependent (Fig. 1B). In contrast, MOLM-14, which had the lowest expression of surface CXCR4 among the cell lines tested, demonstrated minimal chemotaxis. We also found that cell lines with intermediate surface expression of CXCR4 had intermediate SDF-1α-induced chemotaxis (data not shown). We concluded that chemotactic responses to SDF-1α in leukemia cell lines are dependent on levels of surface CXCR4.
Figure 1. Baseline surface CXCR4 levels affect the degree of chemotaxis toward an SDF-1α gradient.
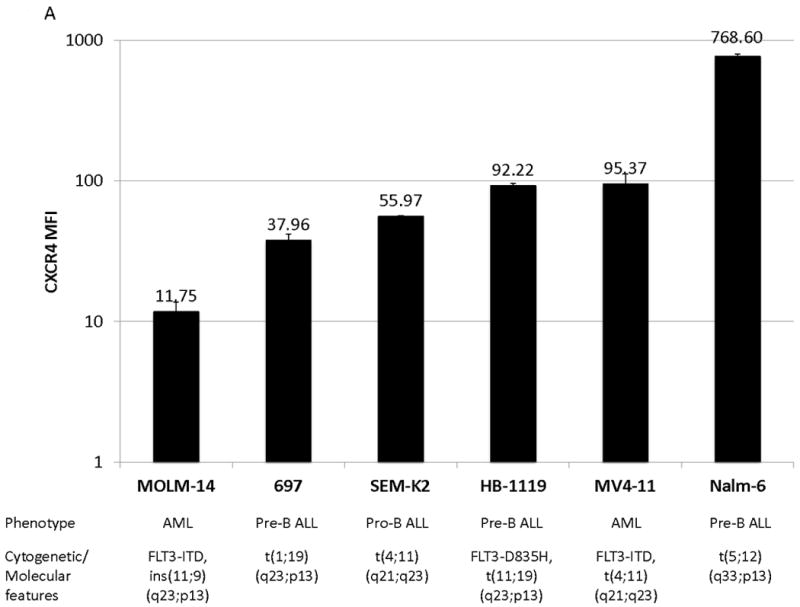
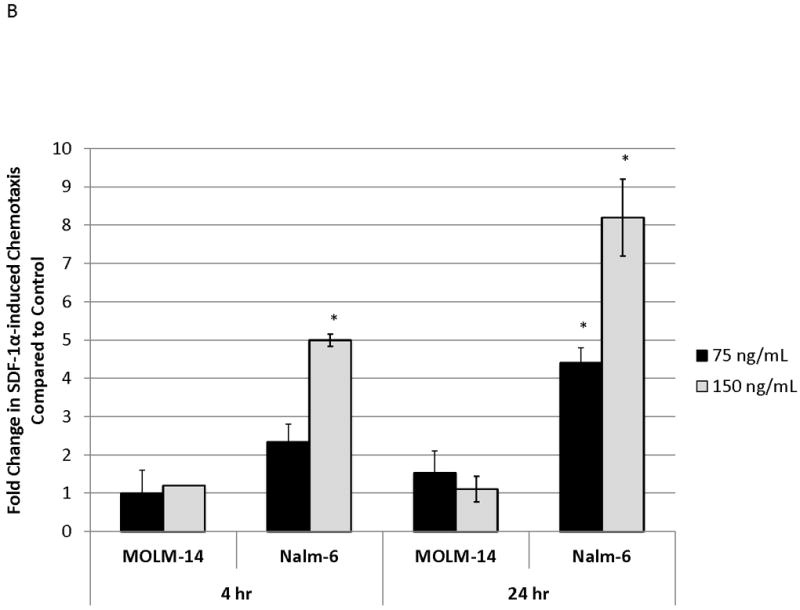
(a) Surface CXCR4 expression in viable, untreated cells was measured by flow cytometry. Results are the averages of triplicate measurements. Error bars in this and subsequent figures represent SEM. (b) MOLM-14 and Nalm-6 were seeded in hanging cell culture inserts and placed into 24-well plates containing supplemented media alone (control) or supplemented media with SDF-1α (75 or 150 ng/mL). Migrated cells were measured in triplicate after 4 and 24 hours. *p<0.05 vs. control.
Surface CXCR4 levels change in response to chemotherapy in leukemia cell lines
We next wanted to determine if surface CXCR4 expression changed in response to chemotherapy. Cell lines were treated with dose ranges of 5 chemotherapeutic agents commonly used in the treatment of either ALL or AML (cytarabine, daunorubicin, etoposide, methotrexate, and vincristine). After 48 hours of treatment, flow cytometry was performed to measure surface CXCR4 expression (Fig. 2A for effects of daunorubicin on MOLM-14). To determine the overall effect of chemotherapy on surface CXCR4 expression, we recorded the maximal change in CXCR4 for each drug-cell line combination. Treatment with chemotherapy resulted in CXCR4 upregulation in 697, MOLM-14, and MV4-11, and downregulation in HB-1119, Nalm-6, and SEM-K2 (Fig. 2B). Interestingly, each cell line responded to all chemotherapeutic agents in a consistent manner (i.e., either up- or downregulation). These results suggest that expression of surface CXCR4 changes dynamically in response to chemotherapy in leukemia cell lines and that these changes in surface CXCR4 expression are consistent within a given cell line from agent to agent.
Figure 2. Surface CXCR4 levels change consistently in response to chemotherapy and cell lines that upregulate CXCR4 are protected by stroma from chemotherapy-induced apoptosis.
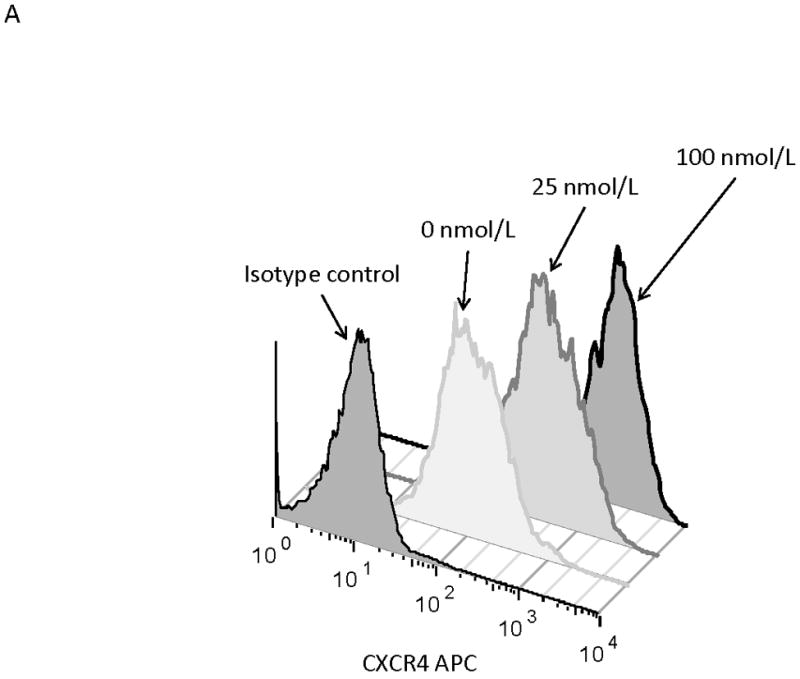
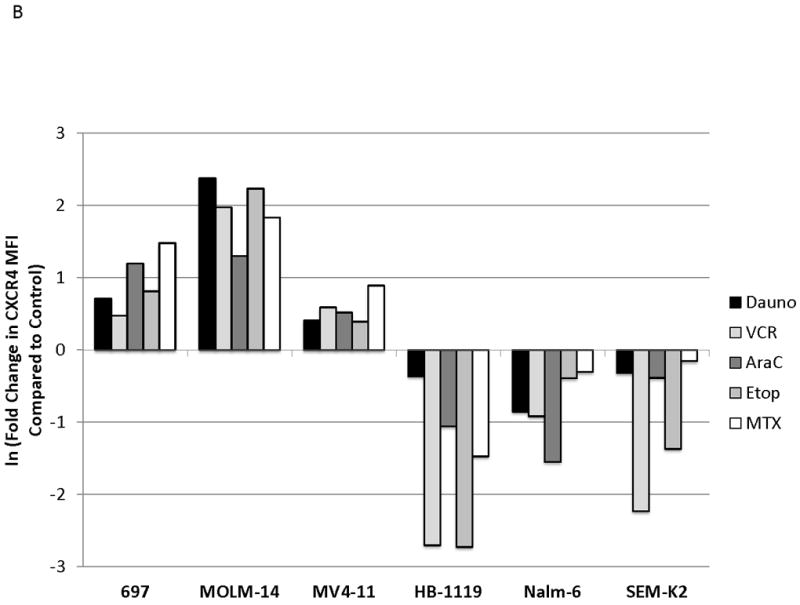
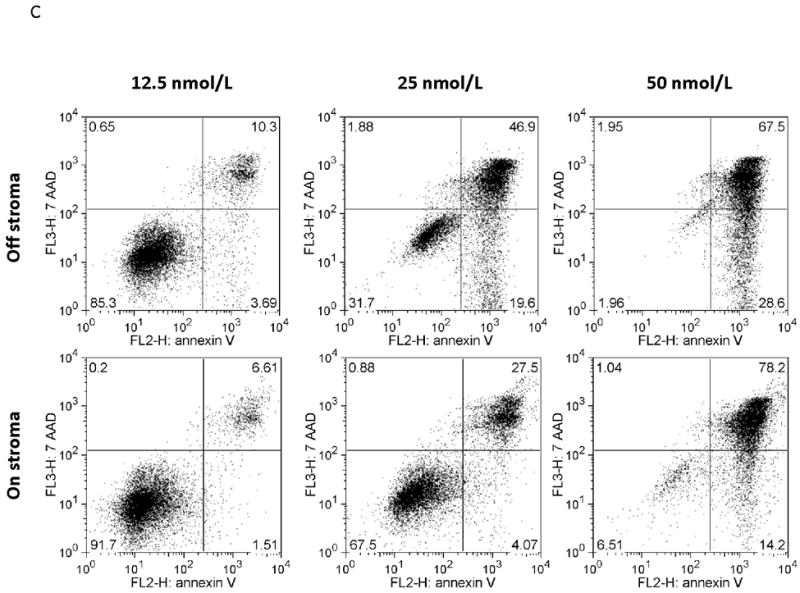
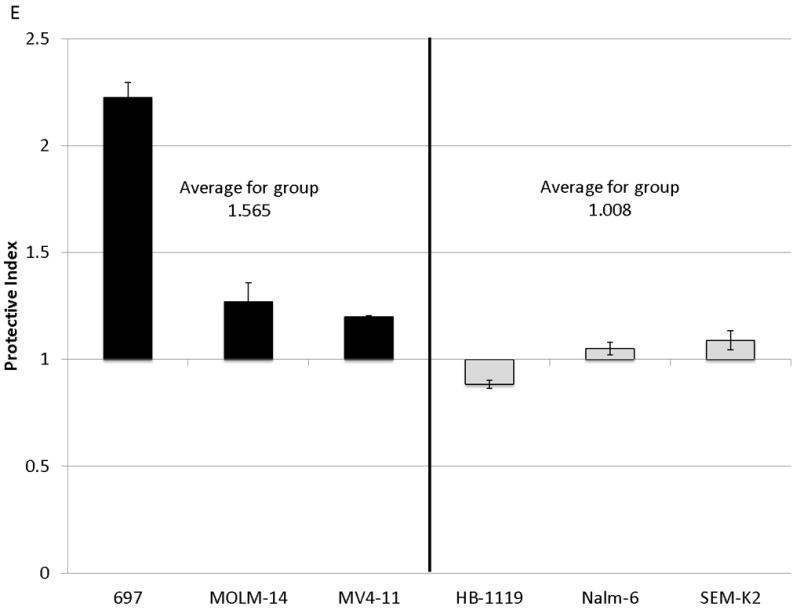
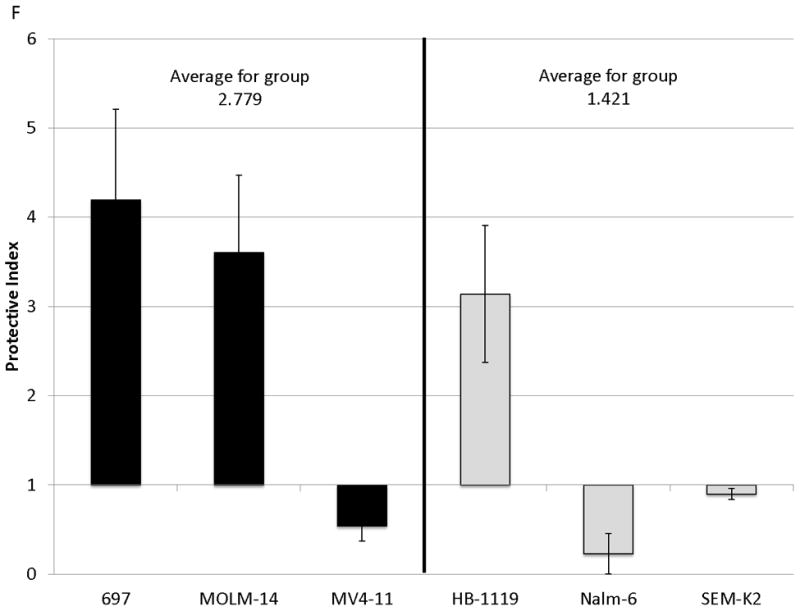
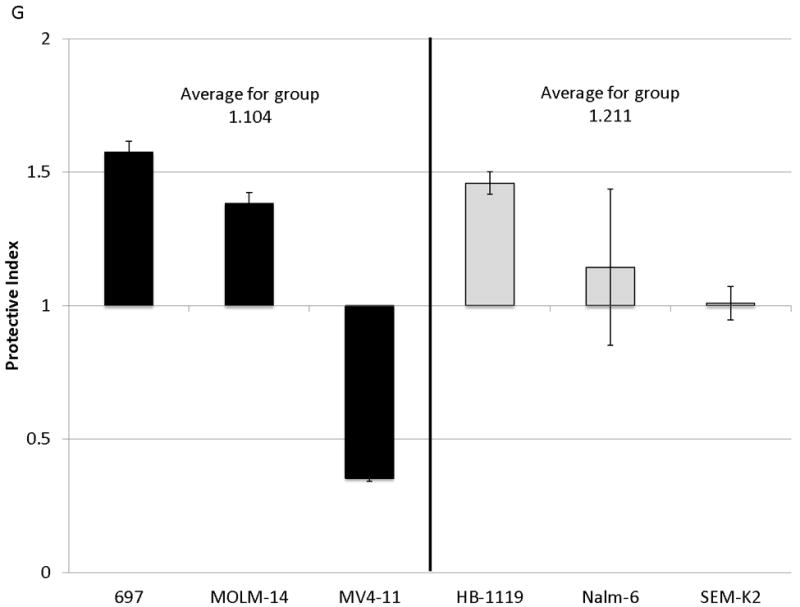
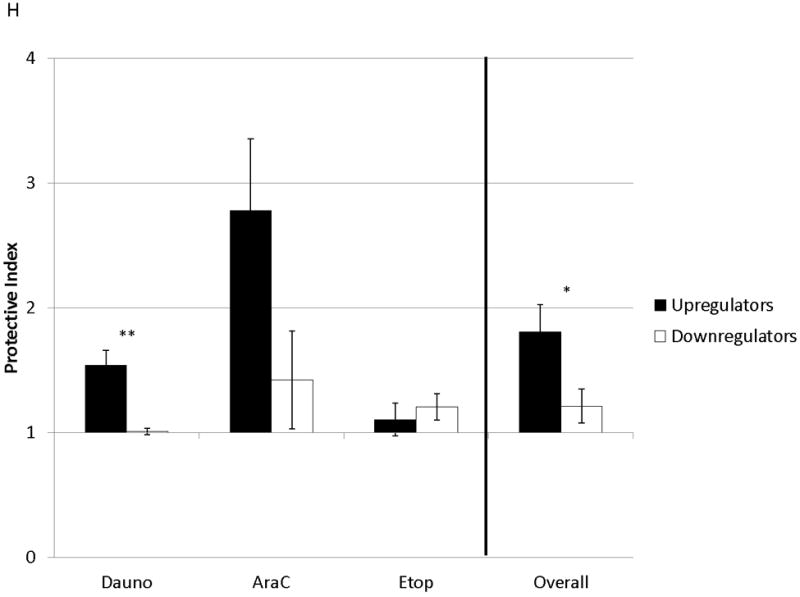
Cell lines were treated for 48 hours with dose ranges of 5 chemotherapeutic agents: daunorubicin (Dauno), vincristine (VCR), cytarabine (AraC), etoposide (Etop), and methotrexate (MTX). Surface CXCR4 expression was measured in the live populations by flow cytometry. Live gates were drawn using forward and side scatter characteristics. (a) Histograms of surface CXCR4 expression for MOLM-14 after treatment for 48 hours with daunorubicin are shown as representative examples of primary analysis. (b) Maximal changes in surface CXCR4 expression from baseline after treatment with all 5 agents are represented in the 6 cell lines. (c-h) Cells were treated with daunorubicin, cytarabine, or etoposide for 48 hours in the presence or absence of normal human bone marrow stroma feeder layers. Apoptosis was measured by Annexin V and 7-AAD staining. Examples of primary analysis after daunorubicin treatment of (c) 697 and (d) MOLM-14 are shown. To quantify the protective effect of stroma, the protective index (PI) after treatment with (e) daunorubicin, (f) cytarabine, and (g) etoposide was calculated for each cell line. (h) Collectively, CXCR4-upregulating cell lines (697, MOLM-14, and MV4-11) were preferentially protected by stroma from chemotherapy-induced apoptosis compared to CXCR4-downregulating cell lines (HB-1119, Nalm-6, and SEM-K2). *p<0.05, **p<0.01 for upregulators vs. downregulators.
Stroma provides protection from chemotherapy-induced apoptosis in cell lines that upregulate surface CXCR4
SDF-1α produced by normal bone marrow stromal cells has been shown to mediate the migration and survival of CXCR4+ HSCs.(42, 43) Studies have also suggested that patients with leukemias with higher surface expression of CXCR4 have a poorer outcome than those with lower surface expression.(22-26) Therefore, we hypothesized that upregulation of surface CXCR4 by leukemia cell lines could lead to stromal protection from chemotherapy-induced apoptosis due to increased leukemia-stromal interactions. Cells were treated for 48 hours with a dose range of daunorubicin, cytarabine, or etoposide in the presence or absence of normal human bone marrow stroma feeder layers. These agents were selected because all are used in the treatment of both ALL and AML. Apoptosis was quantified by flow cytometry, after staining with Annexin V and 7-AAD (Figs. 2C and 2D for examples of primary analysis). We then calculated a range of inhibitory concentrations (IC10 through IC90) using Calcusyn software (Biosoft, Cambridge, United Kingdom). To quantify the protective effect of stroma, we defined the protective index (PI) as the average IC values on stroma divided by the average IC values off stroma: therefore, PI >1 indicated stromal protection, PI ≤1 indicated no stromal protection. In general, cell lines that upregulated surface CXCR4 were protected from chemotherapy-induced apoptosis when plated on stroma, while those that downregulated CXCR4 showed no or minimal protection (average PI for all chemotherapeutic agents 1.807 vs. 1.211, p=0.025) (Figs. 2E-2H). These data suggest that modulation of surface CXCR4 helps to mediate stromal protection from chemotherapy.
Modulation of surface CXCR4 is driven by multiple mechanisms
To determine the kinetics and mechanism of surface CXCR4 modulation, cell lines were treated with chemotherapy for 96 hours. Doses of chemotherapy used in these experiments were the IC50 doses at 48 hours for each cell line-drug combination. The cell line-drug combinations were selected based on which combinations resulted in either a two-fold increase in surface CXCR4 expression after treatment with the 48-hour IC50 (MOLM-14 with daunorubicin, NALM-6 with cytarabine) or a 50 percent decrease in surface CXCR4 expression after treatment with the 48-hour IC50 (SEM-K2 with vincristine, HB-1119 with vincristine). Aliquots of cells were harvested at 10 time points for flow cytometric analysis and RNA isolation. Surface CXCR4 expression was measured by flow cytometry and mRNA expression of CXCR4 and its positively-regulating transcription factor NRF-1 was measured by qRT-PCR. The changes in surface CXCR4 were time-, drug-, and cell line-dependent. Simultaneous qRT-PCR measurement of CXCR4 and NRF-1 suggested specific molecular mechanisms of surface CXCR4 modulation. For example, daunorubicin-induced upregulation of surface CXCR4 in MOLM-14 started at 3 hours, but NRF-1-driven CXCR4 transcription did not increase until 48 hours (Fig. 3A), suggesting that changes in intracellular CXCR4 localization were responsible for early increases in surface CXCR4. Cytarabine-induced upregulation of surface CXCR4 in Nalm-6, on the other hand, correlated temporally with increases in NRF-1-driven CXCR4 transcription (Fig. 3B). Vincristine produced increases in NRF-1-driven CXCR4 transcription in SEM-K2 and simultaneous decreases in surface CXCR4 (Fig. 3C), suggesting CXCR4 degradation prior to cell surface localization. CXCR4 transcription also increased despite decreases in surface CXCR4 in HB-1119, also suggesting CXCR4 degradation (Fig. 3D). Next, we performed additional time course experiments to verify mechanistic patterns of surface CXCR4 upregulation. Specifically, aliquots of cells were harvested at multiple time points for flow cytometric analysis to measure expression of surface and intracellular CXCR4. As suggested by the qRT-PCR data, increases in surface CXCR4 were higher than increases in intracellular CXCR4 in daunorubicin-treated MOLM-14 cells, suggesting that there may have been cytoplasmic relocalization of CXCR4 to the cell surface that accounted for surface CXCR4 upregulation at early time points (Fig. 3E). In addition, cytarabine-induced surface CXCR4 upregulation in Nalm-6 was driven by increases in both surface and intracellular levels of CXCR4 (Fig. 3F). These experiments suggest that chemotherapy-induced modulation of CXCR4 may be driven by a number of factors and that these factors appear to vary by drug, time, and cell line.
Figure 3. Treatment of leukemia cell lines resulted in modulation of CXCR4 cell surface expression, transcription, and localization.
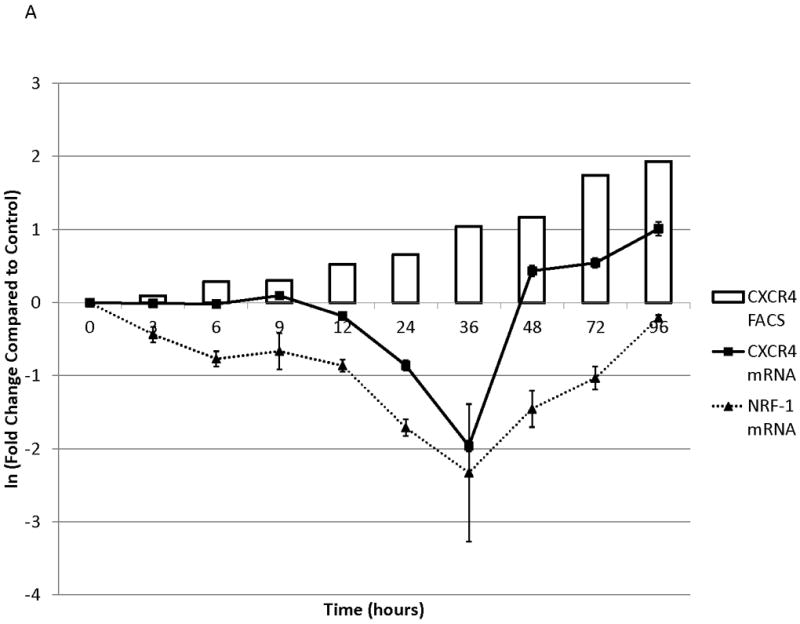
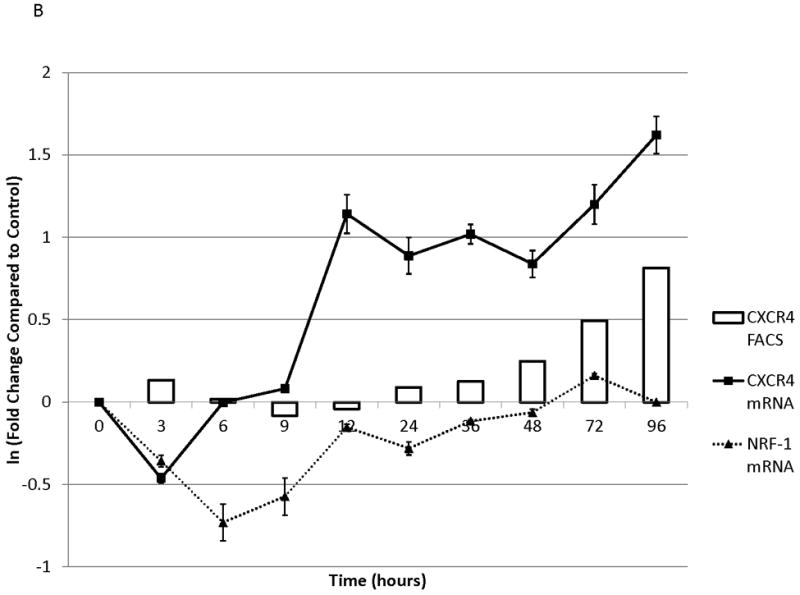
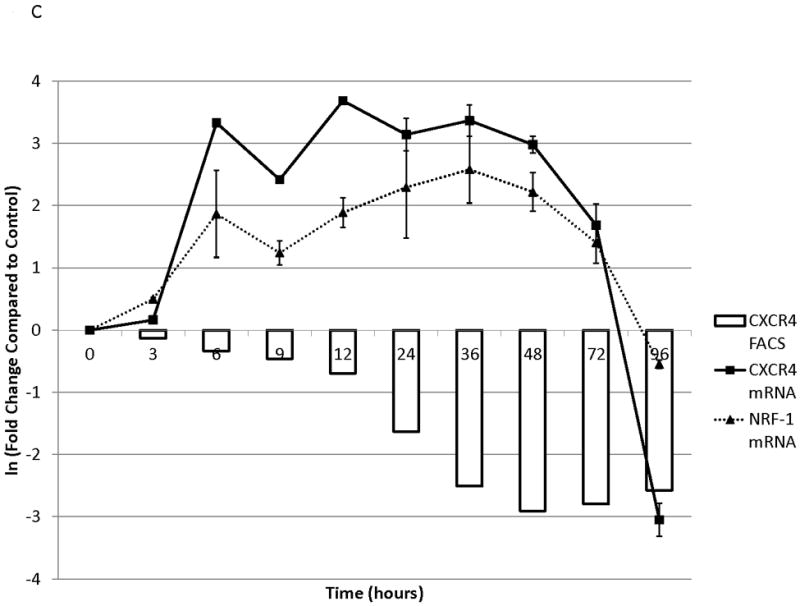
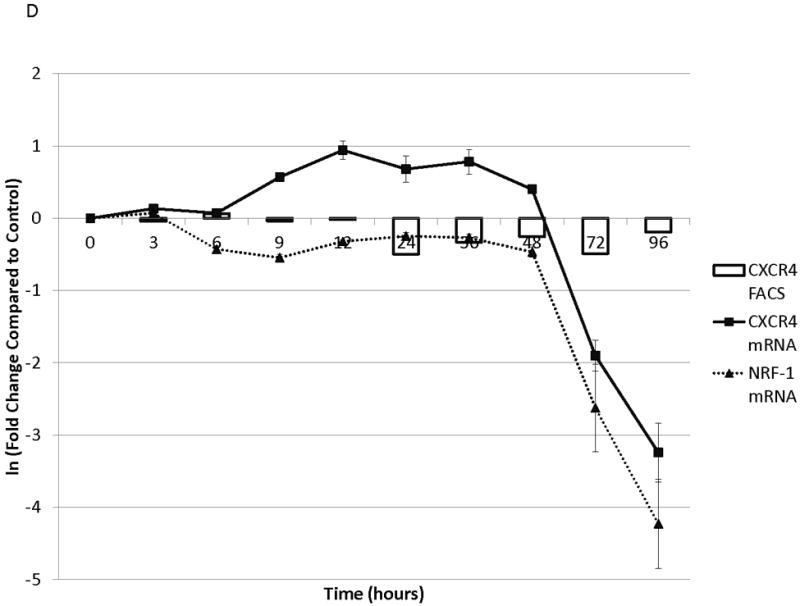
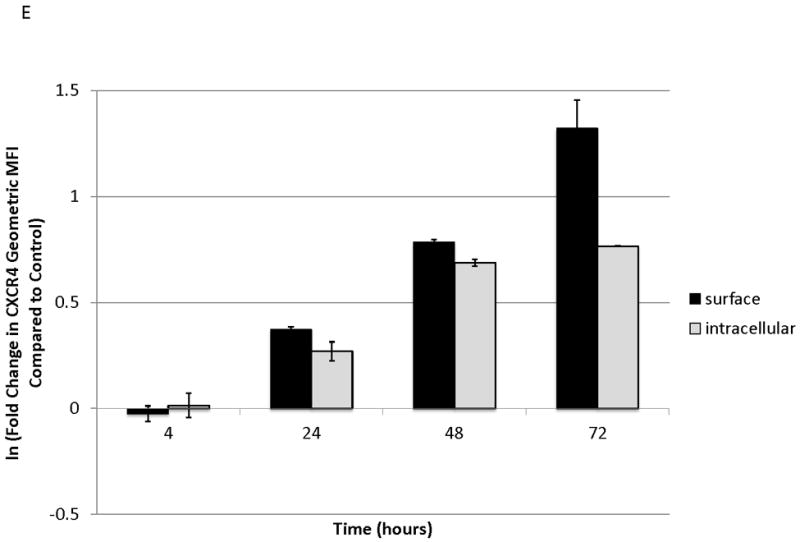
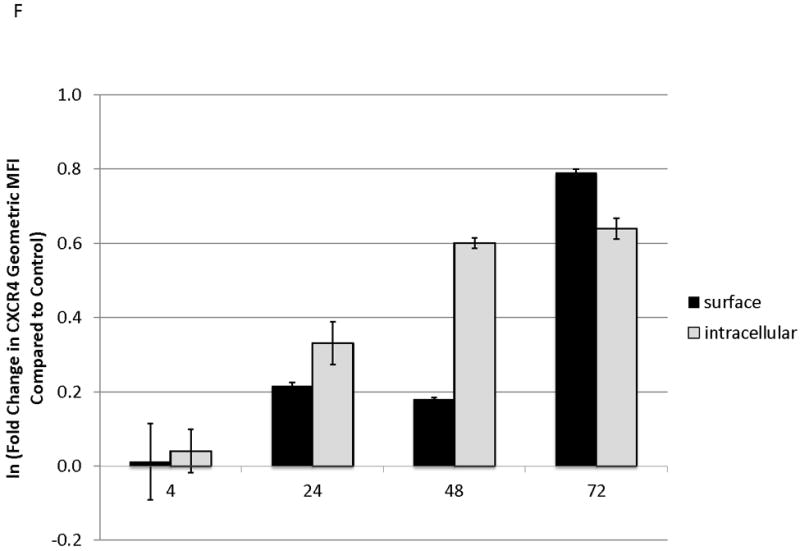
Cell lines were treated with chemotherapy and harvested at 10 time points over 96 hours. Surface CXCR4 and mRNA expression of CXCR4 and NRF1 were measured simultaneously by flow cytometry and qRT-PCR, respectively. The effects of (a) daunorubicin on MOLM-14, (b) cytarabine on Nalm-6, (c) vincristine on SEM-K2, and (d) vincristine on HB-1119 are shown. In additional experiments, changes in surface and intracellular CXCR4 induced by chemotherapy were measured by flow cytometry. (e) Surface and intracellular changes in CXCR4 induced by daunorubicin in MOLM-14 and (f) changes in CXCR4 induced by cytarabine in Nalm-6 are shown.
Chemotherapy-induced upregulation of surface CXCR4 results in increased SDF-1α-mediated chemotaxis and a stromal-mediated survival advantage
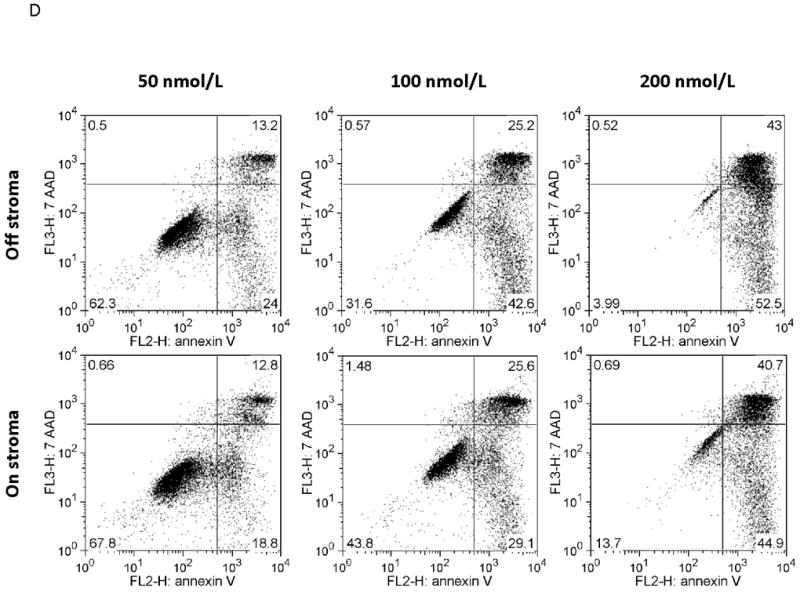
MOLM-14, an AML cell line, was the most consistent upregulator of surface CXCR4 in the above experiments. Therefore, we used MOLM-14 to test the functionality of chemotherapy-induced upregulation of surface CXCR4. Specifically, MOLM-14 cells were split into 3 aliquots and pretreated for 72 hours with chemotherapy at IC50 doses or vehicle control (Fig. 4A). Viable cells were then isolated using density centrifugation and surface CXCR4 was measured by flow cytometry. After 72 hours of pretreatment, viable chemotherapy-pretreated MOLM-14 cells had higher levels of surface CXCR4 compared to control-pretreated cells (Fig. 4B). As activation of CXCR4 leads to phosphorylation of downstream targets, including members of the PI3K/Akt and MAPK/ERK pathways,(44) we wanted to determine if increased surface CXCR4 expression translated to increased CXCR4 activation. Chemotherapy pretreatment did in fact result in higher levels of phosphorylated Akt and phosphorylated ERK 1/2, as measured by flow cytometry (Figs. 4B-4D). Viable cells from each pretreatment aliquot were then used for chemotaxis and apoptosis assays. In the chemotaxis assays, twice as many cytarabine-pretreated cells migrated toward an SDF-1α gradient compared to control-treated cells (Fig. 4E). In the apoptosis assays, cells from each pretreatment aliquot were plated on and off stroma and treated for an additional 72 hours with a full dose range of daunorubicin. We chose daunorubicin as a post-treatment because it is commonly used in AML. Pretreatment with cytarabine and vincristine resulted in striking stromal protection from daunorubicin-induced apoptosis (Fig. 4F). Collectively, these experiments demonstrate that chemotherapy-induced upregulation of surface CXCR4 results in increased SDF-1α-mediated chemotaxis and a stromal-mediated survival advantage.
Figure 4. Chemotherapy-induced upregulation of surface CXCR4 results in functional advantages, including increased chemotaxis and stromal protection from chemotherapy-induced apoptosis.
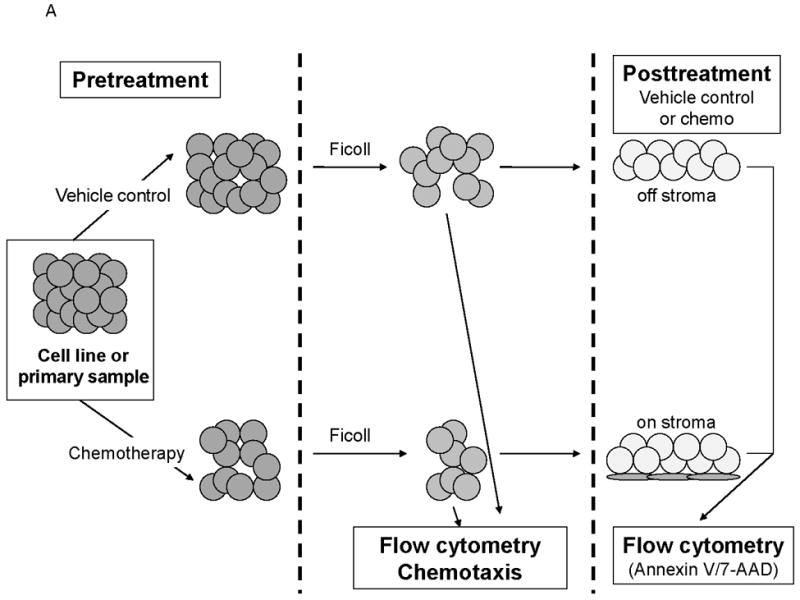
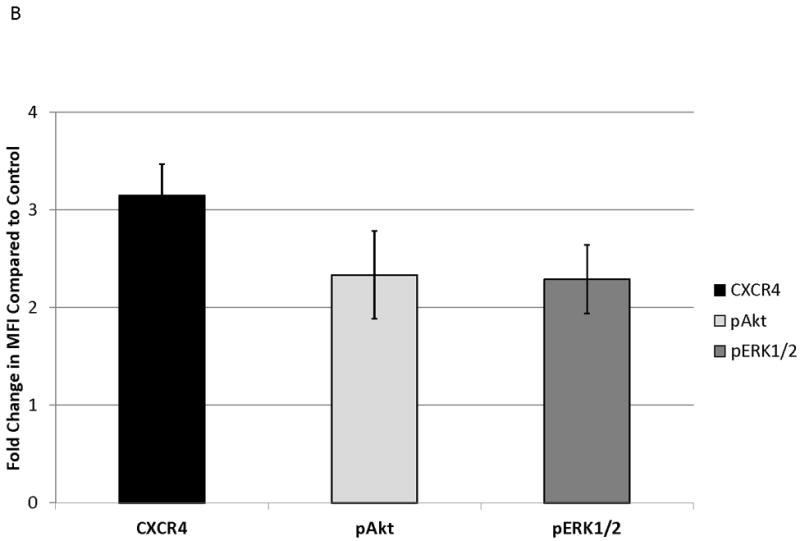
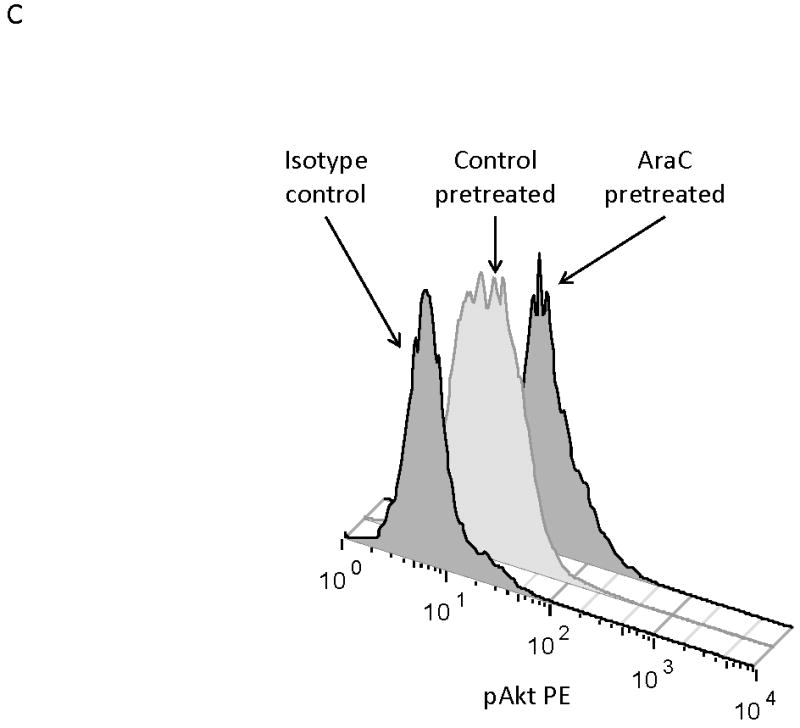
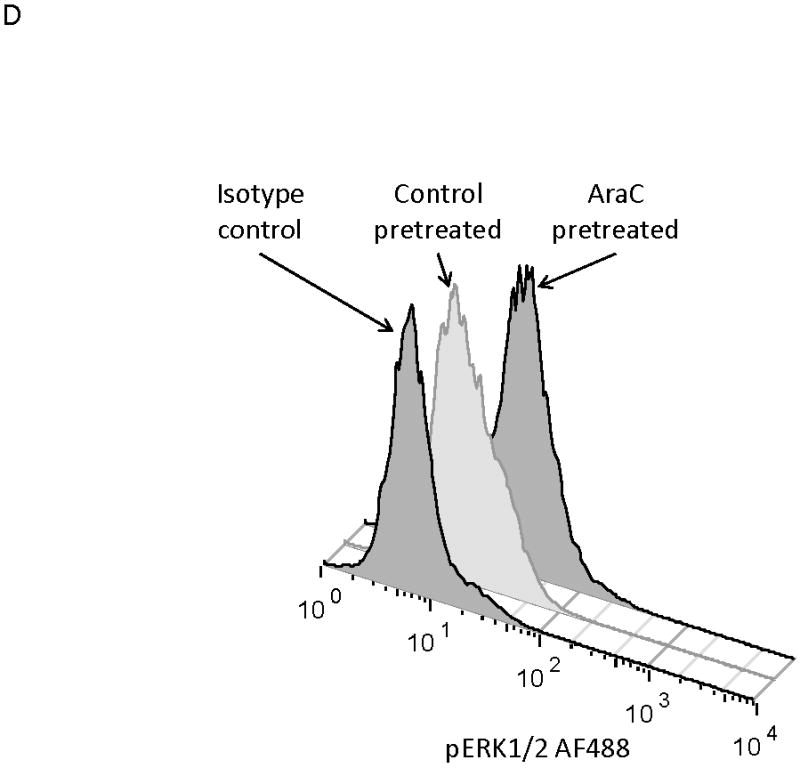
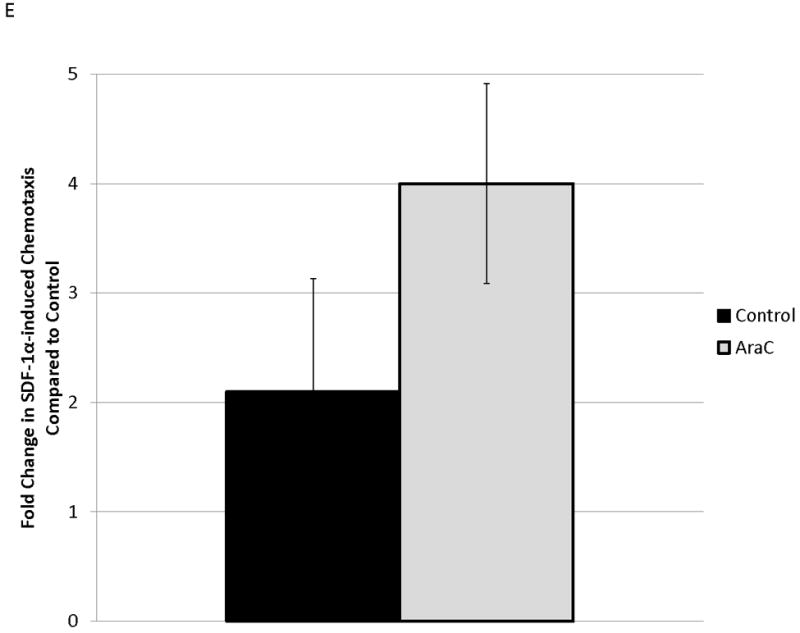
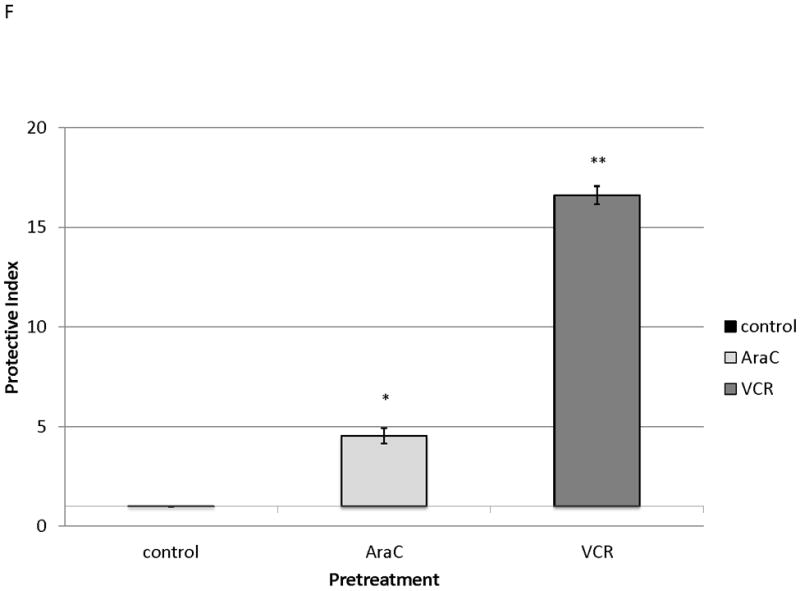
(a) This treatment schema was used for experiments described in figures 4 and 5 to assess functionality of chemotherapy-induced modulation of surface CXCR4. In figure 4, MOLM-14 cells were pretreated with vehicle control or chemotherapy for 72 hours. Viable cells were isolated for flow cytometry, chemotaxis, and apoptosis assays. (b) Expression of surface CXCR4, phosphorylated Akt, and phosphorylated ERK1/2 was measured in duplicate by flow cytometry after pretreatment with vehicle control or cytarabine. Representative histograms of (c) phosphorylated Akt and (d) phosphorylated ERK1/2 expression after control or cytarabine pretreatment are depicted. (e) Control- and cytarabine-pretreated MOLM-14 cells were seeded in hanging cell culture inserts and placed into 24-well plates containing supplemented media alone (control) or supplemented media with SDF-1α (75 ng/mL). Migrated cells were measured in triplicate after 4 hours. (f) Viable cells from each pretreatment aliquot were plated on and off stroma and treated for an additional 72 hours with a dose range of daunorubicin. PI was calculated to quantify the protective effect of stroma. *p<0.05, **p<0.01 vs. control.
Chemotherapy modulates surface CXCR4 expression in primary pediatric AML samples
We observed functional advantages conferred by upregulation of CXCR4 in MOLM-14, an AML cell line. We therefore sought to determine if similar phenomena occurred in primary samples of pediatric AML. We first tested whether surface CXCR4 expression changed in response to chemotherapy. Primary AML samples (n=8, characteristics listed in Table 1) were treated for 48 hours with dose ranges of 3 chemotherapeutic agents used in the treatment of AML (cytarabine, daunorubicin, and etoposide). Baseline levels of surface CXCR4 expression were variable by primary sample (Fig. 5A). Maximal changes in surface CXCR4 were variable by sample and by drug, and individual samples did not always respond to all drugs in the same manner (Fig. 5B).
Table 1.
Characteristics of pediatric AML patient samples.
| Sample | Age at diagnosis (years) | WBC at diagnosis (×1000/uL) | FAB subtype | Cytogenetics/Molecular features |
|---|---|---|---|---|
| 1 | 16 | 155 | M2 | monosomy 7, t(9;22)(q34;q11.2) |
| 2 | 4 | 236 | M4Eo | inv(16)(p13.1q22) |
| 3 | 13 | 100 | M4 | inv(16)(p13.1q22) |
| 4 | 12 | 155 | M4 | t(6;11)(q27;q23) |
| 5 | 17 | 69 | M5 | t(11;22;17)(q23;q13;q11.2) |
| 6 | 7 | 130 | M4 | t(5;11)(q35;p11.2) |
| 7 | 13 | 353 | M5 | FLT3/ITD |
| 8 | 17 | 173 | M2 | inv(3)(q21q26.2) |
Age, white blood cell count, French-American-British (FAB) subtype, and molecular features of the pediatric AML patient samples are listed.
Figure 5. Chemotherapy modulates surface CXCR4 levels and affects the degree of stromal protection in primary pediatric AML samples.
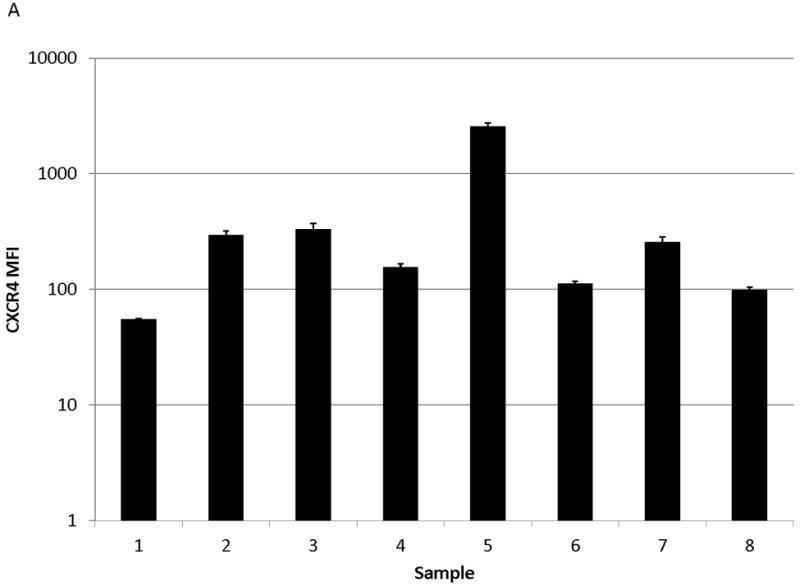
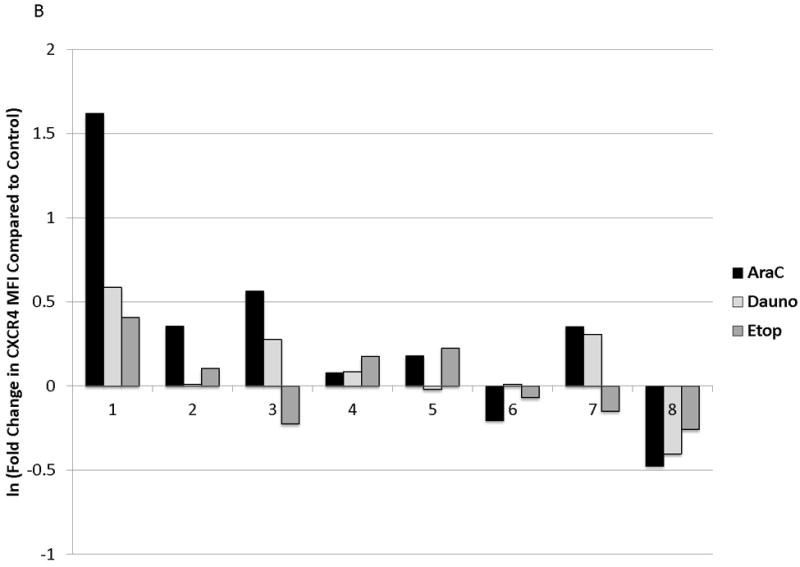
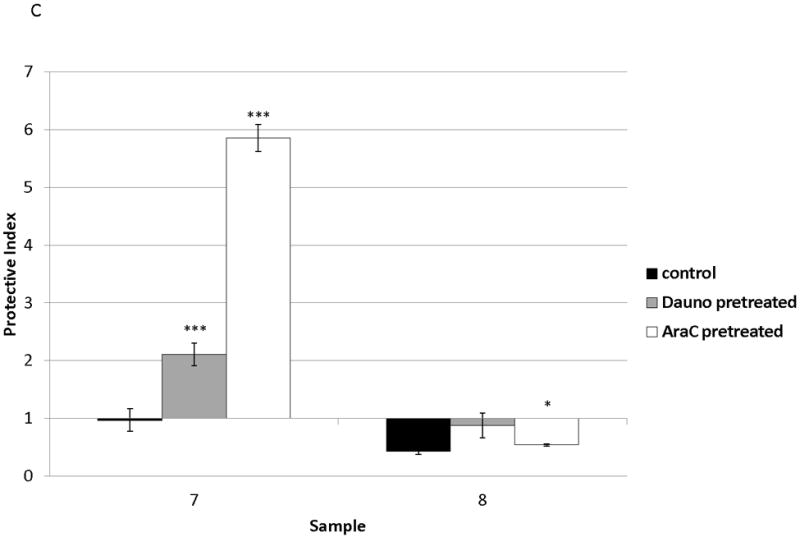
(a) Surface CXCR4 expression in viable, untreated primary samples was measured in triplicate by flow cytometry. (b) Primary pediatric AML samples were treated for 48 hours with dose ranges of chemotherapy. Changes in surface CXCR4 in response to IC50 doses of chemotherapy was measured in the live populations by flow cytometry. (c) Primary samples 7 and 8 were pretreated with vehicle control, daunorubicin, or cytarabine for 48 hours. Viable cells from each pretreatment aliquot were plated on and off stroma and treated for an additional 48 hours with a dose range of daunorubicin. PI was calculated to quantify the protective effect of stroma. *p<0.05, ***p<0.0001 vs. control.
Chemotherapy-induced changes in surface CXCR4 affect stromal protection of primary pediatric AML samples
We sought to determine the effect of changes in surface CXCR4 on stromal protection from chemotherapy-induced apoptosis in primary pediatric AML samples. We chose to study primary samples 7 and 8 because we had enough cells from these samples to perform the treatment schema depicted in Fig. 4A. Pretreatment of primary samples with IC50 doses of chemotherapy was used to induce changes in surface CXCR4. Pretreatment of sample 7 with daunorubicin or cytarabine for 48 hours resulted in 20 and 36 percent increases in surface CXCR4, respectively, compared to control-pretreated cells (Fig. 5B). In contrast, pretreatment of sample 8 with daunorubicin and cytarabine caused 33 and 50 percent decreases in surface CXCR4, compared with control-pretreated cells (Fig. 5B). Viable cells were then isolated, plated on or off stroma, and treated with a dose range of daunorubicin. We again chose daunorubicin as a post-treatment because it is commonly used in AML. Stroma conferred protection from daunorubicin-induced apoptosis in sample 7, while sample 8 was not protected by stroma (Fig. 5C). These results suggest that increases or decreases in surface CXCR4 induced by chemotherapy may modulate the survival advantage conferred by stroma in pediatric AML.
Plerixafor decreases stromal protection and increases chemosensitivity in AML with chemotherapy-induced upregulation of surface CXCR4
We and others have demonstrated that combining chemotherapy or TKIs with a CXCR4 inhibitor decreases the protective effect of stroma in ALL and AML.(10, 27-31) Therefore, to determine if stromal protection in AML cells with chemotherapy-induced upregulation of surface CXCR4 expression was mediated by CXCR4, we cultured MOLM-14 on stroma with or without the CXCR4 inhibitor plerixafor and compared chemotherapy-induced apoptosis by culture and pretreatment conditions. Similar to our previous experiments, we pretreated MOLM-14 with chemotherapy at IC50 doses (cytarabine, daunorubicin, and vincristine) or vehicle control for 72 hours. Viable cells were isolated using density centrifugation and increases in surface CXCR4 expression in chemotherapy pretreated cells was verified by FACS (Fig. 6A). Cells from each pretreatment were then plated on stroma and treated with a dose range of daunorubicin with or without plerixafor. Following treatment, apoptosis was measured by flow cytometry after staining for Annexin V and 7-AAD. Control-pretreated MOLM-14 had a minimal increase in chemosensitivity with plerixafor (Fig. 6B). MOLM-14 pretreated with daunorubicin showed some induction of chemosensitivity with plerixafor at intermediate doses of daunorubicin (Fig. 6C). Interestingly, plerixafor was able to induce the greatest chemosensitization effect in MOLM-14 pretreated with either vincristine (Fig. 6D) or cytarabine (Fig. 6E). As vincristine and cytarabine induced the greatest increases in surface CXCR4 expression (Fig. 6A), these results suggest that plerixafor diminishes stromal protection more effectively in leukemias that upregulate surface CXCR4 in response to chemotherapy.
Figure 6. Plerixafor decreases stromal protection and increases chemosensitivity in MOLM-14 with chemotherapy-induced upregulation of surface CXCR4.
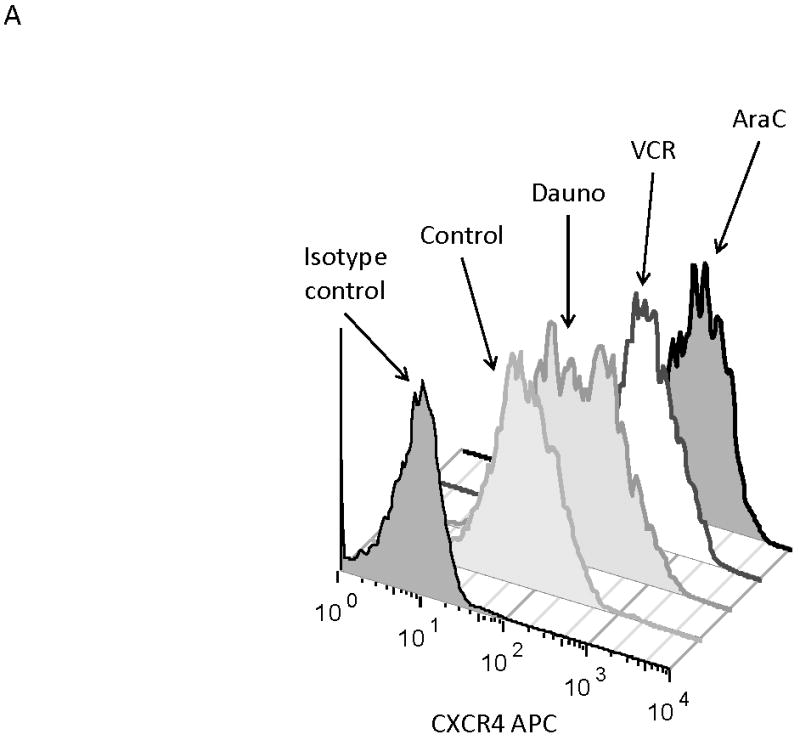
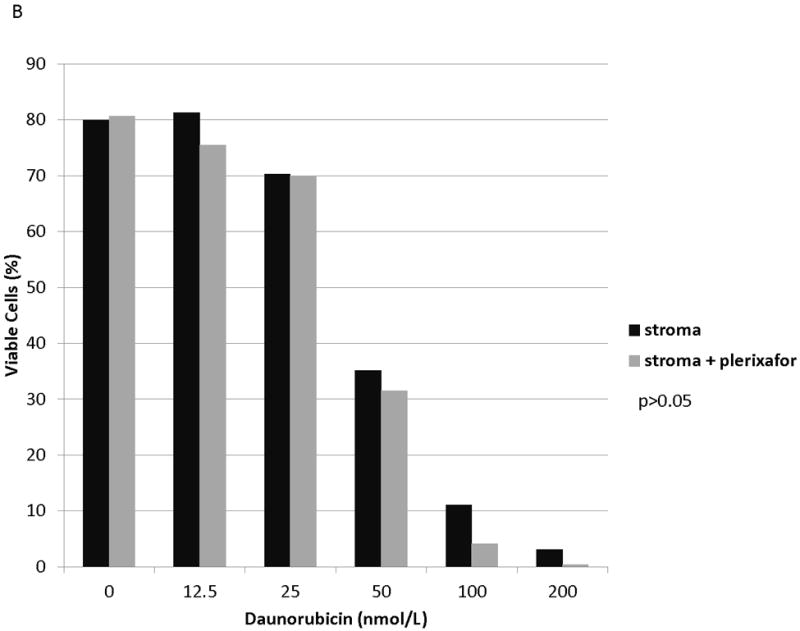
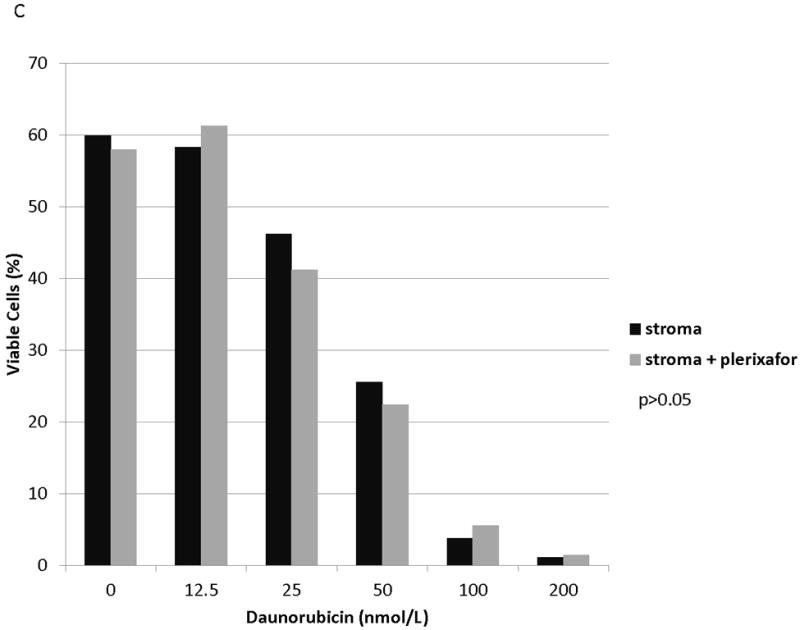
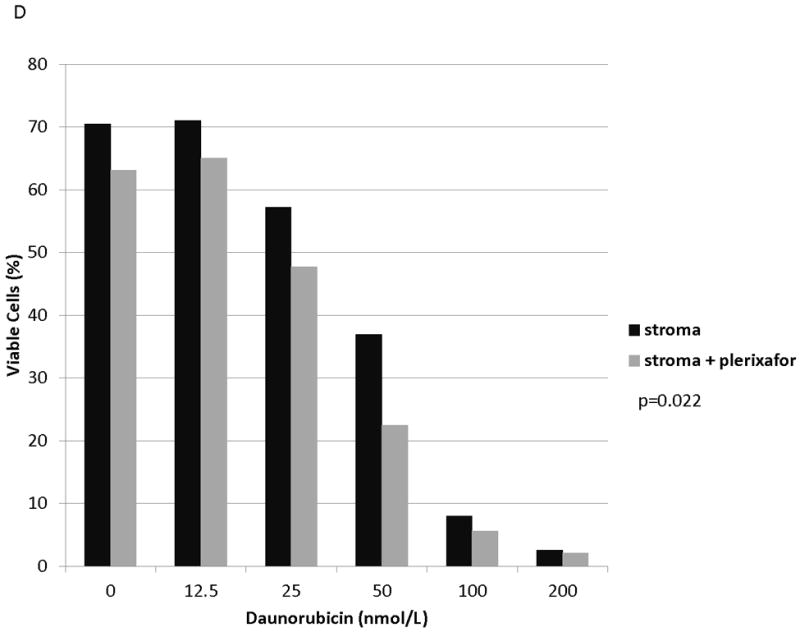
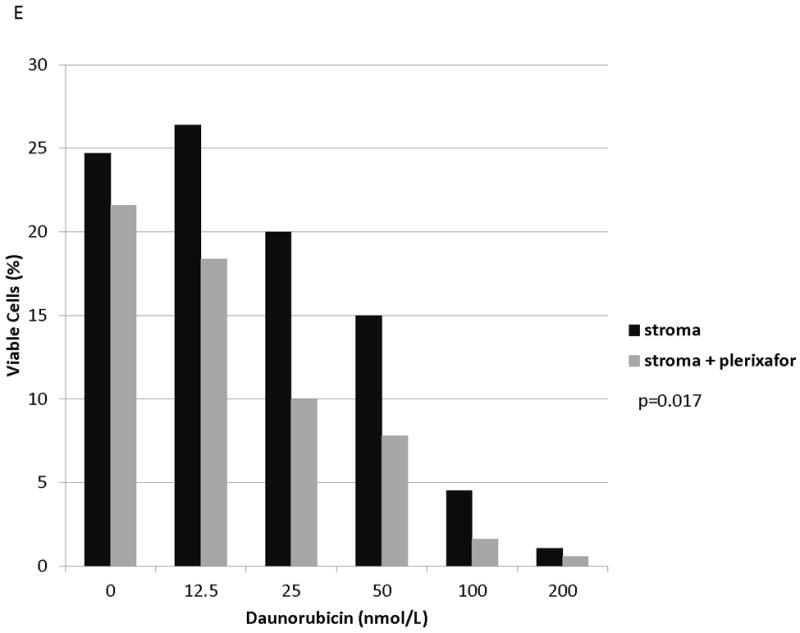
(a) MOLM-14 cells were pretreated with vehicle control or chemotherapy for 72 hours. Surface CXCR4 expression in live cells was measured by flow cytometry. (b-e) Viable cells were plated on stroma and treated with a dose range of daunorubicin with or without 5 μmol/L plerixafor. Following treatment, cells were harvested and stained with Annexin V and 7-AAD. Viable cells are negative for both Annexin V and 7-AAD. Results after pretreatment with (b) vehicle control, (c) daunorubicin, (d) vincristine, and (e) cytarabine are shown. P values compare overall treatment effects by chemotherapeutic agent of the stroma vs. stroma + plerixafor culture conditions.
Discussion
Pediatric AML continues to be difficult to cure in spite of improvements in treatment, including the use of intensive combination chemotherapeutic treatment regimens.(1) For reasons that are not fully understood, a number of leukemias, including leukemias with low-risk cytogenetic features, relapse or are refractory to current therapies. It is possible that survival signals from the bone marrow microenvironment allow subpopulations of leukemia cells to evade chemotherapy-induced death. Therefore, chemotherapy resistance and support of leukemogenesis may not be mediated exclusively by the leukemia itself: its environment may also play a significant role. This idea is substantiated by recent evidence that suggests that the presence of an altered microenvironment can initiate the development of dysplasia and leukemia.(45)
The SDF-1/CXCR4 axis is an important pathway in hematopoiesis and hematologic malignancies. Overexpression of CXCR4 in CD34+ HSCs enhances SDF-1α-mediated chemotaxis, in vitro survival, and engraftment,(42) and increased CXCR4 expression mediates migration and engraftment of AML in a xenograft mouse model.(16) Several studies have reported that increased CXCR4 expression by leukemic blasts portends a poor prognosis in AML,(24-26) suggesting that CXCR4 expression impacts response to therapy. CXCR4 also mediates pseudoemperipolesis, or migration under a monolayer of bone marrow stromal cells, of AML blasts.(21) Pseudoemperipolesis is thought to emulate migration of leukemia cells to microscopic niches in the bone marrow microenvironment and demonstrates the importance of receptor-ligand interactions in leukemia. Our experiments supported these findings, as a leukemia cell line with high surface expression of CXCR4 had increased SDF-1α-induced chemotaxis compared to a cell line with low surface expression of CXCR4.
In addition, CXCR4 expression is affected by anti-cancer agents, suggesting that residual leukemic blasts may have different interactions with the microenvironment after treatment than at diagnosis. For example, the HDAC inhibitor suberoylanilide hydroxamic acid caused decreased CXCR4 expression in chronic lymphocytic leukemia (CLL) primary samples, which resulted in decreased SDF-1α-induced chemotaxis.(33) In contrast, valproic acid induced increased CXCR4 expression, causing increased SDF-1α-mediated chemotaxis.(32) Similarly, imatinib caused increased CXCR4 in chronic myelogenous leukemia cell lines, which functionally resulted in increased stroma-induced migration and quiescence,(34) and dasatinib induced upregulation of surface CXCR4 in Philadelphia-chromosome positive ALL cell lines.(35) Our experiments add to the evidence that anti-cancer agents can modulate surface CXCR4 expression. It is clear that the presence of minimal residual disease after induction therapy portends a poor prognosis in pediatric ALL and AML. To simulate this situation in vitro, we purposely pretreated our cell lines and primary samples with suboptimal doses of chemotherapy in our chemotaxis and apoptosis experiments so as not to completely eradicate the leukemias. In those experiments, treatment with suboptimal doses of chemotherapy led to increased surface CXCR4 expression in cell lines and primary samples, suggesting that inadequate doses of chemotherapy may enhance leukemia-microenvironment interactions. We recognize that CXCR4 modulation and the subsequent effects on stromal protection were variable by drug and cell line. For example, etoposide had a lesser effect on stromal protection than daunorubicin and cytarabine, and HB-1119 and MV4-11 did not always show stromal protection consistent with the direction of CXCR4 modulation. It is certainly possible that differences in each cell line and/or the mechanisms of action of each chemotherapeutic agent led to variations in the degree of stromal protection. We also acknowledge that our experiments were performed using cell lines and bulk leukemia primary samples, and it would be interesting to learn what effects chemotherapy has on CXCR4 expression in LSCs. These are interesting issues that we are beginning to explore experimentally. Ultimately, our results highlight that standard chemotherapy may increase leukemia-microenvironment interactions in residual, viable blasts, which in turn leads to chemotherapy resistance.
Previous work has suggested that leukemia-microenvironment interactions may play an important role in the pathogenesis of high-risk pediatric acute leukemias. For example, we previously demonstrated that stroma preferentially protects MLL-R ALL patient samples and that ex vivo culture of MLL-R primary samples with stroma prior to transplantation into immunodeficient mice promotes the survival of LSCs.(10) In addition, poor clinical outcome(46) and poor cytogenetic risk features(47) have been shown to be determinants of AML patient sample engraftment in immunodeficient mice. However, the role of CXCR4 in the engraftment of AML primary samples remains unclear. Some studies have suggested that higher CXCR4 expression is associated with engraftment of HSCs(42) and AML primary samples(16, 48) in immunodeficient mice, while others have suggested that engraftment of AML primary samples is CXCR4-independent.(46, 47) It appears that leukemia-stromal interactions are multifactorial but it is clear that CXCR4 is a major element.
There is evidence from the literature that FLT3/ITD+ AML cell lines demonstrate increased chemotaxis toward SDF-1α compared to FLT3-wild type cell lines, suggesting that the presence of FLT3/ITD enhances SDF-1/CXCR4 signaling.(49) An additional study found that stromal co-culture results in higher rates of proliferation for FLT3/ITD+ AML blasts compared to FLT3-wild type AML blasts,(31) indicating that leukemia cell interactions with bone marrow stromal cells impart a survival advantage in FLT3/ITD+ leukemia. Both of the AML cell lines (MOLM-14 and MV4-11) tested in our experiments upregulated surface CXCR4 expression in response to chemotherapy. Interestingly, both AML cell lines harbor a FLT3/ITD mutation, which is a high-risk cytogenetic feature in AML. Upregulation of surface CXCR4 in MOLM-14 led to functional advantages, as measured by chemotaxis and stromal protection from additional treatment with chemotherapy. Therefore, we chose to study AML primary samples to determine if these phenomena could be demonstrated using primary samples. Sample 7, which harbored a FLT3/ITD mutation, upregulated surface CXCR4 in response to treatment with daunorubicin and cytarabine and chemotherapy-pretreated cells were protected from daunorubicin-induced apoptosis by stromal co-culture. In contrast, Sample 8, which did not have a low- or high-risk cytogenetic feature, downregulated surface CXCR4 in response to chemotherapy and chemotherapy-pretreated cells were not protected from daunorubicin-induced apoptosis by stromal co-culture. Therefore, it is conceivable that FLT3/ITD+ AML is high-risk and difficult to treat in part because of increased interactions with the bone marrow stromal microenvironment mediated by CXCR4 and SDF-1α. This hypothesis certainly warrants further study.
Our data not only suggest that upregulation of surface CXCR4 is a mechanism of stromal-mediated chemotherapy resistance but that this resistance can be reversed with CXCR4 inhibition. Preclinical studies of CXCR4 inhibition in AML have demonstrated that small molecules and antibodies against CXCR4 causes decreased survival and proliferation and increased sensitivity to anti-cancer therapies both in vitro and in vivo.(16, 28-30) This treatment strategy has recently also moved into the clinic. Most notably, a phase 1/2 trial of plerixafor in combination with mitoxantrone, etoposide, and cytarabine in adults with relapsed or refractory AML demonstrated tolerability, mobilization of AML blasts into the peripheral blood, and resulted in a rate of complete remission (with or without complete blood count recovery) of 46 percent.(50) Ongoing clinical trials of CXCR4 antagonism as a chemosensitization strategy in hematologic malignancies include phase 1 trials of the anti-CXCR4 antibody MDX-1338 alone or in combination with chemotherapy in patients with relapsed or refractory AML, CLL, diffuse large B cell lymphoma, follicular lymphoma, and multiple myeloma; a phase 1 trial of plerixafor in combination with cytarabine and daunorubicin in newly-diagnosed adults with AML; and a phase 1 trial of plerixafor in combination with high-dose cytarabine and etoposide in children and young adults with relapsed or refractory ALL, AML, MDS, and acute leukemias of ambiguous lineage. Inhibition of leukemia-microenvironment interactions is an attractive therapeutic strategy and may enhance the effectiveness of chemotherapy and targeted agents. Our data suggest that the degree of surface CXCR4 upregulation may be a biomarker with the potential to identify optimal patients for CXCR4 inhibition.
We have demonstrated that surface CXCR4 expression is dynamically altered by chemotherapy exposure in ALL and AML cells, and may be driven by transcription, changes in localization of CXCR4, CXCR4 degradation, or a combination of mechanisms. Chemotherapy-induced upregulation of surface CXCR4 results in increased SDF-1α-mediated chemotaxis and a stromal-mediated survival advantage in pediatric AML. Notably, plerixafor decreased stromal protection in chemotherapy-pretreated cells that upregulated surface CXCR4, suggesting that stromal protection is at least in part mediated by CXCR4. Upregulation of surface CXCR4 may therefore represent a mechanism of chemotherapy resistance in pediatric AML.
Acknowledgments
Financial support: This work was supported by a Leukemia and Lymphoma Society (LLS) Translational Research Program Grant, LLS Scholar in Clinical Research Award, and American Cancer Society Research Scholar Grant to Patrick Brown; and by a National Institutes of Health grant T32 CA60441, St. Baldrick’s Foundation Post-Doctoral Fellowship for Childhood Cancer Research, and Conquer Cancer Foundation of the American Society of Clinical Oncology Young Investigator Award to Edward Allan R. Sison.
Authorship
E.A.S., E.M., and D.M. performed the research; E.A.S. and P.B. designed the research study, analyzed the data, and wrote the paper.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: An update. J Clin Oncol. 2011;29(5):551–65. doi: 10.1200/JCO.2010.30.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown P, McIntyre E, Rau R, Meshinchi S, Lacayo N, Dahl G, et al. The incidence and clinical significance of nucleophosmin mutations in childhood AML. Blood. 2007;110(3):979–85. doi: 10.1182/blood-2007-02-076604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ho PA, Alonzo TA, Gerbing RB, Pollard J, Stirewalt DL, Hurwitz C, et al. Prevalence and prognostic implications of CEBPA mutations in pediatric acute myeloid leukemia (AML): A report from the children’s oncology group. Blood. 2009;113(26):6558–66. doi: 10.1182/blood-2008-10-184747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yin T, Li L. The stem cell niches in bone. J Clin Invest. 2006;116(5):1195–201. doi: 10.1172/JCI28568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Méndez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466(7308):829–34. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008;322(5909):1861–5. doi: 10.1126/science.1164390. [DOI] [PubMed] [Google Scholar]
- 7.Ayala F, Dewar R, Kieran M, Kalluri R. Contribution of bone microenvironment to leukemogenesis and leukemia progression. Leukemia. 2009;23(12):2233–41. doi: 10.1038/leu.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manabe A, Coustan-Smith E, Behm FG, Raimondi SC, Campana D. Bone marrow-derived stromal cells prevent apoptotic cell death in B-lineage acute lymphoblastic leukemia. Blood. 1992;79(9):2370–7. [PubMed] [Google Scholar]
- 9.Iwamoto S, Mihara K, Downing JR, Pui CH, Campana D. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J Clin Invest. 2007;117(4):1049–57. doi: 10.1172/JCI30235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sison EA, Rau RE, McIntyre E, Li L, Small D, Brown P. MLL-rearranged acute lymphoblastic leukaemia stem cell interactions with bone marrow stroma promote survival and therapeutic resistance that can be overcome with CXCR4 antagonism. Br J Haematol. 2013;160(6):785–97. doi: 10.1111/bjh.12205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, Fujimi A, et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. 2003;9(9):1158–65. doi: 10.1038/nm909. [DOI] [PubMed] [Google Scholar]
- 12.Peled A, Kollet O, Ponomaryov T, Petit I, Franitza S, Grabovsky V, et al. The chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34+ cells: Role in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood. 2000;95(11):3289–96. [PubMed] [Google Scholar]
- 13.Walter RB, Alonzo TA, Gerbing RB, Ho PA, Smith FO, Raimondi SC, et al. High expression of the very late antigen-4 integrin independently predicts reduced risk of relapse and improved outcome in pediatric acute myeloid leukemia: A report from the children’s oncology group. J Clin Oncol. 2010;28(17):2831–8. doi: 10.1200/JCO.2009.27.5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loetscher M, Geiser T, O’Reilly T, Zwahlen R, Baggiolini M, Moser B. Cloning of a human seven-transmembrane domain receptor, LESTR, that is highly expressed in leukocytes. J Biol Chem. 1994;269(1):232–7. [PubMed] [Google Scholar]
- 15.Peled A, Petit I, Kollet O, Magid M, Ponomaryov T, Byk T, et al. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science. 1999;283(5403):845–8. doi: 10.1126/science.283.5403.845. [DOI] [PubMed] [Google Scholar]
- 16.Tavor S, Petit I, Porozov S, Avigdor A, Dar A, Leider-Trejo L, et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 2004;64(8):2817–24. doi: 10.1158/0008-5472.can-03-3693. [DOI] [PubMed] [Google Scholar]
- 17.Tashiro K, Tada H, Heilker R, Shirozu M, Nakano T, Honjo T. Signal sequence trap: A cloning strategy for secreted proteins and type I membrane proteins. Science. 1993;261(5121):600–3. doi: 10.1126/science.8342023. [DOI] [PubMed] [Google Scholar]
- 18.Nagasawa T, Kikutani H, Kishimoto T. Molecular cloning and structure of a pre-B-cell growth-stimulating factor. Proc Natl Acad Sci U S A. 1994;91(6):2305–9. doi: 10.1073/pnas.91.6.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bleul CC, Fuhlbrigge RC, Casasnovas JM, Aiuti A, Springer TA. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1) J Exp Med. 1996;184(3):1101–9. doi: 10.1084/jem.184.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell’Aquila M, Kipps TJ. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood. 2000 Oct 15;96(8):2655–63. [PubMed] [Google Scholar]
- 21.Burger JA, Spoo A, Dwenger A, Burger M, Behringer D. CXCR4 chemokine receptors (CD184) and alpha4beta1 integrins mediate spontaneous migration of human CD34+ progenitors and acute myeloid leukaemia cells beneath marrow stromal cells (pseudoemperipolesis) Br J Haematol. 2003;122(4):579–89. doi: 10.1046/j.1365-2141.2003.04466.x. [DOI] [PubMed] [Google Scholar]
- 22.Crazzolara R, Kreczy A, Mann G, Heitger A, Eibl G, Fink FM, et al. High expression of the chemokine receptor CXCR4 predicts extramedullary organ infiltration in childhood acute lymphoblastic leukaemia. Br J Haematol. 2001;115(3):545–53. doi: 10.1046/j.1365-2141.2001.03164.x. [DOI] [PubMed] [Google Scholar]
- 23.Schneider P, Vasse M, Al Bayati A, Lenormand B, Vannier JP. Is high expression of the chemokine receptor CXCR-4 of predictive value for early relapse in childhood acute lymphoblastic leukaemia? Br J Haematol. 2002;119(2):579–80. doi: 10.1046/j.1365-2141.2002.03835_6.x. [DOI] [PubMed] [Google Scholar]
- 24.Rombouts EJ, Pavic B, Löwenberg B, Ploemacher RE. Relation between CXCR-4 expression, Flt3 mutations, and unfavorable prognosis of adult acute myeloid leukemia. Blood. 2004;104(2):550–7. doi: 10.1182/blood-2004-02-0566. [DOI] [PubMed] [Google Scholar]
- 25.Konoplev S, Rassidakis GZ, Estey E, Kantarjian H, Liakou CI, Huang X, et al. Overexpression of CXCR4 predicts adverse overall and event-free survival in patients with unmutated FLT3 acute myeloid leukemia with normal karyotype. Cancer. 2007;109(6):1152–6. doi: 10.1002/cncr.22510. [DOI] [PubMed] [Google Scholar]
- 26.Spoo AC, Lübbert M, Wierda WG, Burger JA. CXCR4 is a prognostic marker in acute myelogenous leukemia. Blood. 2007;109(2):786–91. doi: 10.1182/blood-2006-05-024844. [DOI] [PubMed] [Google Scholar]
- 27.Juarez J, Bradstock KF, Gottlieb DJ, Bendall LJ. Effects of inhibitors of the chemokine receptor CXCR4 on acute lymphoblastic leukemia cells in vitro. Leukemia. 2003;17(7):1294–300. doi: 10.1038/sj.leu.2402998. [DOI] [PubMed] [Google Scholar]
- 28.Nervi B, Ramirez P, Rettig MP, Uy GL, Holt MS, Ritchey JK, et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood. 2009;113(24):6206–14. doi: 10.1182/blood-2008-06-162123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tavor S, Eisenbach M, Jacob-Hirsch J, Golan T, Petit I, Benzion K, et al. The CXCR4 antagonist AMD3100 impairs survival of human AML cells and induces their differentiation. Leukemia. 2008;22(12):2151–5158. doi: 10.1038/leu.2008.238. [DOI] [PubMed] [Google Scholar]
- 30.Zeng Z, Shi YX, Samudio IJ, Wang RY, Ling X, Frolova O, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113(24):6215–24. doi: 10.1182/blood-2008-05-158311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacobi A, Thieme S, Lehmann R, Ugarte F, Malech HL, Koch S, et al. Impact of CXCR4 inhibition on Flt3-ITD-positive human AML blasts. Exp Hematol. 2010;38(3):180–90. doi: 10.1016/j.exphem.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gul H, Marquez-Curtis LA, Jahroudi N, Lo J, Turner AR, Janowska-Wieczorek A. Valproic acid increases CXCR4 expression in hematopoietic stem/progenitor cells by chromatin remodeling. Stem Cells Dev. 2009;18(6):831–8. doi: 10.1089/scd.2008.0235. [DOI] [PubMed] [Google Scholar]
- 33.Stamatopoulos B, Meuleman N, De Bruyn C, Delforge A, Bron D, Lagneaux L. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces apoptosis, down-regulates the CXCR4 chemokine receptor and impairs migration of chronic lymphocytic leukemia cells. Haematologica. 2010;95(7):1136–43. doi: 10.3324/haematol.2009.013847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin L, Tabe Y, Konoplev S, Xu Y, Leysath CE, Lu H, et al. CXCR4 up-regulation by imatinib induces chronic myelogenous leukemia (CML) cell migration to bone marrow stroma and promotes survival of quiescent CML cells. Mol Cancer Ther. 2008;7(1):48–58. doi: 10.1158/1535-7163.MCT-07-0042. [DOI] [PubMed] [Google Scholar]
- 35.Fei F, Stoddart S, Müschen M, Kim YM, Groffen J, Heisterkamp N. Development of resistance to dasatinib in Bcr/Abl-positive acute lymphoblastic leukemia. Leukemia. 2010;24(4):1318–27. doi: 10.1038/leu.2009.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hurwitz R, Hozier J, LeBien T, Minowada J, Gajl-Peczalska K, Kubonishi I, et al. Characterization of a leukemic cell line of the pre-B phenotype. Int J Cancer. 1979;23(2):174–80. doi: 10.1002/ijc.2910230206. [DOI] [PubMed] [Google Scholar]
- 37.Findley HWJ, Cooper MD, Kim TH, Alvarado C, Ragab AH. Two new acute lymphoblastic leukemia cell lines with early B-cell phenotypes. Blood. 1982;60(6):1305–9. [PubMed] [Google Scholar]
- 38.Lange B, Valtieri M, Santoli D, Caracciolo D, Mavilio F, Gemperlein I, et al. Growth factor requirements of childhood acute leukemia: Establishment of GM-CSF-dependent cell lines. Blood. 1987;70(1):192–9. [PubMed] [Google Scholar]
- 39.Tkachuk DC, Kohler S, Cleary ML. Involvement of a homolog of drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell. 1992;71(4):691–700. doi: 10.1016/0092-8674(92)90602-9. [DOI] [PubMed] [Google Scholar]
- 40.Pocock CF, Malone M, Booth M, Evans M, Morgan G, Greil J, et al. BCL-2 expression by leukaemic blasts in a SCID mouse model of biphenotypic leukaemia associated with the t(4;11)(q21;q23) translocation. Br J Haematol. 1995;90(4):855–67. doi: 10.1111/j.1365-2141.1995.tb05207.x. [DOI] [PubMed] [Google Scholar]
- 41.Matsuo Y, MacLeod RA, Uphoff CC, Drexler HG, Nishizaki C, Katayama Y, et al. Two acute monocytic leukemia (AML-M5a) cell lines (MOLM-13 and MOLM-14) with interclonal phenotypic heterogeneity showing MLL-AF9 fusion resulting from an occult chromosome insertion, ins(11;9)(q23;p22p23) Leukemia. 1997;11(9):1469–77. doi: 10.1038/sj.leu.2400768. [DOI] [PubMed] [Google Scholar]
- 42.Kahn J, Byk T, Jansson-Sjostrand L, Petit I, Shivtiel S, Nagler A, et al. Overexpression of CXCR4 on human CD34+ progenitors increases their proliferation, migration, and NOD/SCID repopulation. Blood. 2004;103(8):2942–9. doi: 10.1182/blood-2003-07-2607. [DOI] [PubMed] [Google Scholar]
- 43.D’Apuzzo M, Rolink A, Loetscher M, Hoxie JA, Clark-Lewis I, Melchers F, et al. The chemokine SDF-1, stromal cell-derived factor 1, attracts early stage B cell precursors via the chemokine receptor CXCR4. Eur J Immunol. 1997;27(7):1788–93. doi: 10.1002/eji.1830270729. [DOI] [PubMed] [Google Scholar]
- 44.Busillo JM, Benovic JL. Regulation of CXCR4 signaling. Biochim Biophys Acta. 2007;1768(4):952–63. doi: 10.1016/j.bbamem.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464(7290):852–7. doi: 10.1038/nature08851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Monaco G, Konopleva M, Munsell M, Leysath C, Wang RY, Jackson CE, et al. Engraftment of acute myeloid leukemia in NOD/SCID mice is independent of CXCR4 and predicts poor patient survival. Stem Cells. 2004;22(2):188–201. doi: 10.1634/stemcells.22-2-188. [DOI] [PubMed] [Google Scholar]
- 47.Pearce DJ, Taussig D, Zibara K, Smith LL, Ridler CM, Preudhomme C, et al. AML engraftment in the NOD/SCID assay reflects the outcome of AML: Implications for our understanding of the heterogeneity of AML. Blood. 2006;107(3):1166–73. doi: 10.1182/blood-2005-06-2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Patel S, Abdelouahab H, Wittner M, Willekens C, Shen S, et al. CXCR4 inhibitors selectively eliminate CXCR4-expressing human acute myeloid leukemia cells in NOG mouse model. Cell Death Dis. 2012;3:e396. doi: 10.1038/cddis.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fukuda S, Broxmeyer HE, Pelus LM. Flt3 ligand and the Flt3 receptor regulate hematopoietic cell migration by modulating the SDF-1alpha(CXCL12)/CXCR4 axis. Blood. 2005;105(8):3117–26. doi: 10.1182/blood-2004-04-1440. [DOI] [PubMed] [Google Scholar]
- 50.Uy GL, Rettig MP, Motabi IH, McFarland K, Trinkaus KM, Hladnik LM, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119(17):3917–24. doi: 10.1182/blood-2011-10-383406. [DOI] [PMC free article] [PubMed] [Google Scholar]