

Abstract
The recently discovered hexanucleotide repeat expansion, C9ORF72, has been shown to be among the most common cause of familial behavioural variant frontotemporal dementia (bvFTD) and to be present in a significant minority of apparently sporadic cases. While mounting evidence points to prominent episodic memory dysfunction in bvFTD cases, recent reports have also suggested an amnestic profile in C9ORF72 mutation carriers. No study to date, however, has formally characterised the extent to which episodic memory is impaired in C9ORF72 mutation versus sporadic cases, or the underlying neural substrates of such deficits. We conducted a comparison of C9ORF72 (n = 8) and sporadic (n = 15) bvFTD cases using a battery of verbal and visual episodic memory tasks, and contrasted their performance with that of Alzheimer's disease (AD, n = 15) and healthy older control (n = 15) participants. Behaviourally, the two bvFTD groups displayed comparable episodic memory profiles, irrespective of task administered, with prominent impairments evident relative to Controls. Whole-brain voxel-based morphometry analyses revealed distinct neural correlates of episodic memory dysfunction in each patient group. Widespread atrophy in medial prefrontal, medial and lateral temporal cortices correlated robustly with episodic memory dysfunction in sporadic bvFTD cases. In contrast, atrophy in a distributed set of regions in the frontal, temporal, and parietal lobes including the posterior cingulate cortex, was implicated in episodic memory dysfunction in C9ORF72 cases. Our results demonstrate that while episodic memory is disrupted to the same extent irrespective of genetic predisposition in bvFTD, distinct neural changes specific to each patient group are evident. The involvement of medial and lateral parietal regions in episodic memory dysfunction in C9ORF72 cases is of particular significance and represents an avenue of considerable interest for future studies.
Keywords: Episodic memory, Frontotemporal dementia, Alzheimer's disease, C9ORF72 mutation, Neuroimaging
Highlights
-
•
We assessed episodic memory in bvFTD patients with and without C9ORF72 mutations.
-
•
Episodic memory deficits were present in C9ORF72 cases relative to Controls.
-
•
C9ORF72 and sporadic bvFTD cases showed equivalent episodic memory profiles.
-
•
Neural substrates of memory disruption differed contingent on mutation status.
-
•
Medial and lateral parietal involvement in C9ORF72 memory deficits is notable.
1. Introduction
Frontotemporal dementia (FTD) refers to a progressive neurodegenerative disorder which accounts for a significant proportion of young onset dementia (Ratnavalli et al., 2002), and is characterised by clinical and pathological heterogeneity. Patients with the behavioural variant of FTD (bvFTD) present with florid changes in behaviour and interpersonal functioning, manifesting in reduced motivation and disinhibition (Piguet et al., 2011), which is associated with prefrontal, insular and temporal cortical degeneration (Rabinovici et al., 2007; Seeley et al., 2008). The recently discovered hexanucleotide repeat expansion in the C9ORF72 gene has been shown to be among the most common causes of familial FTD (Boeve et al., 2012; DeJesus-Hernandez et al., 2011; Mahoney et al., 2012a; Simon-Sanchez et al., 2012) and motor neurone disease (MND) (Byrne et al., 2012; Chio et al., 2012; Cooper-Knock et al., 2012; Renton et al., 2011). Importantly, the mutation has also been found in around 2–4% of FTD cases with no apparent family history (DeJesus-Hernandez et al., 2011; Mahoney et al., 2012a; Simon-Sanchez et al., 2012). Not surprisingly, current research efforts are focused on discriminating C9ORF72 mutation carriers from non-carriers in vivo at the earliest disease stage.
On a clinical level, demographic features, such as age of onset, sex, and disease duration at presentation, do not seem to differentiate between carriers and non-carriers of the mutation, however, a number of distinctive clinical features have been observed in C9ORF72 mutation-positive cases. The most striking feature is a high rate of psychosis in C9ORF72 compared to sporadic cases (Boeve et al., 2012; Sha et al., 2012; Snowden et al., 2012). In addition, a high proportion of C9ORF72 cases have been reported to display features of ALS (Boeve et al., 2012; Hsiung et al., 2012; Mahoney et al., 2012a; Sha et al., 2012; Simon-Sanchez et al., 2012; Snowden et al., 2012), which is in keeping with the observation of a family history of motor neurone disease in this cohort (Snowden et al., 2012). Evidence of Parkinsonism has also been reported, giving rise to a mild akinetic syndrome without the presence of tremor (Boeve et al., 2012; Hsiung et al., 2012; Sha et al., 2012; Simon-Sanchez et al., 2012; Snowden et al., 2012).
Of particular interest in this context is the observation of severe anterograde amnesia in C9ORF72 cases, leading to potential misdiagnosis as Alzheimer's disease (Dobson-Stone et al., 2012; Mahoney et al., 2012a; van Swieten and Grossman, 2012). Accordingly, the nature of episodic memory impairment represents an important line of enquiry for determining which patients should be considered candidates for screening for C9ORF72 expansions (van Swieten and Grossman, 2012). Evidence regarding the extent to which episodic memory processes are compromised in C9ORF72 mutation cases is limited. While significant episodic memory impairment was a prominent feature of the C9ORF72 case series reported from London (Mahoney et al., 2012a), this observation was made on the basis of a retrospective analysis of case notes. In fact, the majority of studies reporting on the clinical characteristics of C9ORF72 cases hinge on retrospective clinical case note reviews, which likely underestimate the rate and extent of cognitive dysfunction (Hodges, 2012).
The present study therefore sought to formally characterise the extent to which episodic memory is disrupted in C9ORF72 in comparison with sporadic bvFTD cases by (i) conducting an analysis of C9ORF72 and sporadic bvFTD cases who were all assessed prospectively using a battery of verbal and visual episodic memory tasks, and (ii) exploring the neuroanatomical contributions to these episodic memory impairments using whole-brain voxel-based morphometry analyses based on structural scans. We hypothesised that C9ORF72 and sporadic bvFTD cases would show largely similar behavioural profiles of episodic memory dysfunction, in keeping with previous reports in the literature (Dobson-Stone et al., 2012; Mahoney et al., 2012a; van Swieten and Grossman, 2012), with both patient groups displaying prominent memory deficits relative to Controls. Given recent evidence which points to a possible neuroanatomical signature of the C9ORF72 mutation (Whitwell et al., 2012), we predicted that distinct neural correlates would underpin episodic memory dysfunction contingent on bvFTD subgroup. Specifically, we hypothesised that the origin of episodic memory dysfunction in sporadic bvFTD cases would stem from atrophy overwhelmingly in medial prefrontal and anterior temporal regions, in line with previous studies (Frisch et al., 2013; Irish et al., 2013; Pennington et al., 2011). In contrast, for C9ORF72 cases, we predicted that episodic memory deficits would relate to atrophy in frontal, lateral temporal, and parietal regions, given recent evidence of extensive parietal atrophy in this cohort (Sha et al., 2012).
2. Methods
2.1. Participants
A total of 53 subjects participated in this study: 8 behavioural variant frontotemporal dementia (bvFTD) cases identified as C9ORF72 mutation carriers (referred to as C9 cases), 15 bvFTD cases who lack mutations in C9ORF72 (sporadic bvFTD), 15 cases with Alzheimer's disease (AD), and 15 age-matched healthy Controls, all selected from the FRONTIER database, at Neuroscience Research Australia, Sydney. All dementia patients met clinical diagnostic criteria for bvFTD (Rascovsky et al., 2011) or AD (McKhann et al., 2011), with 2 of the C9 cases showing FTD-MND features with disease progression (Strong et al., 2009). Within the C9 group, 3 patients had a family history of MND, 2 patients had a family history of early-onset dementia, and 1 patient had a family history of Parkinson's disease. For sporadic bvFTD cases, 3 patients were found to have a family history of early-onset dementia, with no family history of MND or Parkinsonism evident in any of the sporadic cases. Blood collection for genetic screening had been conducted during a previous clinical visit, following informed consent. The repeat primed polymerase chain reaction (PCR) was performed using the procedure described previously (Dobson-Stone et al., 2012), based on the protocol of Renton and colleagues (Renton et al., 2011). A patient's DNA sample was deemed positive for the C9ORF72 repeat expansion if it contained an allele with > 30 repeats. All bvFTD patients were screened for other common genetic mutations (GRN, MAPT), however, no positive cases were identified.
Diagnosis was established by consensus among a senior neurologist (JRH), neuropsychologist, and occupational therapist based on clinical investigations, cognitive assessment, carer interviews, and evidence of atrophy on structural neuroimaging. Briefly, bvFTD patients presented with insidious onset, decline in personal conduct and social functioning, displaying emotional blunting, loss of insight, and increased apathy. Only dementia patients with evidence of definite progression over time as reported by the caregivers, and atrophy on structural MRI scans were included in this study. This was to exclude potential phenocopy cases in the FTD group (Kipps et al., 2010) and to confirm the diagnosis for all cases. AD patients displayed significant episodic memory loss, in the context of preserved behaviour and personality.
Healthy Controls were patients' family and friends, and individuals from local community clubs. All Controls scored 0 on the Clinical Dementia Rating scale (CDR) (Morris, 1997), and 88 or above on the Addenbrooke's Cognitive Examination — Revised (ACE-R) (Mioshi et al., 2006). Exclusion criteria included prior history of mental illness, significant head injury, movement disorders, cerebrovascular disease, alcohol and other drug abuse, and limited English proficiency. Ethical approval for this study was obtained from the South Eastern Sydney and Illawarra Area Health Service and the University of New South Wales ethics committees. All participants, or their person responsible, provided informed written consent in accordance with the Declaration of Helsinki.
2.2. General cognitive testing
All participants completed a comprehensive neuropsychological test battery as part of their diagnostic work-up. Briefly, global cognitive functioning was assessed using Addenbrooke's Cognitive Examination Revised (ACE-R) (Mioshi et al., 2006), which comprises subscales measuring attention and orientation, memory, fluency, language and visuospatial functioning. The Trail Making Test (Reitan, 1958), was administered to all participants as an index of attention, speed, and mental flexibility. Here, we were interested in set-switching and divided attention and thus used a Trail B − Trail A difference score (Strauss et al., 2006). Verbal letter fluency (F, A, S) (Strauss et al., 2006) was administered as an index of strategic search processes, and Digit span forwards and backwards tasks of the Wechsler Memory Scale (WMS3) (Wechsler, 1997) were used as indices of verbal working memory. Finally, the Hayling test (Burgess and Shallice, 1997) was used to assess behavioural regulation, specifically response inhibition (Strauss et al., 2006). Carers rated the behavioural changes of patients on the Cambridge Behavioural Inventory (CBI) (Wedderburn et al., 2008), in terms of memory decline, and loss of motivation.
2.3. Episodic memory testing
The procedure for this study has been described in detail elsewhere (Irish et al., 2013). Briefly, we administered a battery of verbal and visual episodic memory tests to all participants. The Rey Auditory Verbal Learning Test (RAVLT) (Schmidt, 1996) was used to assess the immediate recall of 15 words (List A), learned over five consecutive trials. Each trial is followed by a free recall test. An interference trial comprising 15 new words (List B) and a free recall test is then presented, following which, the participant is required to retrieve words from the original List A (immediate recall). Following a 30-minute delay, the participant is required to recall List A (delayed recall). We extracted the following scores from the RAVLT: immediate recall following the interference trial (maximum score: 15) and delayed recall following 30 min (maximum score: 15).
The Rey-Osterrieth Complex Figure (RCF) (Meyers and Meyers, 1995) was administered as an index of visual recall of a complex design following a 3-minute delay. Participants were instructed to copy a complex figure as accurately as possible and, following the delay, were required to reproduce the figure from memory. The maximum score for both the copy and recall trials is 36 points. For all experimental scores, a percentage correct score was calculated (i.e., raw score/total test score ∗ 100).
2.4. Statistical analyses
Cognitive data were analysed using IBM SPSS Statistics (Version 20.0). Multivariate analyses of variance (MANOVA) with Sidak post hoc tests were used to explore main effects of group (Controls, C9, sporadic bvFTD, AD) for all episodic memory tests. The rationale for using Sidak modification of the traditional Bonferroni post hoc test is that the statistical power of the analyses is not affected, while the flexibility of the original Bonferroni method is maintained (Keppel and Wickens, 2004). Chi-squared tests (χ2), based on the frequency patterns of dichotomous variables, were also used. To investigate relationships between patterns of grey matter intensity and episodic memory performance, all experimental scores (RAVLT Immediate Recall, RAVLT delayed recall, RCF recall) were entered into a single design matrix as covariates in the neuroimaging analyses.
2.5. Image acquisition
Voxel-based morphometry (VBM) was used to identify grey matter intensity changes across groups on a voxel-by-voxel basis using structural MRI data. Participants underwent whole-brain T1-weighted imaging using a 3 T Philips MRI scanner with standard quadrature head coil (8 channels), using the following sequences: coronal orientation, matrix 256 × 256, 200 slices, 1 × 1 mm2 in-plane resolution, slice thickness 1 mm, echo time/repetition time = 2.6/5.8 ms, flip angle ɑ = 19°. MRI data were analysed with FSL-VBM, a voxel-based morphometry analysis (Ashburner and Friston, 2000; Mechelli et al., 2005) using the FSL-VBM toolbox from the FMRIB software package (http://www.fmrib.ox.ac.uk/fsl/fslvbm/index.html) (Smith et al., 2004). Briefly, structural images were extracted using the brain extraction tool (BET) (Smith, 2002). Tissue segmentation was then carried out on the brain extracted images using FMRIB's Automatic Segmentation Tool (FAST) (Zhang et al., 2001). The resulting grey matter partial volumes were then aligned to the Montreal Neurological Institute standard space (MNI152) using the FMRIB non-linear registration approach (FNIRT) (Andersson et al., 2007a, 2007b), which uses a b-spline representation of the registration warp field (Rueckert et al., 1999). A study-specific template was created, combining AD, C9, sporadic bvFTD, and Control images, to which the native grey matter images were re-registered nonlinearly. The registered partial volume maps were then modulated by dividing by the Jacobian of the warp field. This step was carried out to correct for local expansion or contraction. The modulated segmented images were then smoothed with an isotropic Gaussian kernel with a sigma of 3 mm.
2.6. Voxel-based morphometry analysis
A voxel-wise general linear model was applied to investigate grey matter intensity differences via permutation-based non-parametric testing (Nichols and Holmes, 2002) with 5000 permutations per contrast. In a first step, differences in cortical grey matter intensities between each patient group (C9, sporadic bvFTD, and AD) and Controls were assessed. Next, correlations between performance on experimental episodic memory tests and regions of grey matter atrophy were investigated in each of the patient groups contrasted with controls. This procedure serves to increase the statistical power to detect brain–behaviour relationships across the entire brain by achieving greater variance in behavioural scores (see Irish et al., 2012a; Sollberger et al., 2009). For statistical power, a covariate only statistical model with a [1] contrast was used, providing an index of association between grey matter intensity and episodic memory. In line with a previous study by our group (Irish et al., 2013), the three episodic memory task scores were included separately in the model, and education was included as a covariate in all analyses given the difference between Controls and C9 patients (see below). Anatomical locations of significant results were overlaid on the MNI standard brain, with maximum coordinates provided in MNI stereotaxic space. Anatomical labels were determined with reference to the Harvard–Oxford probabilistic cortical atlas.
3. Results
3.1. Demographics
Demographic and clinical information is presented in Table 1. The participant groups were well matched for age (p = .467) but not for years in education (F(3, 49) = 4.370, p = .008) as Controls spent on average 4 years longer in formal education in comparison to the C9 cases (p = .007). Sex was evenly distributed between the participant groups (p = .843). The patient groups were matched for disease duration (i.e., the number of months elapsed between onset of symptoms and cognitive testing, p = .151).
Table 1.
Demographic and clinical characteristics of study participantsa,b.
| C9 | Sporadic | AD | Controls | F test | C9 vs. Sporadic | |
|---|---|---|---|---|---|---|
| N | 8 | 15 | 15 | 15 | ||
| Sex (m: f) | 6: 2 | 9: 6 | 11: 4 | 10: 5 | n/s | n/s |
| Age (years) | 60.4 (4.6) | 63.9 (9.3) | 64.3 (5.5) | 64.6 (4.0) | n/s | n/s |
| Education (years) | 10.4 (1.8) | 12.3 (3.1) | 12.1 (2.8) | 14.3 (2.2) | n/s | |
| Disease duration (months) | 61.1 (38.0) | 36.5 (21.9) | 42.2 (26.1) | n/a | n/s | n/s |
| ACE-R total (100) | 78.5 (8.2) | 78.5 (9.1) | 73.3 (8.8) | 95.9 (3.5) | ⁎⁎⁎ | n/s |
| Trails (B–A in s) | 121.6 (71.4) | 145.5 (98.5) | 119.2 (72.5) | 39.9 (23.9) | ⁎⁎ | |
| Letter fluency | 23.2 (9.8) | 23.3 (10.4) | 29.3 (13.3) | 47.2 (11.3) | ⁎⁎⁎ | |
| Digit span forwards | 8.1 (1.8) | 9.7 (1.8) | 8.2 (2.8) | 11.5 (2.5) | ⁎ | n/s |
| Digit span backwards | 4.7 (1.6) | 5.2 (2.5) | 4.4 (1.8) | 8.5 (2.4) | ⁎⁎⁎ | n/s |
| Hayling Scaled score |
3.1 (1.8) | 2.9 (2.1) | 3.8 (1.9) | 6.5 (0.6) | ⁎⁎⁎ | n/s |
| CBI Memory (100) | 38.7 (24.4) | 41.2 (22.6) | 40.6 (22.8) | n/a | n/s | n/s |
| CBI Motivation (100) | 41.2 (36.0) | 58.6 (36.4) | 25.7 (25.8) | n/a | n/s | n/s |
CBI information available for 14 Sporadic cases. Hayling data available for 14 Sporadic cases. Digit span data available for 7 C9 and 12 Sporadic cases.
Standard deviations in brackets, maximum score for tests shown in brackets.
Trail Making test data for 11 Sporadic and 9 AD cases.
p < .05.
p < .005.
p < .0001.
3.2. General cognitive screening
Significant group differences were evident for the total ACE-R score (F(3, 49) = 24.493, p < .0001), with all patient groups scoring significantly lower than Controls (all p values < .0001). No significant differences were found between patient groups for total ACE-R score (all p values > .3). The digit span forwards (F(3, 42) = 4.210, p = .011) and backwards (F(3, 42) = 9.282, p < .0001) tasks revealed significant group differences. AD patients showed significantly poorer forwards (p = .013) and backwards (p < .0001) digit span in comparison with Controls. C9 cases displayed a tendency towards compromised forwards digit span (p = .060) with clear deficits emerging on the backwards span task (p = .004) with respect to Controls. Finally, sporadic bvFTD cases demonstrated intact forwards span (p = .459) but impairments for backward span (p = .007) in comparison with Controls. No significant differences were observed between the FTD subgroups for forwards or backwards digit span (all p values > .7).
All patient groups performed significantly poorer than Controls on the Hayling test (F(3, 38) = 11.914, p < .0001; C9 p < .0001; Sporadic bvFTD p < .0001; AD p = .011). For letter fluency, again, significant impairments were evident in the patient groups relative to Controls (F(3, 42) = 10.326, p < .0001) with no significant differences between the patient groups (all p values > .7). On the Trail Making Task, all patients were impaired in comparison with Controls (F(3, 42) = 5.429, p = .003; C9 p = .059; Sporadic bvFTD p = .006; AD p = .056), again with no significant differences between patient groups evident (all p values > .9). Finally, no significant differences were observed between patient groups for caregiver ratings of memory impairment (all p values > .9) or loss of motivation (all p values > .1) on the CBI.
3.3. Episodic memory performance
Fig. 1 displays the performance of the patient groups (C9, sporadic bvFTD, AD) and Controls on each of the episodic memory recall tasks. A multivariate analysis of variance (MANOVA), with education included as a covariate, revealed a main effect of group for immediate and delayed recall on the RAVLT (Immediate, F(3, 47) = 19.552, p < .0001; Delayed, F(3, 47) = 21.797, p < .0001). Sidak post hoc tests revealed Controls significantly outperformed patient groups for Immediate (C9, p = .011; Sporadic, p < .0001; AD, p < .0001) and Delayed (C9, p = .004; Sporadic, p < .0001; AD, p < .0001) recall. Importantly, no significant differences were evident among the patient groups for any of the RAVLT subscales (all p values > .1).
Fig. 1.
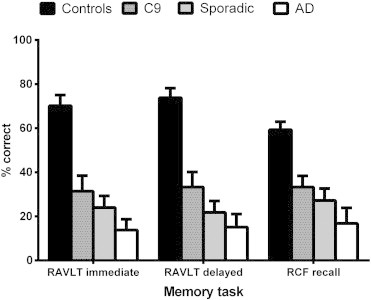
Barchart showing percentage correct scores for C9 mutation carriers, sporadic behavioural variant FTD and Alzheimer's disease participants in comparison with healthy Controls on each of the episodic memory tasks. Error bars represent standard error of the mean.
Further, a main effect was found for RCF recall (F(3, 47) = 9.660, p < .0001). Sidak post hoc tests revealed that while Controls performed significantly higher than Sporadic (p = .002) and AD (p < .0001) patients, no significant difference was evident between Controls and C9 patients on this task (p = .105). Finally, no significant differences were observed between the patient groups (all p values > .5).
3.4. Voxel-based morphometry results
3.4.1. Patterns of atrophy in study samples
Fig. 2 displays the patterns of atrophy in C9, Sporadic, and AD patients in comparison to healthy Control participants, corrected for Family Wise Error (FWE) multiple comparisons at p < .05. The C9 mutation group showed widespread atrophy predominantly in the frontal, temporal and parietal cortices, bilaterally (Fig. 2A). Notably the regions affected included the frontal poles and orbitofrontal cortices bilaterally, extending into the temporal poles, and medial temporal structures including the amygdala, hippocampus and the thalamus bilaterally. Atrophy was also pronounced in the precentral gyrus bilaterally, the parietal opercular cortex and superior parietal lobule bilaterally, the posterior cingulate cortex bilaterally, and the bilateral occipital poles. These patterns of atrophy largely replicate those reported previously (Sha et al., 2012; Whitwell et al., 2012), although we did not find evidence of cerebellar atrophy in our C9 mutation group as has been noted in previous studies (Mahoney et al., 2012a; Sha et al., 2012; Whitwell et al., 2012).
Fig. 2.
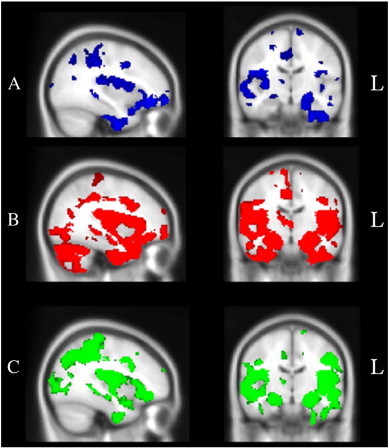
Voxel-based morphometry analyses showing brain areas of decreased grey matter intensity corrected for Family-Wise Error at p < .05 in (A) C9 mutation carriers in comparison with Controls (blue), (B) sporadic bvFTD patients in comparison with Controls (red), and (C) Alzheimer's disease patients in comparison with Controls (green). Coloured voxels show regions that were significant in the analysis using the threshold-free cluster enhancement method (tfce), and overlaid on the MNI standard brain. All clusters reported t > 2.07.
In contrast, the sporadic bvFTD group showed striking and widespread bilateral atrophy, more extensive on the right hand side, emanating from the medial prefrontal cortices to encompass the temporal pole, insular cortex, OFC, anterior cingulate cortex, and frontal poles bilaterally. Marked atrophy was also evident in the cerebellum, lateral temporal, and occipital lobes, as well as in subcortical structures including the hippocampus, amygdala, thalamus, and caudate, bilaterally (Fig. 2B).
Finally, AD patients showed widespread atrophy, compared to Controls, in medial temporal regions including the bilateral hippocampii and amygdalae, extending into bilateral frontal, temporal and parietal regions, and extending posteriorly into the lateral occipital cortices. Further atrophy was evident in the right lateral temporal cortices (Fig. 2C).
Direct comparisons between the C9 and sporadic bvFTD cases revealed that sporadic cases showed significantly more atrophy in the cerebellum bilaterally, as well as in medial temporal regions including the right hippocampus, right amygdala, and right temporal pole. Further, sporadic cases showed significantly more atrophy in the right paracingulate and anterior cingulate gyri and the right lateral occipital cortex (p < .05). Given the small C9 sample, and the ensuing reduced detection power, we used a less conservative threshold to investigate the reverse contrast (C9 versus Sporadic). This contrast failed to reveal any significant regions of greater atrophy in the C9 group compared with the sporadic group (p < .01, uncorrected).
3.4.2. Neural correlates of episodic memory performance
Fig. 3 shows the brain regions associated with episodic memory performance in C9 (A), Sporadic (B) and AD (C) cases. Given the smaller sample of C9 patients, and the ensuing reduction in detection power, we lowered the cluster height threshold for our analyses (p < .01 uncorrected) to explore the neural substrates of recall dysfunction in this group. Recall performance in C9 patients was associated with grey matter intensity decrease in a distributed set of frontal, and temporal, regions including the bilateral temporal cortices extending to the bilateral parahippocampal cortices and notably the right posterior hippocampus. Importantly, a distinct set of parietal regions were implicated in episodic memory dysfunction in the C9 group including the bilateral superior parietal lobule, the right supramarginal gyrus and right precuneus, and the bilateral posterior cingulate cortices (Fig. 3A, Table 2).
Fig. 3.
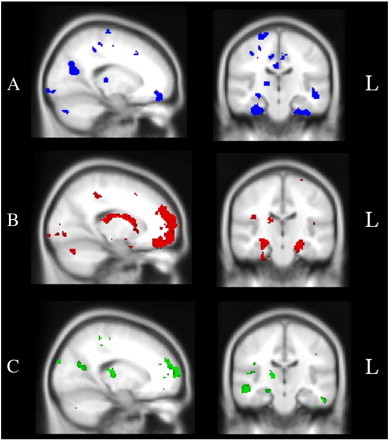
Voxel-based morphometry analyses showing brain regions in which grey matter intensity correlates significantly with episodic recall performance in (A) C9 mutation carriers in comparison with Controls (blue), (B) sporadic bvFTD patients compared with Controls (red), and (C) Alzheimer's disease patients in comparison with Controls (green). Coloured voxels show regions that were significant in the analysis with p < .001 uncorrected for sporadic bvFTD and AD contrasts, and p < .01 for C9 contrasts. All clusters reported t > 2.82. Clusters are overlaid on the MNI standard brain.
Table 2.
Voxel-based morphometry results showing regions of significant grey matter intensity decrease that covary with episodic memory recall performance for C9 patients contrasted with Controls.
| Regions | Hemisphere | MNI coordinates |
Number of voxels |
||
|---|---|---|---|---|---|
| x | y | z | |||
| Temporal fusiform cortex, parahippocampal cortex, inferior temporal gyrus | Left | − 30 | − 12 | − 42 | 805 |
| Parietal operculum cortex, superior parietal lobule | Left | − 46 | − 40 | 32 | 640 |
| Temporal fusiform cortex, parahippocampal cortex, hippocampus | Right | 34 | − 2 | − 50 | 637 |
| Orbitofrontal cortex, inferior frontal gyrus | Left | − 48 | 30 | − 12 | 550 |
| Supramarginal gyrus, superior parietal lobule | Right | 42 | − 30 | 34 | 495 |
| Inferior temporal gyrus | Right | 58 | − 40 | − 26 | 384 |
| Middle temporal gyrus | Left | − 50 | − 18 | − 16 | 301 |
| Supracalcarine cortex, precuneus | Right | 18 | − 70 | 16 | 286 |
| Precentral gyrus | Left | − 22 | − 28 | 52 | 250 |
| Temporal pole | Left | − 28 | 18 | − 30 | 236 |
| Frontal pole | Right | 18 | 48 | − 10 | 204 |
| Middle frontal gyrus | Left | − 38 | 10 | 54 | 179 |
| Superior temporal gyrus | Right | 46 | − 32 | 8 | 177 |
| Insular cortex | Left | − 26 | 12 | 14 | 175 |
| Frontal operculum cortex | Right | 36 | 22 | 14 | 169 |
| Temporal pole | Right | 52 | 24 | − 18 | 147 |
| Frontal pole | Left | − 16 | 52 | − 10 | 143 |
| Posterior cingulate cortex | Bilateral | − 2 | − 18 | 44 | 124 |
| Postcentral gyrus | Right | 18 | − 38 | 50 | 122 |
| Orbitofrontal cortex | Left | − 10 | 32 | − 22 | 122 |
| Occipital pole | Right | 20 | − 104 | − 8 | 104 |
All results uncorrected at p < .01; for brevity only clusters with at least 100 contiguous voxels are reported here. Education is included as a covariate in all contrasts. All clusters reported t > 2.82.
In contrast, for sporadic FTD cases, recall performance correlated extensively with right medial prefrontal and bilateral orbitofrontal cortices extending to the right insular cortex. Lateral and medial temporal regions were also implicated, including the temporal poles bilaterally, the hippocampus and amygdala bilaterally, as well as bilateral cerebellum, left parietal operculum cortex, and left lateral occipital cortex (Fig. 3B, Table 3).
Table 3.
Voxel-based morphometry results showing regions of significant grey matter intensity decrease that covary with recall performance for sporadic bvFTD patients compared with Controls.
| Regions | Hemisphere | MNI coordinates |
Number of voxels |
||
|---|---|---|---|---|---|
| x | y | z | |||
| Frontal pole, medial PFC, anterior cingulate cortex, paracingulate gyrus | Right | 8 | 38 | − 30 | 5012 |
| Orbitofrontal cortex, inferior frontal gyrus, insular cortex | Right | 48 | 28 | − 12 | 1586 |
| Temporal fusiform cortex (anterior), parahippocampal gyrus, hippocampus, amygdala | Left | − 30 | − 6 | − 52 | 1028 |
| Cerebellum | Right | 50 | − 50 | − 52 | 660 |
| Temporal pole, orbitofrontal cortex | Left | − 38 | 8 | − 30 | 496 |
| Parahippocampal gyrus, hippocampus | Right | 28 | − 22 | − 34 | 489 |
| Inferior temporal gyrus (anterior), temporal pole | Right | 38 | − 8 | − 50 | 469 |
| Parietal operculum cortex | Left | − 46 | − 30 | 14 | 355 |
| Cerebellum | Left | − 38 | − 58 | − 42 | 327 |
| Planum temporale | Right | 48 | − 30 | 14 | 282 |
| Precentral gyrus, middle frontal gyrus | Right | 44 | 2 | 22 | 261 |
| Lateral occipital cortex | Left | − 30 | − 82 | − 6 | 123 |
| Middle temporal gyrus | Left | − 70 | − 30 | 0 | 103 |
All results uncorrected at p < .001; only clusters with at least 100 contiguous voxels are included. Education is included as a covariate in all contrasts. All clusters reported t > 3.40.
Finally for AD cases, recall performance was associated with a distributed set of frontal, temporal and parietal regions including the bilateral precuneus, bilateral lateral temporal cortices, left hippocampus, bilateral frontal poles, as well as the bilateral angular gyrus and bilateral occipital cortices (Fig. 3C, Inline Supplementary Table S1).
Inline Supplementary Table S1.
Table S1.
Voxel-based morphometry results showing regions of significant grey matter intensity decrease that covary with episodic memory recall performance for AD patients and Controls.
| Regions | Hemisphere | MNI coordinates |
Number of voxels |
||
|---|---|---|---|---|---|
| x | y | z | |||
| Precuneus | Bilateral | 14 | − 66 | − 20 | 818 |
| Postcentral gyrus, superior parietal lobule | Right | 34 | − 30 | 38 | 446 |
| Inferior temporal gyrus (posterior) | Right | 56 | − 38 | − 28 | 387 |
| Middle temporal gyrus, angular gyrus | Right | 46 | − 52 | − 2 | 376 |
| Angular gyrus, lateral occipital cortex | Left | − 40 | − 58 | 30 | 367 |
| Frontal pole | Right | 18 | 62 | 4 | 314 |
| Orbitofrontal cortex | Left | − 42 | 28 | − 10 | 312 |
| Inferior temporal gyrus (posterior) | Left | − 54 | − 26 | − 28 | 300 |
| Occipital pole | Right | 10 | − 90 | 16 | 197 |
| Frontal pole | Left | − 20 | 58 | 6 | 187 |
| Middle temporal gyrus | Right | 56 | − 20 | − 16 | 181 |
| Temporal fusiform cortex | Left | − 28 | − 10 | − 46 | 152 |
| Angular gyrus | Right | 52 | − 50 | 38 | 127 |
| Precentral gyrus | Left | − 42 | − 8 | 28 | 120 |
| Hippocampus | Left | − 24 | − 36 | − 8 | 120 |
All results uncorrected at p < .001; only clusters with at least 100 contiguous voxels included. Education included as a covariate in all contrasts. All clusters reported t > 3.40.
Finally for AD cases, recall performance was associated with a distributed set of frontal, temporal and parietal regions including the bilateral precuneus, bilateral lateral temporal cortices, left hippocampus, bilateral frontal poles, as well as the bilateral angular gyrus and bilateral occipital cortices (Fig. 3C, Inline Supplementary Table S1).
Inline Supplementary Table S1 can be found online at http://dx.doi.org/10.1016/j.nicl.2013.06.005.
4. Discussion
The recent discovery of the C9ORF72 repeat expansion has led to a surge of research seeking to establish clinical hallmark features of cases with this mutation, with an amnestic profile proposed as a potentially distinguishing feature of this syndrome. The goal of this study was to characterise the extent to which episodic memory is compromised in C9ORF72 bvFTD cases and to elucidate the neural correlates of episodic memory dysfunction in this group. Behaviourally, C9ORF72 cases showed substantial episodic memory impairments relative to age-matched Controls, scoring at similar levels to sporadic bvFTD and AD participants. Notably, a distinctive neuroanatomical signature of memory disruption in C9ORF72 cases was found, involving frontal and lateral temporal, as well as lateral and medial parietal regions. Our findings point to important differences in terms of the underlying regions implicated in episodic memory dysfunction in patients with and without the C9ORF72 mutation, despite behaviourally indistinguishable memory profiles.
Converging evidence points to the existence of marked episodic memory dysfunction in the behavioural variant of frontotemporal dementia, attributable to widespread degeneration of the medial prefrontal, and anterior lateral temporal cortices (Hornberger et al., 2010, 2012; Irish et al., 2013; Pennington et al., 2011). Studies investigating the clinical profile of C9ORF72 mutation carriers have suggested that an amnestic profile represents a characteristic feature of this FTD subgroup (Dobson-Stone et al., 2012; Mahoney et al., 2012a; van Swieten and Grossman, 2012), particularly with disease progression (Mahoney et al., 2012b). Importantly, our current findings resonate well with the position that severe episodic memory deficits are present in C9 cases, however, it is unlikely that C9 mutation carriers can be differentiated from sporadic cases on the basis of episodic memory profiles alone. Behaviourally, it was not possible to distinguish between C9 and sporadic bvFTD cases using our episodic memory battery, despite the suggestion that non-verbal memory might be somewhat less affected in C9 cases, who scored in line with Controls for recall of a complex figure. It may be that visual and verbal memory processes are differentially impaired in C9 mutation carriers, and this proposal is certainly worthy of future investigation.
The most striking finding to emerge from this study concerns the neural substrates of episodic memory deficits in the C9 group. Whole-brain voxel based morphometry analyses revealed that atrophy in a distributed network of frontal, lateral and medial temporal, and lateral and medial parietal regions underpinned episodic memory dysfunction in C9 mutation carriers. Importantly, considerable overlap was observed between the neural substrates in the C9 group with those of the sporadic bvFTD and AD patient groups. Notably, regions commonly implicated in memory dysfunction in C9 and sporadic bvFTD cases included the lateral and medial temporal cortices including the right hippocampus, the right frontal pole, and the bilateral orbitofrontal cortices.
It is interesting to observe that the neuroanatomical signature of episodic memory dysfunction in C9 mutation carriers lies somewhat midway on a continuum between the more anterior network commonly reported in bvFTD and the more posteriorly-oriented substrates of AD (Frisch et al., 2013; Irish et al., 2013). Perhaps most intriguing is the finding of considerable parietal involvement in the C9 group, including the bilateral precuneal/posterior cingulate cortices, and the bilateral superior parietal lobule, mirroring those observed in the AD group. Extensive lateral and medial parietal lobe atrophy has been documented in C9 cases, with the suggestion that diffuse parietal atrophy may prove useful in discriminating between carriers and non-carries of the C9ORF72 mutation (Whitwell et al., 2012). Our results mesh well with this assertion, and point to the involvement of parietal regions in the genesis of episodic memory deficits in C9 mutation carriers. Interestingly, we found only minimal evidence of cerebellar involvement in episodic memory impairment in C9 cases (cluster size of 60 contiguous voxels at p < .01 uncorrected) despite recent suggestions that cerebellar pathology may be a characteristic feature of this group (Whitwell et al., 2012). In the context of the current sample, lateral and medial parietal contributions to memory impairment represent a defining feature of C9ORF72 mutation carriers, and may serve as a useful means of differentiating this cohort from sporadic bvFTD cases.
The integrity of interactions between prefrontal cortical structures and the medial temporal lobes is pivotal for successful episodic memory retrieval (Simons and Spiers, 2003). Indeed, our findings from the sporadic bvFTD group strongly support this position, given that extensive atrophy was associated with memory disruption in this patient group. Our results from the sporadic bvFTD group replicate previous studies demonstrating that damage to the prefrontal cortex, and medial temporal lobes, including the hippocampus, are strongly associated with the amnestic profile in bvFTD (Frisch et al., 2013; Irish et al., 2013; Pennington et al., 2011). The role of the prefrontal cortex in episodic memory performance, however, remains comparatively poorly understood (Simons and Spiers, 2003). Current evidence from neuroimaging and lesion studies suggests that strategic aspects of episodic memory recall are mediated by prefrontal brain structures (Becker and Lim, 2003; Kramer et al., 2005), in particular the dorsolateral prefrontal cortex (Long et al., 2010). Accordingly, it has been proposed that episodic memory deficits in bvFTD may stem from compromised strategic retrieval processes due to difficulties with planning and organisation of information during encoding and/or retrieval (Kramer et al., 2005; Wicklund et al., 2006). Given the substantial impairments in executive functioning in bvFTD, emanating from the progressive deterioration of the frontal lobes (Collette et al., 2010; Piolino et al., 2007), and the finding of marked impairments of bvFTD patients on source memory tasks specifically stressing prefrontal cortical processes (Irish et al., 2012b; Söderlund et al., 2008), the strategic retrieval hypothesis appears particularly relevant to memory dysfunction in this group. Importantly, our results lend further weight to the proposition that episodic memory dysfunction in neurodegenerative disorders reflects widespread degeneration of complex neural networks, in which the hippocampus is but one participating structure (Irish et al., 2012c; Simons and Spiers, 2003). This position is underscored by recent findings from bvFTD patients, in which specific nodes of the Papez circuit, including the mammillary bodies and the anterior thalamus, have been shown to modulate the amnestic presentation (Hornberger et al., 2012).
Finally, our AD findings support the large body of evidence in which a widespread network of medial temporal, frontal, and parietal structures are implicated in episodic memory dysfunction (Buckner et al., 2005; Irish et al., 2012a, 2013). Importantly, our behavioural data reinforce the inherent difficulty in distinguishing between clinical AD and FTD patients, but also between C9 and sporadic FTD cases, based on measures of episodic memory alone (Frisch et al., 2013; Irish et al., 2013). From a clinical perspective, the utility of episodic memory tests to determine which patients should undergo screening for genetic mutations does not appear to represent a promising line of enquiry and we suggest that focusing on the possible neuropsychiatric disturbance in C9 carriers may represent a more fruitful approach.
A number of methodological issues warrant consideration in the current context. Firstly, our sample of C9 patients who had undergone structural neuroimaging and memory testing is relatively small, and limits the extent to which we can draw conclusions from these findings. That said, it is likely that replication of this study in a larger cohort of C9 cases would reveal interesting group differences, particularly on the visual memory task. While our neuroimaging analyses on the whole replicated previous reports in the literature, we may have lacked sufficient power to detect brain–behaviour relationships. Our sporadic bvFTD and AD patient sample have not yet come to autopsy, and thus we do not have access to neuropathological data to confirm the disease pathology in these groups. It is important to note, however, that our findings converge well with a growing number of studies highlighting severe memory disruption in pathologically confirmed cases of bvFTD (Graham et al., 2005; Hornberger et al., 2010, 2012) and in cases with genetically confirmed C9ORF72 mutations (Galimberti et al., 2013; Mahoney et al., 2012b). Replication of our current results in a larger sample, in conjunction with neuropathological data, represents an important avenue for future research.
In summary, we have confirmed the presence of marked episodic memory disturbance in carriers of the C9ORF72 genetic mutation, in keeping with previous reports in the literature pointing to an amnestic profile in this group. For C9ORF72 mutation carriers, degeneration of a distributed set of frontal, lateral and medial temporal, and lateral and medial parietal regions results in episodic memory deficits, comparable to that seen in disease-matched sporadic bvFTD and AD cases. Our results have important clinical implications in that memory impairment may not represent a useful construct to distinguish between carriers and non-carriers of the C9ORF72 mutation. Given the distinctive neural substrates underlying episodic memory dysfunction in C9 versus sporadic bvFTD cases, we suggest that the next challenge lies in devising clinically sensitive tools which differentially stress posterior versus anterior aspects of episodic retrieval, respectively. This approach may aid the clinician to identify those candidates who should undergo genetic screening, with the ultimate goal of creating tailored interventions to assist in the long-term management of these patients.
Role of the funding source
This work was supported in part by an Australian Research Council (ARC) Discovery Project (DP1093279); the ARC Centre of Excellence in Cognition and its Disorders (CE110001021); MI is supported by an ARC Discovery Early Career Research Award (DE130100463); MH is supported by an ARC Research Fellowship (DP110104202); OP is supported by a National Health and Medical Research Council of Australia Career Development Fellowship (NHMRC, APP 1022684); JBK is supported by an NHMRC Project Grant (APP1005769); CDS is supported by an NHMRC Project Grant (APP630428). These sources had no role in the study design, collection, analyses and interpretation of data, writing of the manuscript, or in the decision to submit the paper for publication. The authors report no conflict of interest.
Acknowledgements
We are grateful to the patients and their families for their support of our research.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- Andersson J.L.R., Jenkinson M., Smith S. University of Oxford FMRIB Centre; 2007. Non-linear Optimisation. [Google Scholar]
- Andersson J.L.R., Jenkinson M., Smith S. University of Oxford FMRIB Centre; 2007. Non-linear Registration, aka Spatial Normalisation. [Google Scholar]
- Ashburner J., Friston K.J. Voxel-based morphometry — the methods. NeuroImage. 2000;11:805–821. doi: 10.1006/nimg.2000.0582. [DOI] [PubMed] [Google Scholar]
- Becker S., Lim J. A computational model of prefrontal control in free recall: strategic memory use in the California Verbal Learning Task. Journal of Cognitive Neuroscience. 2003;15:821–832. doi: 10.1162/089892903322370744. [DOI] [PubMed] [Google Scholar]
- Boeve B.F., Boylan K.B., Graff-Radford N.R., DeJesus-Hernandez M., Knopman D.S. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain. 2012;135:765–783. doi: 10.1093/brain/aws004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner R.L., Snyder A.Z., Shannon B.J., LaRossa G., Sachs R. Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. The Journal of Neuroscience. 2005;25:7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess P., Shallice T. Thames Valley Test Company; Thurston Suffolk: 1997. The Hayling and Brixton Tests. [Google Scholar]
- Byrne S., Elamin M., Bede P., Shatunov A., Walsh C. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurology. 2012;11:232–240. doi: 10.1016/S1474-4422(12)70014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chio A., Borghero G., Restagno G., Mora G., Drepper C. Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain. 2012;135:784–793. doi: 10.1093/brain/awr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collette F., Van der Linden M., Salmon E. Dissociation between controlled and automatic processes in the behavioral variant of fronto-temporal dementia. Journal of Alzheimer's Disease. 2010;22(3):897–907. doi: 10.3233/JAD-2010-100042. [DOI] [PubMed] [Google Scholar]
- Cooper-Knock J., Hewitt C., Highley J.R., Brockington A., Milano A. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain. 2012;135:751–764. doi: 10.1093/brain/awr365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M., Mackenzie I.R., Boeve B.F., Boxer A.L., Baker M. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson-Stone C., Hallupp M., Bartley L., Shepherd C.E., Halliday G.M. C9ORF72 repeat expansion in clinical and neuropathologic frontotemporal dementia cohorts. Neurology. 2012;79:995–1001. doi: 10.1212/WNL.0b013e3182684634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch S., Dukart J., Vogt B., Horstmann A., Becker G. Dissociating memory networks in early Alzheimer's disease and frontotemporal lobar degeneration — a combined study of hypometabolism and atrophy. PLoS One. 2013;8:e55251. doi: 10.1371/journal.pone.0055251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galimberti D., Fenoglio C., Serpente M., Villa C., Bonsi R. Autosomal dominant frontotemporal lobar degeneration due to the C9ORF72 hexanucleotide repeat expansion: late-onset psychotic clinical presentation. Biological Psychiatry. 2013 doi: 10.1016/j.biopsych.2013.01.031. [DOI] [PubMed] [Google Scholar]
- Graham A., Davies R.R., Xuereb J.H., Halliday G.M., Kril J.J. Pathologically proven frontotemporal dementia presenting with severe amnesia. Brain. 2005;128:597–605. doi: 10.1093/brain/awh348. [DOI] [PubMed] [Google Scholar]
- Hodges J.R. Familial frontotemporal dementia and amyotrophic lateral sclerosis associated with the C9ORF72 hexanucleotide repeat. Brain. 2012;135:652–655. doi: 10.1093/brain/aws033. [DOI] [PubMed] [Google Scholar]
- Hornberger M., Piguet O., Graham A.J., Nestor P.J., Hodges J.R. How preserved is episodic memory in behavioral variant frontotemporal dementia? Neurology. 2010;74:472–479. doi: 10.1212/WNL.0b013e3181cef85d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornberger M., Wong S., Tan R., Irish M., Piguet O. In vivo and post-mortem memory circuit integrity in frontotemporal dementia and Alzheimer's disease. Brain. 2012;135:3015–3025. doi: 10.1093/brain/aws239. [DOI] [PubMed] [Google Scholar]
- Hsiung G.Y., DeJesus-Hernandez M., Feldman H.H., Sengdy P., Bouchard-Kerr P. Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain. 2012;135:709–722. doi: 10.1093/brain/awr354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irish M., Addis D.R., Hodges J.R., Piguet O. Considering the role of semantic memory in episodic future thinking: evidence from semantic dementia. Brain. 2012;135:2178–2191. doi: 10.1093/brain/aws119. [DOI] [PubMed] [Google Scholar]
- Irish M., Graham A., Graham K.S., Hodges J.R., Hornberger M. Differential impairment of source memory in progressive versus non-progressive behavioral variant frontotemporal dementia. Archives of Clinical Neuropsychology. 2012;27:338–347. doi: 10.1093/arclin/acs033. [DOI] [PubMed] [Google Scholar]
- Irish M., Piguet O., Hodges J.R. Self-projection and the default network in frontotemporal dementia. Nature Reviews. Neurology. 2012;8:152–161. doi: 10.1038/nrneurol.2012.11. [DOI] [PubMed] [Google Scholar]
- Irish M., Piguet O., Hodges J.R., Hornberger M. Common and unique grey matter correlates of episodic memory dysfunction in frontotemporal dementia and Alzheimer's disease. Human Brain Mapping. 2013 doi: 10.1002/hbm.22263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppel G., Wickens T.D. A Researcher's Handbook. Prentice Hall; Englewood Cliffs (NJ): 2004. Design and analysis. [Google Scholar]
- Kipps C.M., Hodges J.R., Hornberger M. Nonprogressive behavioural frontotemporal dementia: recent developments and clinical implications of the ‘bvFTD phenocopy syndrome’. Current Opinion in Neurology. 2010;23:628–632. doi: 10.1097/WCO.0b013e3283404309. [DOI] [PubMed] [Google Scholar]
- Kramer J.H., Rosen H.J., Du A.T., Schuff N., Hollnagel C. Dissociations in hippocampal and frontal contributions to episodic memory performance. Neuropsychology. 2005;19:799–805. doi: 10.1037/0894-4105.19.6.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long N.M., Oztekin I., Badre D. Separable prefrontal cortex contributions to free recall. The Journal of Neuroscience. 2010;30:10967–10976. doi: 10.1523/JNEUROSCI.2611-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney C.J., Beck J., Rohrer J.D., Lashley T., Mok K. Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain. 2012;135:736–750. doi: 10.1093/brain/awr361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney C.J., Downey L.E., Ridgway G.R., Beck J., Clegg S. Longitudinal neuroimaging and neuropsychological profiles of frontotemporal dementia with C9ORF72 expansions. Alzheimer's Research & Therapy. 2012;4:41. doi: 10.1186/alzrt144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann G.M., Knopman D.S., Chertkow H., Hyman B.T., Jack C.R., Jr. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's & Dementia. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechelli A., Price C.J., Friston C.J., Ashburner J. Voxel-based morphometry of the human brain: methods and applications. Current Medical Imaging Reviews. 2005;1:1–9. [Google Scholar]
- Meyers J., Meyers K. Psychological Assessment Resources; Odessa, Florida: 1995. The Meyers Scoring System for the Rey Complex Figure and the Recognition Trial: Professional Manual. [Google Scholar]
- Mioshi E., Dawson K., Mitchell J., Arnold R., Hodges J.R. The Addenbrooke's Cognitive Examination Revised (ACE R): a brief cognitive test battery for dementia screening. International Journal of Geriatric Psychiatry. 2006;21:1078–1085. doi: 10.1002/gps.1610. [DOI] [PubMed] [Google Scholar]
- Morris J. Clinical dementia rating: a reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. International Psychogeriatrics. 1997;9:173–176. doi: 10.1017/s1041610297004870. [DOI] [PubMed] [Google Scholar]
- Nichols T.E., Holmes A.P. Nonparametric permutation tests for functional neuroimaging: a primer with examples. Human Brain Mapping. 2002;15:1–25. doi: 10.1002/hbm.1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington C., Hodges J.R., Hornberger M. Neural correlates of episodic memory in behavioural variant frontotemporal dementia. Journal of Alzheimer's Disease. 2011;24:261–268. doi: 10.3233/JAD-2011-101668. [DOI] [PubMed] [Google Scholar]
- Piguet O., Hornberger M., Mioshi E., Hodges J.R. Behavioural-variant frontotemporal dementia: diagnosis, clinical staging, and management. Lancet Neurology. 2011;10:162–172. doi: 10.1016/S1474-4422(10)70299-4. [DOI] [PubMed] [Google Scholar]
- Piolino P., Chetelat G., Matuszewski V., Landeau B., Mezenge F. In search of autobiographical memories: a PET study in the frontal variant of frontotemporal dementia. Neuropsychologia. 2007;45:2730–2743. doi: 10.1016/j.neuropsychologia.2007.04.013. [DOI] [PubMed] [Google Scholar]
- Rabinovici G.D., Seeley W.W., Kim E.J., Gorno-Tempini M.L., Rascovsky K. Distinct MRI atrophy patterns in autopsy-proven Alzheimer's disease and frontotemporal lobar degeneration. American Journal of Alzheimer's Disease and Other Dementias. 2007;22:474–488. doi: 10.1177/1533317507308779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascovsky K., Hodges J.R., Knopman D., Mendez M.F., Kramer J.H. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134:2456–2477. doi: 10.1093/brain/awr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnavalli E., Brayne C., Dawson K., Hodges J. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615–1621. doi: 10.1212/wnl.58.11.1615. [DOI] [PubMed] [Google Scholar]
- Reitan R. Validity of the Trail Making Test as an indicator of organic brain damage. Perceptual and Motor Skills. 1958;8:271–276. [Google Scholar]
- Renton A.E., Majounie E., Waite A., Simon-Sanchez J., Rollinson S. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueckert D., Sonoda L.I., Hayes C., Hill D.L., Leach M.O. Nonrigid registration using free-form deformations: application to breast MR images. IEEE Transactions on Medical Imaging. 1999;18:712–721. doi: 10.1109/42.796284. [DOI] [PubMed] [Google Scholar]
- Schmidt M. Western Psychological Services; Los Angeles: 1996. Rey Auditory and Verbal Learning Test: A handbook. [Google Scholar]
- Seeley W.W., Crawford R., Rascovsky K., Kramer J.H., Weiner M. Frontal paralimbic network atrophy in very mild behavioral variant frontotemporal dementia. Archives of Neurology. 2008;65:249–255. doi: 10.1001/archneurol.2007.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha S.J., Takada L.T., Rankin K.P., Yokoyama J.S., Rutherford N.J. Frontotemporal dementia due to C9ORF72 mutations: clinical and imaging features. Neurology. 2012;79:1002–1011. doi: 10.1212/WNL.0b013e318268452e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons J.S., Spiers H.J. Prefrontal and medial temporal lobe interactions in long-term memory. Nature Reviews. Neuroscience. 2003;4:637–648. doi: 10.1038/nrn1178. [DOI] [PubMed] [Google Scholar]
- Simon-Sanchez J., Dopper E.G., Cohn-Hokke P.E., Hukema R.K., Nicolaou N. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain. 2012;135:723–735. doi: 10.1093/brain/awr353. [DOI] [PubMed] [Google Scholar]
- Smith S.M. Fast robust automated brain extraction. Human Brain Mapping. 2002;17:143–155. doi: 10.1002/hbm.10062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S.M., Jenkinson M., Woolrich M.W., Beckmann C.F., Behrens T.E. Advances in functional and structural MR image analysis and implementation as FSL. NeuroImage. 2004;23(Suppl. 1):S208–219. doi: 10.1016/j.neuroimage.2004.07.051. [DOI] [PubMed] [Google Scholar]
- Snowden J.S., Rollinson S., Thompson J.C., Harris J.M., Stopford C.L. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain. 2012;135:693–708. doi: 10.1093/brain/awr355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderlund H., Black S.E., Miller B.L., Freedman M., Levine B. Episodic memory and regional atrophy in frontotemporal lobar degeneration. Neuropsychologia. 2008;46:127–136. doi: 10.1016/j.neuropsychologia.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollberger M., Stanley C.M., Wilson S.M., Gyurak A., Beckman V. Neural basis of interpersonal traits in neurodegenerative diseases. Neuropsychologia. 2009;47:2812–2827. doi: 10.1016/j.neuropsychologia.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss E., Sherman E.M.S., Spreen O. Oxford University Press; USA: 2006. A Compendium of Neuropsychological Tests: Administration, Norms, and Commentary. [Google Scholar]
- Strong M.J., Grace G.M., Freedman M., Lomen-Hoerth C., Woolley S. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis. 2009;10:131–146. doi: 10.1080/17482960802654364. [DOI] [PubMed] [Google Scholar]
- van Swieten J.C., Grossman M. FTD/ALS families are no longer orphaned: the C9ORF72 story. Neurology. 2012;79:962–964. doi: 10.1212/WNL.0b013e318268471a. [DOI] [PubMed] [Google Scholar]
- Wechsler D. Psychological Corporation; San Antonio, Texas: 1997. Wechsler Memory Scale — Third edition: Administration and Scoring Manual. [Google Scholar]
- Wedderburn C., Wear H., Brown J., Mason S.J., Barker R.A. The utility of the Cambridge Behavioural Inventory in neurodegenerative disease. Journal of Neurology, Neurosurgery, and Psychiatry. 2008;79:500–503. doi: 10.1136/jnnp.2007.122028. [DOI] [PubMed] [Google Scholar]
- Whitwell J.L., Weigand S.D., Boeve B.F., Senjem M.L., Gunter J.L. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain. 2012;135:794–806. doi: 10.1093/brain/aws001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicklund A.H., Johnson N., Rademaker A., Weitner B.B., Weintraub S. Word list versus story memory in Alzheimer disease and frontotemporal dementia. Alzheimer Disease and Associated Disorders. 2006;20:86–92. doi: 10.1097/01.wad.0000213811.97305.49. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Brady M., Smith S. Segmentation of brain MR images through a hidden Markov random field model and the expectation-maximization algorithm. IEEE Transactions on Medical Imaging. 2001;20:45–57. doi: 10.1109/42.906424. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.