

Abstract
Background:
Pt-(GpG) intrastrand crosslinks are the major DNA adducts induced by platinum-based anticancer drugs. In the cell lines and mouse models, the persistence of these lesions correlates significantly with cell damage. Here we studied Pt-(GpG) DNA adducts in circulating tumour cells (CTC) treated with cisplatin in medium upfront to systemic therapy from patients with advanced non-small-cell lung cancer (NSCLC).
Methods:
Blood was drawn before systemic treatment and the CD45/CD15-depleted fraction of mononuclear cells was exposed to cisplatin, verified for the presence of CTC by pan-cytokeratin (pCK) staining and immunoanalysed for the level of Pt-(GpG) in DNA.
Results:
Immunostaining for pCK, CD45 and subsequently for Pt-(GpG) adducts in the cisplatin-exposed cells (ex vivo) at different time points depicted distinct differences for adduct persistence in CTC between responders vs non-responders.
Conclusion:
Pt-(GpG) adducts can be detected in CTC from NSCLC patients and assessing their kinetics may constitute a clinically feasible biomarker for response prediction and dose individualisation of platinum-based chemotherapy. This functional pre-therapeutic test might represent a more biological approach than measuring protein factors or other molecular markers.
Keywords: Pt-(GpG) DNA adducts, circulating tumour cells, pre-therapeutic, ex vivo, non-small-cell lung cancer, platinum-based chemotherapy
Platinum-doublets are the key elements of first-line treatment in lung cancer. The antineoplastic activity of the platinum complexes is mediated by their reaction with nuclear DNA and the formation of structurally defined intra- and interstrand adducts (Kelland, 2007). The sensitivity/resistance of lung cancer cells to treatment with cisplatin is significantly correlated with their ability to remove such damage in their genome, for example, by the nucleotide excision repair (NER) system as shown for human cell lines as well as for primary tumours in a mouse model of lung cancer (Oliver et al, 2010). Though retrospective studies on expression profiles of DNA repair factors like excision repair cross-complementing group 1 (ERCC1) or others have been countless, they did not lead to successful implementation in daily clinical routine so far (Hubner et al, 2011). For none of these many distinct protein factors a rate-limiting role has been demonstrated yet, and no stringent correlation could be shown in human tumour cell lines between functional removal of cisplatin-DNA adducts and the expression profiles of particular repair proteins, neither at the mRNA nor at the protein level (Wang et al, 2011b). Isoforms may even enhance the varying association of single markers to platinum response (Friboulet et al, 2013).
Measuring total platinum concentration or accumulation in biopsies has been discussed as a more mechanistic and therefore reliable approach to individualise treatment and reduce toxicity (Henderson et al, 2011; Kim et al, 2012). Platinum-DNA adducts represent the effective damage leading to cell-cycle arrest and apoptotic death. Olaussen et al (2013) showed that the extent of DNA platination significantly correlated with the sensitivity of human lung cancer cell lines to cisplatin. We previously used monoclonal antibodies against guanine–guanine (Pt-(GpG)) or adenine–guanine (Pt-(ApG)) intrastrand crosslinks to evaluate formation and removal of these adducts with an immunoanalytic method directly in the nucleic DNA of tumour and haematopoietic cells (Liedert et al, 2006; Oliver et al, 2010).
So far, the main obstacle to using this promising method in clinical routine was the need for fresh tissue to test for platinum-DNA adducts. Especially in lung cancer repeated biopsies are associated with an increased patients' risk (Printz, 2010). Repeated samples, however, would be necessary to directly monitor adduct formation and repair in tumour cells. Furthermore, malignancies do not represent a static tissue and their cellular composition changes during treatment, resulting in the development of drug resistance.
Here we intended to scrutinise whether the level of Pt-(GpG) adducts can be analysed quantitatively in the nuclear DNA of circulating tumour cells (CTC) isolated from peripheral blood before systemic therapy and incubated with cisplatin ex vivo. Furthermore, we tested whether such adduct kinetics correlate well with Pt-(GpG) values obtained from the extracted CTC during and after systemic treatment in vivo. Additionally, we examined whether the expression of the DNA repair protein ERCC1 in CTC as measured at the mRNA level correlates with the adduct persistence and shows an association to the therapeutic response.
Materials and methods
Study population, blood samples, informed consent
Patients with non-small-cell lung cancer (NSCLC) who received anti-cancer treatment in our hospital were consecutively included in this study after agreeing and signing a written informed consent in accordance with the requirements of our institution's board of ethics (internal reference number: No. 12-5047-BO). Patient demographics are described in Table 1. Tumour staging was performed according to the criteria of the International Union Against Cancer (Sobin, 2003). Response Evaluation Criteria in Solid Tumors (RECIST) were used to define response or stable disease in patients after receiving two cycles of systemic cytotoxic chemotherapy (Therasse et al, 2000).
Table 1. Patient demographics.
| Patients (n=11) | ||
|---|---|---|
|
Demographic |
No. |
% |
|
Age | ||
| Median, years | 61 | |
| Range |
50–74 |
|
|
Histology | ||
| Adenocarcinoma | 7 | 64 |
| Squamous epithelium |
4 |
36 |
|
Staging | ||
| IIIa/b | 6 | 55 |
| IV |
5 |
45 |
|
Grading | ||
| G2 | 4 | 36 |
| G3 | 2 | 18 |
| Gx |
5 |
45 |
|
Therapy | ||
| Cisplatin/Paclitaxel | 7 | 64 |
| Carboplatin/Paclitaxel | 2 | 18 |
| Cisplatin/Pemetrexed |
2 |
18 |
|
Response | ||
| SD | 6 | 55 |
| PR | 5 | 45 |
Abbreviations: G2=moderately differentiated; G3=poorly differentiated; Gx=grade cannot be assessed; SD=stable disease; PR=partial response.
Preparation of blood samples and CTC enrichment
CTC were isolated from all patients upfront to and during treatment and examined for epithelial characteristics (Nel et al, 2012). At baseline, duplicates of 20 ml citrated peripheral venous blood were drawn (i) for ex vivo examination of Pt-(GpG) adduct persistence before systemic cisplatin-based treatment and (ii) for gene expression analysis. Cells for adduct measurement were incubated with cisplatin under standard cell culture conditions (see below). In some cases, additional blood samples for the analyses of in vivo adduct formation were drawn at 2, 4 and 24 h after intravenous cisplatin administration. Blood samples were processed immediately after collection.
For measuring platinum adduct kinetics, preparation of blood samples and subsequent CTC staining were done as follows: 20 ml of blood was diluted with 10 ml PBS and carefully layered into a Leucosep (Greiner Bio-One, Frickenhausen, Germany) tube containing 16 ml Ficoll-Paque (GE-Healthcare, Buckinghamshire, Great Britain) below a porous barrier. After buoyant density gradient centrifugation (1600 g, 20 °C, 20 min), the interphase consisting of peripheral blood mononuclear cells (PBMNC) and CTC was removed and washed. CTC were enriched in that fraction by depleting as many haematopoietic cells as necessary to allow downstream analysis (i.e., microscopic imaging). For depletion, cells were incubated with a 1 : 1 mixture of anti-CD45 and anti-CD15 antibody-coated immunomagnetic beads (Dynabeads; Invitrogen, Carlsbad, CA, USA) and separated in a magnetic particle processor (King Fisher mL; Thermo Fisher, Waltham, MA, USA) leading to a bead-free CTC-enriched cell fraction.
Cells were suspended in culture medium (DMEM supplemented with 10% fetal bovine serum; 1% gentamicin; 1% L-glutamine; 0.1% MITO serum extender and 0.5% bovine pituitary extract) and exposed to cisplatin for 2 h at 37 °C. Cells were then washed twice with PBS and further incubated in fresh culture media. Immediately after treatment (t0 h) and after 2, 4 and 24 h, aliquots were taken, washed with PBS, spotted on adhesive slides (Superfrost Gold, ThermoFisher) and air-dried. An aliquot of each sample was spotted before cisplatin exposure as a negative control. Cells from each of the five time-points were spotted onto multiple slides and 10 to 15 CTC per time point were evaluated.
For in vivo adduct detection, the depleted cell suspension was spun onto adhesion slides using the Cell Spin II centrifuge (Tharmac, Waldsolms, Germany), air-dried and subsequently fixated with 96% Ethanol. Slides were stored at 4 °C.
RNA extraction and RT-PCR
For ERCC1 expression analysis, CTC were pre-enriched by haematopoietic cell depletion using anti-CD45 immunomagnetic beads. Then epithelial CTC were positively selected using anti-EpCAM immunomagnetic beads (Dynabeads Epithelial Enrich; Invitrogen) resulting in an EpCAM-positive CTC suspension for molecular analysis. Total RNA was extracted from recovered EpCAM-positive tumour cells using MagAttract RNA Cell Mini M48 Kits (Qiagen, Hilden, Germany) and King Fisher mL magnetic particle processor (Thermo Fisher). Nucleic acids were purified from lysates in one step through their binding to the silica surface of the particles in the presence of a chaotropic salt. The particles were separated from the lysates using a magnet, and DNA was removed by treatment with RNase-free DNase. The magnetic particles were washed, and RNA was eluted in the buffer provided. Additionally, remaining DNA was removed using RQ1 RNase free DNase (Promega, Fichtburg, WI, USA). One-step real-time RT-PCR (Roche LightCycler 480, Roche Diagnostics, Basel, Switzerland) was performed using the Precision OneStep qRT-PCR Mastermix Kit with SYBR Green (Primerdesign, Southampton, UK) for gene expression analysis of ERCC1 (Primerdesign). The primers for the reference gene β-actin (Eurofins MWG, Nantes, France) were as follows: forward: 5′-GAGCGCGGCTACAGCTT-3′ and reverse: 5′-TCCTTAATGTCACGCACGATTT-3′. Assays were performed in triplicates to determine the expression levels (Hoffmann et al, 2009). Thermal cycling conditions were 10 min at 50 °C and 5 min at 95 °C for RT and initial denaturation followed by 50 cycles of 95 °C for 10 s and 60 °C for 30 s. Triplicates of A549-RNA (10 ng μl−1) were used as internal standard to control each run. Each primer has been validated in serial dilutions of RNA extracted from different cell lines.
Identification of CTC
Immunofluorescence staining of epithelial and haematopoietic cells was carried out in the CD45-/CD15-depleted cell fraction on microscopic slides. Briefly, the staining method included fixation of the cells in methanol (−20 °C) for 5 min, washing in PBS, blocking in 5% BSA solution for 30 min, binding of primary antibodies (final concentration 5 μg ml−1) with guinea pig anti-(pCK) (ABIN126062, antibodies-online, Atlanta, GA, USA) for CTC and mouse anti-(CD45) (MEM-28) (ab8216, Abcam, Cambridge, UK) for haematopoietic cells overnight at 4 °C. Then slides were washed in PBS containing 0.1% Tween 20, incubated with secondary antibodies (AlexaFlour647-conjugated AffiniPure F(ab')2 fragment goat anti-guinea pig and Cy3-conjugated AffiniPure goat anti-mouse; Jackson Immuno Research, Hamburg, Germany) for 30 min at 37 °C and washed in PBS/0.1% Tween. Subsequently, cells were stained with 4′6-diamidino-2-phenylindole dihydrochloride (1 μg ml−1; DAPI; Sigma-Aldrich, St Louis, MO, USA) for 10 min, washed in PBS and stored in the dark until microscopic evaluation. Detection and localisation of CTC was carried out using the digital Keyence BZ9000 (model BIOREVO, Osaka, Japan) all-in-one fluorescence microscope with integrated camera and BZ-Analyzer Software. Stained slides were manually examined using a × 20 magnification, and objects that stained positive for DAPI (nucleus) and pCK (epithelial marker) and negative for CD45 (haematopoeitic marker) were considered as tumour cells. Images of detected CTC and their position coordinates on the slide were stored to ensure relocation.
Measurement of Pt-(GpG) adducts in the nuclear DNA of CTC
After CTC localisation, Pt-(GpG) intrastrand cross-links in the nuclear DNA of CTC were visualised and measured by an immunocytological assay (ICA) using the adduct-specific monoclonal antibody ‘R-C18' as previously described (Liedert et al, 2006) with some modifications. Briefly, cells either spotted or spun onto adhesion slides were fixed in methanol (−20 °C) followed by alkaline denaturation (70 mM NaOH, 140 mM NaCl in 40% methanol, 0 °C, 5 min) and sequential proteolytic digestion with pepsin (200 μg ml−1) and proteinase K (100 μg ml−1; both Sigma-Aldrich). During this step, fluorescence signals derived from cell type-specific staining based on protein markers such as pCK and CD45 were completely abolished. After blocking with skim milk, DNA adducts were visualised by indirect immunostaining with primary antibody R-C18 (0.2 μg ml−1) and Cy3-labeled rabbit anti-(rat Ig) secondary antibody (Dianova, Hamburg, Germany). Nuclear DNA was counterstained with DAPI (1 μg ml−1). The stored positions on the slides were rescanned for Cy3- and DAPI-derived fluorescence signals from detected and relocated CTC nuclei and measured by Keyence cellcount software. Cy3 signals were normalised to the corresponding DAPI signals from each cell. Per patient and slide (five spots on each of the four slides), at least 15 CTC were calculated and mean adduct levels were expressed as arbitrary fluorescence units (AFU). CTC were analysed in triplicates per time-point and per slide.
Statistical analyses
Spearman's rank correlation was used to examine associations between single parameters of the Pt-(GpG) adduct persistence, such as the highest Pt-(CpG) signal amplitude (AFUmax), slope (between t0 h and t2 h) and area under the curve (AUC). To identify a potential independent factor associated with response, these parameters were tested in a linear multivariate regression model. The adduct persistence of each patient per time-point was then summarised into AUC and analysed statistically with the serial measurements command. We used receiver operating characteristic (ROC) curve analysis to test the ability of Pt-(GpG) adduct kinetics to discriminate stable disease from partial response. Recursive descent partition analysis was used to identify the strongest divisor of all factors, and the most significant split determined by the largest likelihood-ratio chi-square statistic in relation to clinical response as described previously (Hoffmann et al, 2008, 2009). The split was chosen to maximise the difference in the responses between the two branches of the split. The level of significance was set to P<0.05. All P values were based on two-sided tests. All statistical analyses were performed using the Software Packages Medcalc, Version 12.4.0 (MedCalc Software, Ostend, Belgium) and JMP 10.0 (SAS Institute, Cary, NC, USA).
Results
Immunocytochemical identification of CTC and measurement of Pt-(GpG) adducts
We were able to detect epithelial CTC that stained positive for DAPI and pCK and negative for CD45 (Figure 1). The ratio of pCK-positive to CD45-positive cells in the depleted fractions was in the range of ⩽1–5000, resulting in up to three CTC per cell spot on the slides. To ensure CTC relocation, the position coordinates and microscopic images of each detected CTC on the slide were stored electronically. After subsequent immunofluorescence staining for Pt-(GpG) adducts, the slide positions were rescanned, and CTC relocation as well as adduct measurement was technically feasible (Figure 1). CD45-positive cells depicted nearly 10-fold lower levels of DNA platination after exposure to cisplatin as compared with CTC. The same intercellular distribution was observed in all the cell samples, including those isolated under systemic therapy with cisplatin (see below).
Figure 1.
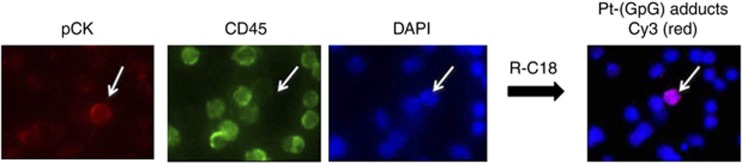
Visualisation of Pt-(GpG) adducts in the nuclear DNA of CTC exposed to cisplatin ex vivo. The mononuclear cell fraction isolated from the peripheral blood of NSCLC patients was depleted for CD45 cells and exposed to cisplatin (20 μg ml−1) for 2 h in liquid holding. Tumour cells positive for pan-cytokeratin, negative for CD45 were identified and the position coordinates were stored to ensure relocation. Pt-(GpG) DNA adducts in CTC nuclei were visualised by indirect immunostaining with the adduct-specific antibody R-C18. The image shows an epithelial pCK-positive/CD45-negative/DAPI-positive CTC.
Pt-(GpG) kinetics in CTC exposed to cisplatin ex vivo
The measurement of Pt-(GpG) adducts in the nuclear DNA of short term-exposed CTC depict a characteristic time course of formation and removal (Figure 2). Adduct kinetics were reproducible when repeated in individual blood samples from the same donor but showed broad variations between CTC from individual patients. Formation and persistence of DNA platination can be characterised by different parameters such as curve inclination between distinct time points (slope), peak adduct level (AFUmax) and area under the curve (AUC) (Figure 2). We have calculated the degree of association between these three values of ex vivo adduct persistence in CTC from 11 patients with their therapeutic response using the Spearman's rho rank correlation coefficient. AFUmax correlated significantly with AUC (P=0.001) and response (P=0.05). The AUC was also significantly correlated with response (P=0.03). A linear backward multivariate regression model with response as the dependent variable identified AUC as the only factor significantly and independently associated with response (P=0.002). Therefore, summary measures of ex vivo Pt-(GpG) adduct levels from patients before treatment were expressed in AUC. Persistence data did not show a normal distribution, and a Mann–Whitney test was performed for a non-parametric test, which revealed a significant difference between patients showing stable disease or partial response 6 weeks after systemic platinum-based treatment (P=0.01; n=11; Figure 3A). We used recursive descent partition tree analysis to find the factor showing the strongest association with response and define the optimal cut-point of this factor. We used response vs non-response as the defining factor and adduct persistence values (AFUmax, AUC, slope) alongside ERCC1 mRNA expression (see below) and clinical factors such as age, histology, staging (TNM) and grading as dividing factors. The strongest divisor in this study group was AUC followed by AFUmax. The most significant split determined by the largest likelihood-ratio chi-square statistic of AUC was the 50th percentile (4.166). According to the descent recursive partition analysis, patients were then split into low and high AUC groups at the 50th percentile. A stepwise linear multivariate regression analysis using AUC as a dichotomous variable confirmed high AUC (>50th percentile) as being independently associated with response with an overall model fit of P=0.036. Patients with a high AUC, and therefore higher ex vivo adduct persistence in tumour cells incubated with cisplatin upfront to intravenous therapy, had a higher chance to benefit from systemic cisplatin treatment. Receiver operating curve (ROC) analysis was used to assess the sensitivity and specificity of high AUC (>50th percentile) as a parameter of ex vivo Pt-(GpG) persistence to predict response from patients upfront to cytotoxic therapy and revealed a sensitivity of 80% and a specificity of 83% with a significance of P=0.01 and an ROC area under the curve of 0.817 (Figure 3B). To test whether adduct kinetics measured in tumour cells after ex vivo exposure to cisplatin mirror the formation and repair rates for Pt-(GpG) in the DNA of those cells in vivo during systemic treatment, blood samples from the same patient were analysed under both the conditions. Specimen drawn at baseline and 2, 4 and 24 h after cisplatin infusion exhibited similarly shaped courses as measured in CTC aliquots ex vivo (Figure 4). In this case, the clinical dose of cisplatin (50 mg m−2) translating into 1.4 mg kg−1 body weight resulted in CTC adduct levels comparable to a short time ex vivo exposure with 20 μg ml−1 for 2 h.
Figure 2.

Formation and repair of Pt-(GpG) in the nuclear DNA of cisplatin-exposed CTC from NSCLC patients. Cells were treated with cisplatin (see Figure 1) for 2 h (t−2 h−t0 h), then transferred to drug-free medium and were harvested at different times (0, 2, 4 and 24 h). Following immunostaining for CTC markers and Pt-adducts, Cy3 and DAPI signals from tumour cells were measured by quantitative image analysis and calculated as mean arbitrary fluorescence units (AFU). Error bars display s.d. Adduct kinetic parameters (AFUmax; slope and area under the curve (AUC)) were determined as described in Material and Methods.
Figure 3.

(A) Illustration of area under the curve (AUC) of ex vivo repair kinetics from patients with either partial response (PR; mean: 37.08) or stable disease (SD; mean: 3.11; P=0.01; n=11) in a box plot with Log scale: the central box represents the values from the 25th to the 75th percentile; middle line equals median; horizontal line represents distribution from minimum to maximum value excluding outliers. (B) Receiver operating characteristic (ROC) analysis was used to assess the sensitivity and specificity of high AUC (>50th percentile) as parameter of repair kinetics to distinguish partial response from stable disease.
Figure 4.

Ex vivo and in vivo Pt-(GpG) repair in CTC of the same patient show similar courses. Cisplatin treatment of isolated CTC ex vivo was as in Figure 2, in vivo adduct kinetics were measured in cells isolated from the peripheral blood during and after systemic treatment with cisplatin (50 mg m−2). −2 h refers to samples taken before treatment.
Adduct persistence and ERCC1 mRNA expression
For ERCC1 gene expression analysis, we used a sequential enrichment strategy to physically isolate tumour cells from PBMNC. After depletion of white blood cells with anti-CD45 and -CD15 immunomagnetic beads, EpCAM+ cells were enriched from the resulting cell suspension for downstream analysis. ERCC1 mRNA levels were measured in selected CTC isolated upfront to cytotoxic chemotherapy. The RT-PCR-based data showed no association with ex vivo Pt-(GpG) adduct persistence values, including slope, AFUmax and AUC (P=0.67, P=0.44 and P=0.78, respectively). Although the mean expression ratio of ERCC1 was nearly twofold higher (1.02±0.58) in the patient group with stable disease as compared with patients with partial response (0.69±0.23), no significant correlation with response was observed (P=0.55; Figure 5).
Figure 5.

Comparison of ERCC1 mRNA expression in CTC from patients with either partial response (PD) or stable disease (SD). The values did not show a significant correlation to response (P=0.55). The mean ERCC1 mRNA expression ratio was 0.69±0.23 (1.02±0.58) in patients with partial response (stable disease). The middle line equals mean. Dots represent distribution from minimum to maximum value excluding outliers.
Discussion
With this pilot study, we examined whether cisplatin-induced DNA adducts can be detected and quantified in extracted CTC after exposure to the drug under standard cell culture conditions and whether the adduct kinetics during a 24-h time period depict significant inter-patient differences. Secondly, we wanted to scrutinise whether the adduct persistence observed in ex vivo-treated tumour cells correlated with values obtained from CTC during and after in vivo (systemic) treatment with cisplatin. Additionally, we tested for an association with mRNA expression of the essential NER component ERCC1, a marker often proposed as a potential predictor of resistance in several solid tumours receiving cisplatin-based treatment (Cobo et al, 2007; Hoffmann et al, 2010).
Though this pilot study encompassed a relatively small number of patients, the persistence values for Pt-(GpG) adducts in DNA already separated clearly into two groups using partition tree analysis. These groups showed a significant correlation with treatment response in uni- and multivariate analysis (P=0.03; P=0.002). The examined group included patients with either response or stable disease but not patients with progressive disease based on RECIST criteria. Levels of ERCC1 mRNA expression did not show statistically significant correlation with either of the adduct parameters AUC, AFUmax or slope of persistence. ERCC1 mRNA expression did, however, show a clear distributional trend—though not statistically significant—between patients with partial response vs stable disease (Figure 5). This conforms not only with a previously published meta-analysis published by Hubner et al (2011) but may also be explained by recent findings of Friboulet et al (2013) showing that only one of the five known ERCC1 isoforms is essential for platinum adduct repair. The RT–PCR primers used in this study were not isoform specific. Our finding that AUC values were applicable to distinguish between stable disease and partial response with high sensitivity (80%) and specificity (83%, P=0.01) supports the validity of the used method and probably the superiority over expression-based approaches. Moreover, we were able to show similarity of ex vivo (pre-therapeutic) and in vivo adduct persistence in CTC from the same patient. Although the correlation of ex vivo and in vivo kinetics requires further validation with a larger patient cohort, the results underline the potential clinical usability of pre-screening CTC for their response to platinum compounds.
Until now, there have been numerous approaches to convert the analysis of DNA platination into a predictive assay for drug sensitivity. Recently, Henderson et al (2011) reported the measurement of 14C-labeled carboplatin-induced DNA-bound monoadducts in cell lines by using a very sensitive accelerated mass spectrometry (AMS)-based method. This study also aimed for a pre-therapeutic approach to administer platinum-based therapy with more efficiency. The authors were able to show that the levels of initially formed and transient DNA monoadducts correlated with the cellular resistance to carboplatin. However, in contrast to the ICA analysis used here, the AMS technique is restricted to the measurement of carboplatin adducts in isolated DNA and is not suited for adduct analysis at the level of single cells. Therefore we feel that the method described here bears a greater potential to be used in clinical routine, especially due to its minimal invasive character. Kim et al (2012) used flameless atomic absorption spectrophotometry (AAS) on stored fresh-frozen NSCLC specimens to test for whole-tissue platinum accumulation. All patients (n=44) received neoadjuvant treatment with platinum before they underwent surgical resection and displayed either response or stable disease. The authors were able to prove that higher total platinum concentration was correlated with longer time to recurrence (P=0.034) and to progression-free survival (P=0.018) in the multivariate Cox regression analysis. Their report also underlines the impact of platinum levels on response and outcome, although the measurement predominantly reflects the import/export activity of the cells rather than formation and persistence of the critical DNA platination as >90% of intracellular platinum is bound to non-DNA target molecules. The main obstacle here seems to be the need for tissue and the use of patients who underwent intravenous administration of several cycles of platinum therapy in a neoadjuvant setting. Though the results of Kim et al (2012) illustrate that platinum persistence resembles a very strong indicator of therapeutic outcome, this information has no clinical consequence after systemic treatment. In accordance to our results, platinum concentration segregated significantly between patients with partial response vs stable disease. This supports our finding that a more functional approach may have higher selectivity than expression-based methods.
Wang et al (2011a) also used a blood-based pre-therapeutic approach using in vitro measurements of DNA repair capacity in stimulated T-lymphocytes from NSCLC patients. By a host cell reactivation assay with BPDE-reacted plasmids, they were able to show an association with patient's overall survival (OS) and therefore concluded that the repair capacity of normal T-lymphocytes may serve as surrogate for platinum-resistance in tumours. In a comment to this report, Olaussen et al (2012) pointed out that the correlation of DNA repair in lymphocytes with OS may represent the combination of response and other factors, especially toxicity, and therefore could be an approach to detect potential haematopoietic toxicity rather than primary tumour response. Moreover, we found that using PBMNC as a surrogate may not be sufficient, as even among haematopoietic cells the levels of platinum uptake and DNA adduct processing vary significantly (Bracker et al, 2006). Using a cisplatin concentration for ex vivo incubation, which induced adduct levels comparable to systemic intensity, we measured much lower levels of DNA platination in haematopoietic cells as compared with CTC in this fraction. This finding is in accordance to the in vivo results and similar observations in mice where T cells exhibited 5–10-fold lower levels of Pt-(GpG) adducts in comparison to, for example, kidney or liver cells (Liedert et al, 2006). Only increasing the cisplatin concentration 10 times delivered quantifiable Pt-(GpG) DNA adducts in PBMNC. Therefore we think that without specific cell type classification one cannot use this approach as a clinical test to anticipate therapeutic response of the tumour. However, with the technique used in our study, we are able to test adduct persistence in extracted epithelial CTC and in white blood cells during a 24-h period upfront to systemic administration. Interestingly, in some of the blood samples analysed, a distinct proportion of cells stained positive for pCK as well as for and CD45, a phenomenon already described by Yu et al (2011). The additional CD45+ staining may not be exclusive for haematopoietic cells but may hypothetically be acquired during the dormant stay in the bone marrow or through effects comparable to trogocytosis, that is, transfer of membrane proteins (Joly and Hudrisier, 2003). These questions need to be addressed in further studies which will include more subtypes of CTC such as mesenchymal and stem cell-like CTC and their reaction to platinum-DNA adducts. Moreover, adding potential modifiers of platinum persistence such as PARP-1 inhibitors (Olaussen et al, 2013) to the cells under standard cell culture conditions upfront to platinum incubation may identify patients who may benefit from this combination.
Conclusion
With this pilot study, we show the feasibility of measuring platinum adduct persistence ex vivo in extracted CTC upfront to systemic therapy. Adduct kinetics resemble formation and repair of DNA platination in CTC exposed in vivo. The resulting values varied greatly between patients and were associated significantly with therapeutic response. Predictive capabilities showed significant sensitivity and specificity. Further studies, including modifiers of platinum sensitivity/repair, as well as careful comparison of adduct kinetics in CTC, including subgroups and putative surrogate blood cells, seem to be warranted.
Acknowledgments
We thank Professor Lang, Institute of Immunology, University Clinic Essen, for granting us access to his microscope. This project was funded by the Dr Werner Jackstädt-Foundation, Wuppertal, Germany.
Statement of translational relevance: This is the first description of the feasibility of measuring platinum-DNA adducts in circulating tumour cells ex vivo and in vivo before and during systemic treatment to predict resistance to platinum in non-small-cell lung cancer (NSCLC). These findings represent an important step towards a biological- and functional-based approach towards the development of biomarkers for individualising treatment decisions in NSCLC patients.
Author contributions
Conception and design: A-CH, JT, and MS. Proof-reading of manuscript: A-CH, JT, TG, WE, and MS. Provision of study materials or patients: A-CH and TG. Collection and assembly of data: IN, A-CN, A-CH. Data analysis and interpretation: IN, JT, A-CH. Manuscript writing: IN, A-CH. Final approval of manuscript: A-CH.
Footnotes
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
First results of this study were presented at the Annual Meeting of the British Thoracic Oncology Group in Dublin, January 2013 (Poster Prize).
References
- Bracker TU, Giebel B, Spanholtz J, Sorg UR, Klein-Hitpass L, Moritz T, Thomale J. Stringent regulation of DNA repair during human hematopoietic differentiation: a gene expression and functional analysis. Stem Cells. 2006;24 (3:722–730. doi: 10.1634/stemcells.2005-0227. [DOI] [PubMed] [Google Scholar]
- Cobo M, Isla D, Massuti B, Montes A, Sanchez JM, Provencio M, Vinolas N, Paz-Ares L, Lopez-Vivanco G, Munoz MA, Felip E, Alberola V, Camps C, Domine M, Sanchez JJ, Sanchez-Ronco M, Danenberg K, Taron M, Gandara D, Rosell R. Customizing cisplatin based on quantitative excision repair cross-complementing 1 mRNA expression: a phase III trial in non-small-cell lung cancer. J Clin Oncol. 2007;25 (19:2747–2754. doi: 10.1200/JCO.2006.09.7915. [DOI] [PubMed] [Google Scholar]
- Friboulet L, Olaussen KA, Pignon JP, Shepherd FA, Tsao MS, Graziano S, Kratzke R, Douillard JY, Seymour L, Pirker R, Filipits M, Andre F, Solary E, Ponsonnailles F, Robin A, Stoclin A, Dorvault N, Commo F, Adam J, Vanhecke E, Saulnier P, Thomale J, Le Chevalier T, Dunant A, Rousseau V, Le Teuff G, Brambilla E, Soria JC. ERCC1 isoform expression and DNA repair in non-small-cell lung cancer. N Engl J Med. 2013;368 (12:1101–1110. doi: 10.1056/NEJMoa1214271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson PT, Li T, He M, Zhang H, Malfatti M, Gandara D, Grimminger PP, Danenberg KD, Beckett L, De Vere White RW, Turteltaub KW, Pan CX. A microdosing approach for characterizing formation and repair of carboplatin-DNA monoadducts and chemoresistance. Int J Cancer. 2011;129 (6:1425–1434. doi: 10.1002/ijc.25814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann AC, Danenberg KD, Taubert H, Danenberg PV, Wuerl P. A three-gene signature for outcome in soft tissue sarcoma. Clin Cancer Res. 2009;15 (16:5191–5198. doi: 10.1158/1078-0432.CCR-08-2534. [DOI] [PubMed] [Google Scholar]
- Hoffmann AC, Mori R, Vallbohmer D, Brabender J, Klein E, Drebber U, Baldus SE, Cooc J, Azuma M, Metzger R, Hoelscher AH, Danenberg KD, Prenzel KL, Danenberg PV. High expression of HIF1a is a predictor of clinical outcome in patients with pancreatic ductal adenocarcinomas and correlated to PDGFA, VEGF, and bFGF. Neoplasia. 2008;10 (7:674–679. doi: 10.1593/neo.08292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann AC, Wild P, Leicht C, Bertz S, Danenberg KD, Danenberg PV, Stohr R, Stockle M, Lehmann J, Schuler M, Hartmann A. MDR1 and ERCC1 expression predict outcome of patients with locally advanced bladder cancer receiving adjuvant chemotherapy. Neoplasia. 2010;12 (8:628–636. doi: 10.1593/neo.10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner RA, Riley RD, Billingham LJ, Popat S. Excision repair cross-complementation group 1 (ERCC1) status and lung cancer outcomes: a meta-analysis of published studies and recommendations. PLoS One. 2011;6 (10:e25164. doi: 10.1371/journal.pone.0025164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly E, Hudrisier D. What is trogocytosis and what is its purpose. Nat Immunol. 2003;4 (9:815–816. doi: 10.1038/ni0903-815. [DOI] [PubMed] [Google Scholar]
- Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7 (8:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- Kim ES, Lee JJ, He G, Chow CW, Fujimoto J, Kalhor N, Swisher SG, Wistuba II, Stewart DJ, Siddik ZH. Tissue platinum concentration and tumor response in non-small-cell lung cancer. J Clin Oncol. 2012;30 (27:3345–3352. doi: 10.1200/JCO.2011.40.8120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedert B, Pluim D, Schellens J, Thomale J. Adduct-specific monoclonal antibodies for the measurement of cisplatin-induced DNA lesions in individual cell nuclei. Nucleic Acids Res. 2006;34 (6:e47. doi: 10.1093/nar/gkl051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nel I, Ertlel JM, Baba HA, Weber F, Sitek B, Stephan C, Meyer H, Schlaak JF, Hoffmann AC. A novel method for the detection of different subgroups of circulating tumor cells in patients with hepatocellular carcinoma. Hepatology. 2012;56:457A–457A. [Google Scholar]
- Olaussen KA, Adam J, Vanhecke E, Vielh P, Pirker R, Friboulet L, Popper H, Robin A, Commo F, Thomale J, Kayitalire L, Filipits M, Le Chevalier T, Andre F, Brambilla E, Soria JC. PARP1 impact on DNA repair of platinum adducts: preclinical and clinical read-outs. Lung Cancer. 2013;80 (2:216–222. doi: 10.1016/j.lungcan.2013.01.014. [DOI] [PubMed] [Google Scholar]
- Olaussen KA, Andre F, Soria JC. DNA repair capacity in circulating lymphocytes and influence on platinum effect in tumor cells. J Clin Oncol. 2012;30 (13:1567–1568,. author reply 1568-9. doi: 10.1200/JCO.2011.40.9888. [DOI] [PubMed] [Google Scholar]
- Oliver TG, Mercer KL, Sayles LC, Burke JR, Mendus D, Lovejoy KS, Cheng MH, Subramanian A, Mu D, Powers S, Crowley D, Bronson RT, Whittaker CA, Bhutkar A, Lippard SJ, Golub T, Thomale J, Jacks T, Sweet-Cordero EA. Chronic cisplatin treatment promotes enhanced damage repair and tumor progression in a mouse model of lung cancer. Genes Dev. 2010;24 (8:837–852. doi: 10.1101/gad.1897010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Printz C. BATTLE to personalize lung cancer treatment. Novel clinical trial design and tissue gathering procedures drive biomarker discovery. Cancer. 2010;116 (14:3307–3308. doi: 10.1002/cncr.25493. [DOI] [PubMed] [Google Scholar]
- Sobin LH. TNM: evolution and relation to other prognostic factors. Semin Surg Oncol. 2003;21 (1:3–7. doi: 10.1002/ssu.10014. [DOI] [PubMed] [Google Scholar]
- Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92 (3:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- Wang LE, Yin M, Dong Q, Stewart DJ, Merriman KW, Amos CI, Spitz MR, Wei Q. DNA repair capacity in peripheral lymphocytes predicts survival of patients with non-small-cell lung cancer treated with first-line platinum-based chemotherapy. J Clin Oncol. 2011a;29 (31:4121–4128. doi: 10.1200/JCO.2010.34.3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QE, Milum K, Han C, Huang YW, Wani G, Thomale J, Wani AA. Differential contributory roles of nucleotide excision and homologous recombination repair for enhancing cisplatin sensitivity in human ovarian cancer cells. Mol Cancer. 2011b;10:24–36. doi: 10.1186/1476-4598-10-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Stott S, Toner M, Maheswaran S, Haber DA. Circulating tumor cells: approaches to isolation and characterization. J Cell Biol. 2011;192 (3:373–382. doi: 10.1083/jcb.201010021. [DOI] [PMC free article] [PubMed] [Google Scholar]