

1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
Idiopathic dilated cardiomyopathy (IDC) is defined by the presence of left ventricular dilatation and systolic dysfunction in the absence of an underlying cause, such as hypertension, valve disease or coronary artery disease, sufficient to cause global systolic impairment.
Synonyms: dilated cardiomyopathy (DCM/CMD), dilated cardiomyopathy with conduction defect and/or arrhythmia, familial DCM/CMD, IDC, familial idiopathic cardiomyopathy and non-ischemic congestive heart failure.
1.2 OMIM# of the disease
115200 Cardiomyopathy, dilated, 1A; CMD1A.
600884 Cardiomyopathy, dilated, 1B; CMD1B.
601493 Cardiomyopathy, dilated, 1C; CMD1C.
601494 Cardiomyopathy, dilated, 1D; CMD1D.
601154 Cardiomyopathy, dilated, 1E; CMD1E.
604145 Cardiomyopathy, dilated, 1G; CMD1G.
604288 Cardiomyopathy, dilated, 1H; CMD1H.
604765 Cardiomyopathy, dilated, 1I; CMD1I.
605362 Cardiomyopathy, dilated, 1J; CMD1J.
605582 Cardiomyopathy, dilated, 1K; CMD1K.
606685 Cardiomyopathy, dilated, 1L; CMD1L.
607482 Cardiomyopathy, dilated, 1M; CMD1M.
607487 Cardiomyopathy, dilated, 1N; CMD1N.
608569 Cardiomyopathy, dilated, 1O; CMD1O.
609909 Cardiomyopathy, dilated, 1P; CMD1P.
609915 Cardiomyopathy, dilated, 1Q; CMD1Q.
613424 Cardiomyopathy, dilated, 1R; CMD1R.
613426 Cardiomyopathy, dilated, 1S; CMD1S.
613740 Cardiomyopathy, dilated, 1T; CMD1T.
613694 Cardiomyopathy, dilated, 1U; CMD1U.
613697 Cardiomyopathy, dilated, 1V; CMD1V.
611407 Cardiomyopathy, dilated, 1W; CMD1W.
611615 Cardiomyopathy, dilated, 1X; CMD1X.
611878 Cardiomyopathy, dilated, 1Y; CMD1Y.
611879 Cardiomyopathy, dilated, 1Z; CMD1Z.
612158 Cardiomyopathy, dilated, 1AA; CMD1AA.
612877 Cardiomyopathy, dilated, 1BB; CMD1BB.
613122 Cardiomyopathy, dilated, 1CC; CMD1CC.
613172 Cardiomyopathy, dilated, 1DD; CMD1DD.
613252 Cardiomyopathy, dilated, 1EE; CMD1EE.
613286 Cardiomyopathy, dilated, 1FF; CMD1FF.
613642 Cardiomyopathy, dilated, 1GG; CMD1GG.
613881 Cardiomyopathy, dilated, 1HH; CMD1HH.
611880 Cardiomyopathy, dilated, 2A; CMD2A.
614672 Cardiomyopathy, dilated, 2B; CMD2B.
302060 Cardiomyopathy, dilated, 3A; CMD3A.
302045 Cardiomyopathy, dilated, 3B; CMD3B.
The symbol CMD1F was formerly used for a disorder later found to be the same as desmin-related myopathy (601419 Myopathy, myofibrillar, 1; MFM1)
1.3 Name of the analysed genes or DNA/chromosome segments.
1. Sarcomeric protein genes:
a. Alpha tropomyosin 1 gene (TPM1) locus 15q22.1-q22.2.
b. Cardiac alpha-myosin heavy chain 6 gene (MYH6) locus 14q11.2-q12.
c. Cardiac beta-myosin heavy chain 7 gene (MYH7) locus 14q11.2-q12.
d. Cardiac myosin-binding protein-C gene (MYBPC3) locus 11p11.2.
e. Cardiac troponin I gene (TNNI3) locus 19q13.42.
f. Cardiac troponin T2 gene (TNNT2) locus 1q32.1.
g. Homolog of rat nexilin gene (NEXN) locus 1p31.1.
h. Slow troponin C gene (TNNC1) locus 3p21.1.
2. Cytoskeletal protein genes:
a. Alpha-actinin 2 gene (ACTN2) locus 1q43.
b. Alpha-cardiac actin 1 gene (ACTC1) locus 15q14.
c. Alpha-crystallin B gene (CRYAB) locus 11q23.1.#
d. Alpha-laminin 4 gene (LAMA4) locus 6q21.#
e. Cysteine- and glycine-rich protein 3 gene (CSRP3) locus 11p15.1.
f. Delta-sarcoglycan gene (SGCD) locus 5q33.3.
g. Desmin gene (DES) locus 2q35.
h. Dystrophin gene (DMD) locus Xp21.2-p21.1.
i. Four-and-a-half LIM domains 2 gene (FHL2) locus 2q12.1-q12.2.#
j. Fukutin gene (FKTN) locus 9q31.2.
k. Fukutin-related protein gene (FKRP) locus 19q13.32.#
l. Integrin-linked kinase gene (ILK) locus 11p15.4.#
m. LIM domain-binding 3 gene (LDB3) locus 10q23.2.
n. Myopalladin gene (MYPN) locus 10q21.1.#
o. PDZ and LIM domain protein 3 gene (PDLIM3) locus 4q35.1.#
p. Titin-cap gene (TCAP) locus 17q12.
q. Titin gene (TTN) locus 2q31.2.
r. Vinculin gene (VCL) locus 10q22.2.
3. Nuclear envelope protein genes:
a. Emerin gene (EMD) locus Xq28.#
b. Lamin A/C gene (LMNA) locus 1q22.
c. Thymopoietin gene (TMPO) locus 12q23.1.
4. Desmosomal protein genes:
a. Desmocollin 2 gene (DSC2) locus 18q12.1.#
b. Desmoglein 2 gene (DSG2) locus 18q12.1.
c. Desmoplakin gene (DSP) locus 6p24.3.#
d. Junction plakoglobin gene (JUP) locus 17q21.2.
e. Plakophilin 2 gene (PKP2) locus 12p11.21.#
5. Calcium/sodium-handling genes:
a. ATP-binding cassette, subfamily C, member 9 gene (ABCC9) locus 12p12.1.
b. Phospholamban gene (PLN) locus 6q22.31.
c. Ryanodine receptor 2 gene (RYR2) locus 1q43.#
d. Sodium channel, voltage-gated, type V, alpha subunit gene (SCN5A) locus 3p22.2.
6. Transcription factor genes:
a. Ankyrin repeat domain-containing protein 1 gene (ANKRD1) locus 10q23.3.#
b. T-box 20 gene (TBX20) locus 7p14.2.#
7. Other genes:
a. BCL2-associated athanogene 3 gene (BAG3) locus 10q26.11.
b. Caveolin 3 gene (CAV3) locus 3p25.3.#
c. Eyes absent 4 gene (EYA4) locus 6q23.2.
d. GATA zinc-finger domain-containing protein 1 gene (GATAD1) locus 7q21.2.
e. Lysosome-associated membrane protein 2 gene (LAMP2) locus Xq24.#
f. Presenilin 1 gene (PSEN1) locus 14q24.2.
g. Presenilin 2 gene (PSEN2) locus 1q42.13.
h. RNA-binding motif protein 20 gene (RBM20) locus 10q25.2.
i. Succinate dehydrogenase complex, subunit A, flavoprotein gene (SDHA) locus 5p15.33.
j. Tafazzin gene (TAZ) locus Xq28.
k. Thioredoxin reductase 2 gene (TXNRD2) locus 22q11.21.#
#=gene not yet annotated as DCM-related in the OMIM database.
Several additional loci for familial dilated cardiomyopathy have been mapped:
CMD1B on 9q13.
CMD1H on 2q14-q22.
CMD1K on 6q12-q16.
CMD1Q on 7q22.3-q31.1.
1.4. OMIM# of the gene(s)
1. Sarcomeric protein genes:
a. *191010 Alpha tropomyosin 1 gene (TPM1) locus 15q22.1-q22.2.
b. *160710 Cardiac alpha-myosin heavy chain 6 gene (MYH6) locus 14q11.2-q12.
c. *160760 Cardiac beta-myosin heavy chain 7 gene (MYH7) locus 14q11.2-q12.
d. *600958 Cardiac myosin-binding protein-C gene, (MYBPC3) locus 11p11.2.
e. *191044 Cardiac troponin I gene (TNNI3) locus 19q13.42.
f. *191045 Cardiac troponin T2 gene (TNNT2) locus 1q32.1.
g. *613121 Homolog of rat nexilin gene (NEXN) locus 1p31.1.
h. *191040 Slow troponin C gene (TNNC1) locus 3p21.1.
2. Cytoskeletal protein genes:
a. *102573 Alpha-actinin 2 gene (ACTN2) locus 1q43.
b. *102540 Alpha-cardiac actin 1 gene (ACTC1) locus 15q14.
c. *123590 Alpha-crystallin B gene (CRYAB) locus 11q23.1.#
d. *600133 Alpha-laminin 4 gene (LAMA4) locus 6q21.#
e. *600824 Cysteine- and glycine-rich protein 3 gene (CSRP3) locus 11p15.1.
f. *601411 Delta-sarcoglycan gene (SGCD) locus 5q33.3.
g. *125660 Desmin gene (DES) locus 2q35.
h. *300377 Dystrophin gene (DMD) locus Xp21.2-p21.1.
i. *602633 Four-and-a-half LIM domains 2 gene (FHL2) locus 2q12.1–q12.2.#
j. *607440 Fukutin gene (FKTN) locus 9q31.2.
k. *606596 Fukutin-related protein gene (FKRP) locus 19q13.32.#
l. *602366 Integrin-linked kinase gene (ILK) locus 11p15.4.#
m. *605906 LIM domain-binding 3 gene (LDB3) locus 10q23.2.
n. *608517 Myopalladin gene (MYPN) locus 10q21.1.#
o. *605889 PDZ and LIM domain protein 3 gene (PDLIM3) locus 4q35.1.#
p. *604488 Titin-cap gene (TCAP) locus 17q12.
q. *188840 Titin gene (TTN) locus 2q31.2.
r. *193065 Vinculin gene (VCL) locus 10q22.2.
3. Nuclear envelope protein genes:
a. *300384 Emerin gene (EMD) locus Xq28.#
b. *150330 Lamin A/C gene (LMNA) locus 1q22.
c. *188380 Thymopoietin gene (TMPO) locus 12q23.1.
4. Desmosomal protein genes:
a. *125645 Desmocollin 2 (DSC2) locus 18q12.1.#
b.*125671 Desmoglein 2 gene (DSG2) locus 18q12.1.
c. *125647 Desmoplakin gene (DSP) locus 6p24.3.#
d. *173325 Junction plakoglobin gene (JUP) locus 17q21.2.
e. *602861 Plakophilin 2 gene (PKP2) locus 12p11.21.#
5. Calcium/sodium-handling genes:
a. *601439 ATP-binding cassette, subfamily C, member 9 gene (ABCC9) locus 12p12.1.
b. *172405 Phospholamban gene (PLN) locus 6q22.31.
c. *180902 Ryanodine receptor 2 gene (RYR2) locus 1q43.#
d. *600163 Sodium channel, voltage-gated type V, alpha subunit gene (SCN5A) locus 3p22.2.
6. Transcription factor genes:
a. *609599 Ankyrin repeat domain-containing protein 1 gene (ANKRD1) locus 10q23.3.#
b. *606061 T-box 20 gene (TBX20) locus 7p14.2.#
7. Other genes:
a. *603883 BCL2-associated athanogene 3 gene (BAG3) locus 10q26.11.
b. *601253 Caveolin 3 gene (CAV3) locus 3p25.3.#
c. *603550 Eyes absent 4 gene (EYA4) locus 6q23.2.
d. *614518 GATA zinc-finger domain-containing protein 1 gene (GATAD1) locus 7q21.2.
e. *309060 Lysosome-associated membrane protein 2 gene (LAMP2) locus Xq24.#
f. *104311 Presenilin 1 gene (PSEN1) locus 14q24.2.
g. *600759 Presenilin 2 gene (PSEN2) locus 1q42.13.
h. *613171 RNA-binding motif protein 20 gene (RBM20) locus 10q25.2.
i. *600857 Succinate dehydrogenase complex, subunit A, flavoprotein gene (SDHA) locus 5p15.33.
j. *300394 Tafazzin gene (TAZ) locus Xq28.
k. *606448 Thioredoxin reductase 2 gene (TXNRD2) locus 22q11.21.#
#=gene not yet annotated as DCM-related in the OMIM database.
1.5 Mutational spectrum
Until now, 51 DCM genes have been reported. The majority of the mutations in these genes are missense mutations. Less frequently, other types of mutations, such as splice site, frameshift (due to small insertions/deletions) and nonsense mutations, and large deletions or insertions are detected. For instance, in TTN, a large number of truncating (splice site, frameshift and nonsense) mutations have recently been reported in about 25% of familial DCM cases, whereas large deletions/insertions of one or more exons have been described in DMD, LMNA and BAG3.1, 2, 3, 4, 5
1.6 Analytical methods
Sanger-sequencing of coding regions and their flanking intronic sequences is used in combination with pre-screening methods, such as DGGE, DHPLC, CSCE and so on. For a subset of genes, the multiplex ligation-dependent probe amplification (MLPA) technique is used in order to identify deletions or duplications of whole exons.
1.7 Analytical validation
Sequencing of both strands. When a mutation is identified, it is recommended to validate this in an independent experiment by direct sequencing or MLPA (in case of a large deletion/duplication) using a freshly prepared DNA dilution.
1.8 Estimated frequency of the disease
Incidence at birth (‘birth prevalence') or population prevalence. If known to be variable between ethnic groups, please report:
An epidemiological study on idiopathic DCM from 1989 estimated the incidence of the disease at 6 per 100 000 per year and the prevalence at 36.5 per 100 000.6 However, a recent review suggests that these numbers are likely to underestimate the condition significantly.7 No additional, up-to-date epidemiological studies are available.
1.9 Diagnostic setting

Comment: The most frequently mutated DCM genes are TTN, LMNA, MYH7 and TNNT2. A recent study of a small cohort of idiopathic DCM patients reported that the frequency of TTN mutations was 25% in familial DCM and 18% in apparently sporadic cases.1 MYH7 and TNNT2 mutations are identified with a frequency of 4–5% and 3%, respectively.8 LMNA mutations are also frequently detected in up to 6% of cases and in particular in the subgroup of idiopathic DCM associated with conduction disease (30–33%).9, 10
In certain populations, specific mutations may be overrepresented due to a common founder mutation, for example, the LMNA p.Ser143Pro in 7% of the Finnish idiopathic DCM population or the PLN p.Arg14del in up to 15% of Dutch idiopathic DCM patients.11, 12
Several studies have reported that DCM patients carrying more than one disease-associated mutation have an early onset, severe disease expression and a bad prognosis, which is most likely due to a gene-dosage effect.13, 14, 15, 16 It is expected that next-generation sequencing techniques will identify an increasing number of patients with such complex genotypes.
2. Test characteristics
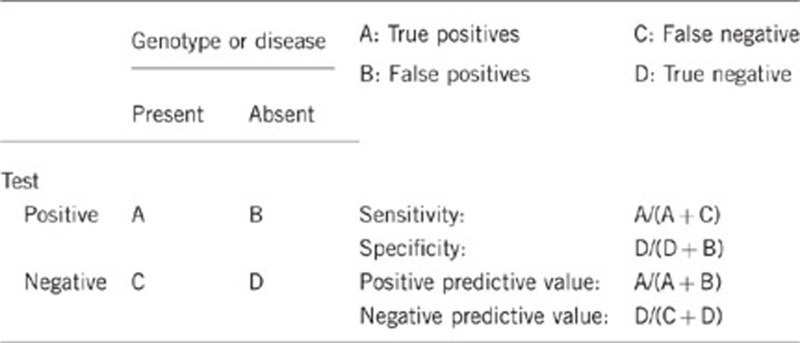
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)
Almost 100%, also depending on the analytical method used. Preferential amplification of one allele may occur when one of the primers is located on a SNP. To prevent this, primers have to be verified using the SNPCheck software (https://ngrl.manchester.ac.uk/SNPCheckV3/snpcheck.htm) yearly.
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)
Almost 100%, depending on the analytical method used.
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present)
The clinical sensitivity is dependent on variable factors such as age, disease manifestation and family history.
Mutational analysis of the 51 currently known DCM genes may explain the genetic background in up to 50% of familial DCM cases.1 The yield of genetic testing varies considerably. The mutation detection rates for the most prevalent DCM-related genes are: TTN: 18–25%, LMNA: 6%, MYH7: 4–5%, MYH6: 3–4%, MYBPC3: 2–4%, TNNT2: 3% and BAG3: 2–3%.5, 8 If cardiac conduction disease accompanies idiopathic DCM, an LMNA gene mutation can be identified in up to one-third of cases.9, 10
However, large differences in the yield of genetic testing have been observed, which are most likely explained by differences in sample size, variability in frequency of founder mutations and ethnicity.
2.4 Clinical specificity
(proportion of negative tests if the disease is not present)
The clinical specificity is dependent on variable factors such as age, disease manifestation and family history.
As the penetrance of inherited DCM is incomplete and age dependent, the clinical specificity is <100%.
2.5 Positive clinical predictive value
(life-time risk to develop the disease if the test is positive)
Probably reaches 100% at advanced age because the clinical penetrance is age dependent (<20 years: 10%, 20–30 years: 34% 30–40 years: 60% >40 years: 90%), with onset generally in the fourth decade of life.17
2.6 Negative clinical predictive value
(probability not to develop the disease if the test is negative)
Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
If a pathogenic mutation is found in the index patient, the risk of being a carrier for a first-degree relative is 50% (assuming autosomal-dominant inheritance of a familial mutation). If a pathogenic mutation is identified in the index patient, and this mutation is absent in another family member, this family member is generally believed not to be at increased risk for developing DCM. However, because carriership of multiple putative disease-causing mutations in more than one gene has been reported, it cannot be excluded that this mutation-negative family member may carry a still unidentified DCM-related mutation. The chance of carrying such an additional DCM-related mutation is believed to be <3%.7, 15, 16
Index case in that family had not been tested:
Idiopathic DCM is believed to be familial in about 30–50% of cases. Therefore, if an index patient has been clinically diagnosed with idiopathic DCM, and no genetic test has been performed, the chance for a first-degree relative to develop DCM may reach up to 15–25%.
3. Clinical utility
3.1 (Differential) diagnostics: the tested person is clinically affected
(To be answered if in 1.9 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?

Comment: The clinical diagnosis of familial DCM is generally made by echocardiography based upon the criteria proposed by Mestroni et al.18; that is, a reduced ejection fraction of the left ventricle (<0.45; >2 SD) and/or fractional shortening (<25% >2 SD); left ventricular end diastolic diameter >117% of the predicted value (corrected for age and body surface area). However, it is of great importance to exclude other disorders that may give rise to phenocopies. Therefore, additional investigations (imaging modalities and blood testing) have to be carried out to exclude, for example, ischemic heart disease, diabetes and thyroid disease.
3.1.2 Describe the burden of alternative diagnostic methods to the patient
Electrocardiography, echocardiography and magnetic resonance imaging are non-invasive imaging techniques with negligible risks and little inconvenience to the patient.
If DCM is diagnosed using these methods, additional techniques (CT-angiography and coronary angiography) have to be used to exclude other causes such as coronary artery disease.
3.1.3 How is the cost-effectiveness of alternative diagnostic methods to be judged?
When a pathogenic mutation is identified in an index patient, cascade genetic screening (=targeted genetic testing for the identified mutation in the family) can be offered to first degree, apparently healthy relatives, to establish whether they are mutation carriers or not.
Mutation-carrying relatives are advised to be screened regularly by a cardiologist to detect early cardiac disease in order to initiate timely treatment. In addition, specific advice regarding physical or occupational activities might be discussed, as well as general lifestyle advices.
Non-mutation carriers can be reassured and dismissed from regular clinical follow-up if the DCM phenotype in that family is consistent with carriership of a single mutation. This method has been shown to be cost-effective for HCM, but no formal cost-effectiveness studies in DCM are available yet.19
3.1.4 Will disease management be influenced by the result of a genetic test?

Possible exceptions are LMNA gene mutation carriers, who may benefit from early implantable cardioverter defibrillator (ICD) therapy if they develop conduction disease.20 Moreover, a recent study showed that male patients and patients with either non-sustained ventricular tachycardias or left ventricular ejection fraction of <45% carrying a non-missense mutation in LMNA have a higher risk for developing malignant ventricular arrhythmias.21
3.2 Predictive Setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.9 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive, regular cardiological evaluation is recommended to detect signs of DCM and initiate early medical treatment when indicated. The treatment might also include pacemaker and/or ICD implantation. Advice regarding lifestyle and physical activity is also provided. In addition, mutation carriers will receive genetic counselling on risks for their offspring and other close relatives. If the genetic test result is negative, family members without the mutation can be discharged from regular cardiological follow-up. However, because carriership of multiple putative disease-causing mutations in more than one gene have been reported, it cannot be excluded that this mutation-negative family member may carry a still unidentified DCM-related mutation (see also 2.6).
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
These are similar to those who have a positive genetic test.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.9 ‘C' was marked)
Predictive genetic testing of asymptomatic relatives is only offered when a pathogenic mutation has been identified in an affected individual. Because the predominant mode of inheritance in idiopathic DCM is autosomal-dominant, first-degree family members of an affected patient generally have a 50% risk of having inherited the same mutation.
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
If a pathogenic mutation is identified in the affected index patient, and the clinical phenotype is in line with what is known about that specific mutation or gene, the genetic situation is resolved.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Yes, a positive test in the index patient enables the possibility of predictive cascade genetic screening (=targeted testing for the identified mutation in the family). Non-carriers can be dismissed from regular cardiological evaluation and this results in saving the costs of cardiological investigations, unless carriership of multiple mutations can be expected (see also 2.6).
When the test in the index patient is negative, all relatives are advised to continue regular cardiological follow-up.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes, once a pathogenic mutation is identified, a predictive genetic test is possible in family members.
3.4 Prenatal diagnosis
(To be answered if in 1.9 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Prenatal genetic diagnosis in DCM is technically possible when a pathogenic mutation has been identified. However, requests for prenatal diagnosis in late-onset genetic disease are uncommon and only rarely performed. It is not actively offered in non-syndromic late-onset DCM. Recently, pre-implantation genetic diagnosis has been reported in a DCM family.22
4. If applicable, further consequences of testing
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe).
The major advantage is that close relatives who are under regular cardiological surveillance can be dismissed from regular follow-up once it has been proven that they do not carry the mutation present in the index patient.
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469) and the European Society of Human Genetics.
The authors declare no conflict of interest.
References
- Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntoni F, Cau M, Ganau A, et al. Brief report: deletion of the dystrophin muscle-promoter region addociated with X-linked dilated cardiomyopathy. N Engl J Med. 1993;329:921–925. doi: 10.1056/NEJM199309233291304. [DOI] [PubMed] [Google Scholar]
- Towbin JA, Hejtmancik JF, Brink P, et al. X-linked dilated cardiomyopathy Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation. 1993;87:1854–1865. doi: 10.1161/01.cir.87.6.1854. [DOI] [PubMed] [Google Scholar]
- van Tintelen JP, Tio RA, Kerstjens-Frederikse WS, et al. Severe myocardial fibrosis caused by a deletion of the 5' end of the lamin A/C gene. J Am Coll Cardiol. 2007;49:2430–2439. doi: 10.1016/j.jacc.2007.02.063. [DOI] [PubMed] [Google Scholar]
- Norton N, Li D, Rieder MJ, et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet. 2011;88:273–282. doi: 10.1016/j.ajhg.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codd MB, Sugrue DD, Gersh BJ, et al. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy: a population-based study in Olmsted, County, Minnesota, 1975-1984. Circulation. 1989;80:564–572. doi: 10.1161/01.cir.80.3.564. [DOI] [PubMed] [Google Scholar]
- Hershberger RE, Morales A, Siegfried JD. Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet Med. 2010;12:655–667. doi: 10.1097/GIM.0b013e3181f2481f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger RE, Siegfried JD. Update 2011: Clinical and genetic issues in familial cardiomyopathy. J Am Coll Cardiol. 2011;57:1641–1649. doi: 10.1016/j.jacc.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbustini E, Pilotto A, Repetto A, et al. Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect-related disease. J Am Coll Cardiol. 2002;39:981–990. doi: 10.1016/s0735-1097(02)01724-2. [DOI] [PubMed] [Google Scholar]
- van Tintelen JP, Hofstra RM, Katerberg H, et al. High yield of LMNA mutations in patients with dilated cardiomyopathy and/or conduction disease referred to cardiogenetics outpatient clinics. Am Heart J. 2007;154:1130–1139. doi: 10.1016/j.ahj.2007.07.038. [DOI] [PubMed] [Google Scholar]
- Kärkkäinen S, Heliö T, Miettinen R, et al. A novel mutation, Ser143Pro, in the lamin A/C gene is common in finnish patients with familial dilated cardiomyopathy. Eur Heart J. 2004;25:885–893. doi: 10.1016/j.ehj.2004.01.020. [DOI] [PubMed] [Google Scholar]
- van der Zwaag PA, van Rijsingen IA, Asimaki A, et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 2012;14:1199–1207. doi: 10.1093/eurjhf/hfs119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingles J, Doolan A, Chiu C, et al. Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J Med Genet. 2005;42:e59. doi: 10.1136/jmg.2005.033886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Yang Z, Vatta M, et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2010;55:587–597. doi: 10.1016/j.jacc.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampersaud E, Siegfried JD, Norton N, et al. Rare variant mutations identified in pediatric patients with dilated cardiomyopathy. Prog Pediatr Cardiol. 2011;31:39–47. doi: 10.1016/j.ppedcard.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller DV, Andersen PS, Hedley P, et al. The role of sarcomere gene mutations in patients with idiopathic dilated cardiomyopathy. Eur J Hum Genet. 2009;17:1241–1249. doi: 10.1038/ejhg.2009.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestroni L, Rocco C, Gregori D, et al. Familial dilated cardiomyopathy: evidence for genetic and phenotypic heterogeneity. J Am Coll Cardiol. 1999;34:181–190. doi: 10.1016/s0735-1097(99)00172-2. [DOI] [PubMed] [Google Scholar]
- Mestroni L, Maisch B, McKenna WJ, et al. Guidelines for the study of familial dilated cardiomyopathies. Collaborative research group of the European Human and Capital Mobility project on familial dilated cardiomyopathy. Eur Heart J. 1999;20:93–102. doi: 10.1053/euhj.1998.1145. [DOI] [PubMed] [Google Scholar]
- Wordsworth S, Leal J, Blair E, et al. DNA testing for hypertrophic cardiomyopathy: a cost-effectiveness model. Eur Heart J. 2010;31:926–935. doi: 10.1093/eurheartj/ehq067. [DOI] [PubMed] [Google Scholar]
- Meune C, Van Berlo JH, Anselme F, et al. Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med. 2006;354:209–210. doi: 10.1056/NEJMc052632. [DOI] [PubMed] [Google Scholar]
- van Rijsingen IA, Arbustini E, Elliott PM, et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J Am Coll Cardiol. 2012;59:493–500. doi: 10.1016/j.jacc.2011.08.078. [DOI] [PubMed] [Google Scholar]
- Kuliev A, Pomerantseva E, Polling D, et al. PGD for inherited cardiac diseases. Reprod Biomed Online. 2012;24:443–453. doi: 10.1016/j.rbmo.2011.12.009. [DOI] [PubMed] [Google Scholar]