

1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
Poikiloderma with Neutropenia (PN, Clericuzio-type Poikiloderma with Neutropenia).
1.2 OMIM# of the disease
#604173.
1.3 Name of the analysed genes or DNA/chromosome segments
C16orf57 (Chromosome 16 open reading frame 57).
1.4 OMIM# of the gene(s)
*613276.
1.5 Mutational spectrum
Poikiloderma with Neutropenia (PN), a very rare autosomal recessive inherited genodermatosis first described in the Navajo tribe of Native Americans,1 has recently been associated with biallelic mutations in the C16orf57 gene.2
So far, 19 different C16orf57 mutations have been detected in 37 PN patients subjected to molecular-genetic testing. Of these 37 patients, 31 (84%) carry homozygous mutations, whereas the other six are compound heterozygous.3 All identified mutations lead to the generation of truncated and most likely non-functional C16orf57 protein. C16orf57 is a 3′–5′ exonuclease essential for the biogenesis of the splicing apparatus.4, 5 Several classes of mutations have been identified, listed here in order of decreasing prevalence: nonsense mutations (c.232C>T, c.243G>A, c.258T>A, c.267T>A, c.415C>T, c.541C>T, c.673C>T); small out-of-frame deletions (c.176_177delG, c.179delC, c.489_492del4, c.496delA, c.531delA, c.683_893+1del12); and splicing alterations, including substitutions at canonical splice junctions or at splice-site consensus sequences (c.265+2T>G, c.266−1G>A, c.450−2A>G, c.502A>G, c.504−2A>C, c.693+1G>T).2, 3, 6, 7, 8, 9, 10 No missense mutations have yet been found; c.502A>G can be categorised as a splicing alteration because it leads to the excision of the fourth exon from the mature C16orf57-001 transcript.2
The most frequent recurrent mutations, c.531delA, c.496delA and c.179delC, reflect three geographical clusters. c.531delA has been recorded in seven patients from the Caucasus region, two of whom are members of unrelated Turkish families; the second most frequent mutation, c.496delA, has been detected in five patients from the Athabaskan ethnic group; and the third most frequent, c.179delC, was identified in four patients of North African origin. Considering the very low frequency of PN syndrome and the prevalence of patients with homozygous mutations, common ancestry is the hypothesis most likely to explain the recurrence of these mutations in specific ethnic groups.3
1.6 Analytical methods
To detect point mutations, PCR amplification is applied to genomic DNA fragments corresponding to all seven exons of the C16orf57-001 isoform and to the alternative exon 4 of the C16orf57-004 isoform, followed by bidirectional Sanger sequencing of amplicons including exon–intron boundaries.
To confirm the effects of splicing mutations, or to clarify the role of apparent missense mutations, RT-PCR is performed on RNA extracted from patients' blood, followed by specific PCR on cDNA. In silico prediction methods can also be applied.
1.7 Analytical validation
Mutations are confirmed by testing an independent biological sample from the proband and his/her unaffected relatives who are potential carriers, especially parents and siblings, using bidirectional sequencing with different sets of primers. If applicable, RFLP (restriction-fragment length polymorphism) can also be applied.
1.8 Estimated frequency of the disease (incidence at birth ("birth prevalence") or population prevalence. If known to be variable between ethnic groups, please report):
The prevalence is unknown. PN is a very rare disorder, and only 37 patients subjected to molecular-genetic testing have been reported in the literature; however, PN is likely to have been under-diagnosed due to the lack of awareness of the disorder and its clinical overlap with better-known diseases such as Rothmund–Thomson syndrome (RTS; #268400) and autosomal recessive Dyskeratosis Congenita (DC; #224230).
1.9 Diagnostic setting

Comment: The main diagnostic signs of PN are poikiloderma and non-cycling neutropenia, which develop during the first year of life.
Skin alterations initially involve the extremities (acral presentation), and then extend to the trunk and face, appearing as acute erythematous rash. These alterations subsequently evolve into poikiloderma, a chronic lesion characterised by reticulated areas of depigmentation and hyperpigmentation associated with telangiectasic lesions and cutaneous atrophy.
Given that many genodermatoses present with poikiloderma,11 the pattern of presentation should be carefully considered, as should the concomitance of other common PN typical signs such as early-onset recurrent infections, driven by non-cyclic neutropenia, pachyonychia, palmo-plantar hyperkeratosis and elevated lactate dehydrogenase (LDH) serum levels. Less common features not displayed by all patients include craniofacial dysmorphisms such as saddle nose, midfacial hypoplasia and retrognatia, defects in teeth, short stature, and bone alterations such as osteoporosis/osteopetrosis and bone fragility. Since the second decade of life, evolution of the disease may be marked by bone marrow failure, myelodysplastic syndrome (MDS) and acute myeloid leukaemia (AML), signs often shared by the related cancer predisposing syndrome DC.
Ambiguity related to the partial clinical overlap with RTS and DC can be resolved by C16orf57 molecular testing. PN does not seem to be a heterogeneous genetic disease. C16orf57 molecular testing can also aid in re-classification of misdiagnosed RTS or DC patients.
2. Test characteristics
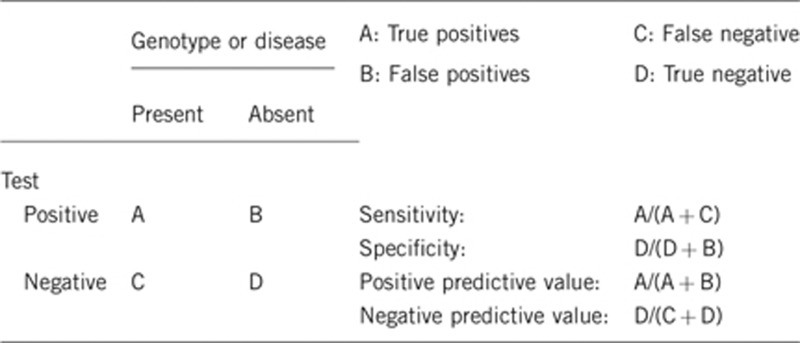
2.1 Analytical sensitivity (proportion of positive tests if the genotype is present)
Depends on the method(s) used. The analytical sensitivity could be close to 100%, provided that DNA testing is targeted on all C16orf57 coding exons and exon–intron boundaries.
2.2 Analytical specificity (proportion of negative tests if the genotype is not present)
Close to 100%.
2.3 Clinical sensitivity (proportion of positive tests if the disease is present)
The clinical sensitivity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
The age of onset and range of clinical features may be variable. Nevertheless, the first presentation of skin involvement is recorded during the first months of life, typically with acral occurrence and progression of lesions to the trunk and face. Based on cutaneous poikiloderma, screening for non-cyclic neutropenia leads to a diagnostic work-up. During childhood, the main clinical signs should be present.
Genome sequencing of all C16orf57 coding exons and exon–intron boundaries enables identification of biallelic disease-causing mutations in nearly 100% of individuals diagnosed with PN. Incomplete clinical sensitivity could be explained by mutations located in C16orf57 regulatory regions that have not yet been reported.
2.4 Clinical specificity (proportion of negative tests if the disease is not present)
100%.
2.5 Positive clinical predictive value (lifetime risk to develop the disease if the test is positive)
100% for pathogenic mutations. Based on the literature, all patients reported so far develop the hallmark features of PN during the first year of life. In particular, the skin features may be apparent from the first months of life,3, 8 whereas other signs, such as pachyonychia, may develop later. Non-cyclic neutropenia, the hallmark haematologic sign, may be suspected due to susceptibility to recurrent infections (mainly pulmonary) during infancy, and should be confirmed by neutrophil count. Penetrance is complete at early childhood, with variable expressivity. For patients positive for C16orf57 mutations, genetic counselling and surveillance should be provided due to the increased risk of developing MDS/AML before the second decade of life (12 cases); PN is therefore classified as a bone marrow failure syndrome.2, 3, 7, 10, 12, 13, 14 To date, solid cancers have only been reported in two PN patients, both of whom developed squamous cell carcinoma of the skin.10, 15 Prevalences of bone marrow failure (including pre-dysplastic anomalies evolving in MDS/AML) and skin cancer in PN are 48% and 5%, respectively.2, 3, 7, 10, 12, 13, 14, 15, 16
2.6 Negative clinical predictive value (probability of not developing the disease if the test is negative)
Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
If the index case in that family has been tested and found positive, a negative test in a non-affected family member would exclude the increased risk of disease (negative clinical predictive value close to 100%).
Index case in that family had not been tested:
Under this condition, it would be inappropriate to test potentially at-risk family members at early infancy, when the full spectrum of features may not be manifest. A preliminary step would be to test both parents of the index patient to assess their carrier status for mutations in C16orf57, the only gene associated with PN to date.
3. Clinical utility
3.1 (Differential) diagnostics: The tested person is clinically affected (To be answered if in 1.9 ‘A' was marked)
In classic PN patients, the correct diagnosis is based on clinical presentation with early-onset poikiloderma and non-cyclic neutropenia unveiled by recurrent infections. Rarely, the neutrophil defect is absent. Haematologic screening must be performed, specifically in patients with additional dermatologic and bone signs typical of PN. In borderline patients (poikiloderma syndromes), differential diagnosis with RTS and DC should be considered in order to orient genetic testing. Absence of severe short stature and cataracts, together with the specific presence of neutropenia, should help to orient the molecular strategy to PN and to C16orf57 gene testing.
3.1.1 Can a diagnosis be made other than through a genetic test?

3.1.2 Describe the burden of alternative diagnostic methods to the patient
No single alternative diagnostic method can be envisaged. A potential panel of clinical-instrumental exams to formulate a correct diagnosis would include: (1) skin inspection by a dermatologist experienced in genodermatoses, to detect the onset and the pattern of poikiloderma during the first year of life (progressive truncal and facial presentation over time), palmo-plantar keratoderma and nail dystrophy/pachyonychia; (2) laboratory investigations: neutrophil count to detect moderate (500–1000 cells/μl) to severe (200–500 cells/μl) non-cyclic neutropenia, and LDH to detect increasing serum levels (>500 U/l); (3) detailed clinical evaluation by an expert dysmorphologist; and (4) skeletal radiographs to detect bone alterations and fragility.
A list of major and minor criteria has been proposed for PN patients,6 but their specificity, sensitivity, and positive and negative predictive values have not been determined and validated.
Pathological analysis of bone marrow smears is mandatory in order to monitor accumulation of dysplastic features: abnormal maturation of the neutrophil lineage, increased numbers of immature cells or increased numbers of myeloid precursors.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
A genetic diagnosis is a more satisfactory tool for the patient and his/her family, and more cost-effective than all aforementioned alternative diagnostic methods. Such a diagnosis is conclusive and allows the patient to be referred to those specialists best suited to manage his/her care and guarantee appropriate follow-up.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive setting: The tested person is clinically unaffected but carries an increased risk based on family history (To be answered if in 1.9 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe)
If the test result is negative (please describe)
3.2.2 Which options in view of lifestyle and prevention does a person at risk have if no genetic test has been done (please describe)?
3.3 Genetic risk assessment in family members of a diseased person (To be answered if in 1.9 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes, if biallelic loss-of-function causative mutations have been identified in the C16orf57 gene. It is possible to assess the carrier status of all unaffected family members and to offer genetic counselling to the family.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Yes. However, if the result is negative, testing of family members is not recommended.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Infrequently, given that the disease has an early onset, ie, during infancy or even soon after birth.
3.4 Prenatal diagnosis (To be answered if in 1.9 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes, provided that both disease-causing alleles have been identified in an affected family member and their segregation from obligate carrier parents has been traced.
4. IF APPLICABLE, FURTHER CONSEQUENCES OF TESTING
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe)
For clinically affected individuals, identification of both mutations (positive genetic test) allows confirmation of the diagnosis and significantly increases the likelihood of long term follow-up, which in turn helps to prevent medical complications; this is especially true in the context of oncological surveillance. Genetic testing has no immediate medical consequences for healthy carriers; however, carriers' awareness of their genetic status is important for family planning.
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469) and the European Society of Human Genetics.
The authors declare no conflict of interest.
References
- Clericuzio C, Hoyme HE, Asse JM. Immune deficient poikiloderma: a new genodermatosis. Am J Hum Genet. 1991;49 ((Suppl:A661. [Google Scholar]
- Volpi L, Roversi G, Colombo EA, et al. Targeted next-generation sequencing appoints C16orf57 as Clericuzio-type poikiloderma with neutropenia gene. Am J Hum Genet. 2010;86:72–76. doi: 10.1016/j.ajhg.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo EA, Bazan JF, Negri G, et al. Novel C16orf57 mutations in patients with poikiloderma with neutropenia: bioinformatic analysis of the protein and predicted effects of all reported mutations. Orphanet J Rare Dis. 2012;23:7–7. doi: 10.1186/1750-1172-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mroczek S, Krwawicz J, Kutner J, et al. C16orf57, a gene mutated in poikiloderma with neutropenia, encodes a putative phosphodiesterase responsible for the U6 snRNA 3′ end modification. Genes Dev. 2012;26:1911–1925. doi: 10.1101/gad.193169.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shchepachev V, Wischnewski H, Missaglia E, et al. Mpn1, Mutated in Poikiloderma with Neutropenia, is a conserved 3′-5′ RNA exonuclease processing U6 snRNA. Cell Rep. 2012;2:855–865. doi: 10.1016/j.celrep.2012.08.031. [DOI] [PubMed] [Google Scholar]
- Arnold AW, Itin PH, Pigors M, et al. Poikiloderma with neutropenia: a novel C16orf57 mutation and clinical diagnostic criteria. Br J Dermatol. 2010;163:866–869. doi: 10.1111/j.1365-2133.2010.09929.x. [DOI] [PubMed] [Google Scholar]
- Clericuzio C, Harutyunyan K, Jin W, et al. Identification of a novel C16orf57 mutation in Athabaskan patients with poikiloderma with neutropenia. Am J Med Genet. 2011;155A:337–342. doi: 10.1002/ajmg.a.33807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piard J, Holder-Espinasse M, Aral B, et al. Systematic search for neutropenia should be part of the first screening in patients with poikiloderma. Eur J Med Genet. 2012;55:8–11. doi: 10.1016/j.ejmg.2011.07.004. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Morice-Picard F, Lacombe D, et al. Identification of a homozygous deletion mutation in C16orf57 in a family with Clericuzio-type poikiloderma with neutropenia. Am J Med Genet. 2010;152A:1347–1348. doi: 10.1002/ajmg.a.33455. [DOI] [PubMed] [Google Scholar]
- Walne AJ, Vulliamy T, Beswick R, et al. Mutations in C16orf57 and normal-length telomeres unify a subset of patients with dyskeratosis congenita, poikiloderma with neutropenia and Rothmund-Thomson syndrome. Hum Mol Genet. 2010;19:4453–4461. doi: 10.1093/hmg/ddq371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet J Rare Dis. 2010;5:2. doi: 10.1186/1750-1172-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concolino D, Roversi G, Muzzi GL, et al. Clericuzio type poikiloderma with neutropenia syndrome in three sibs with mutations in the C16orf57 gene: delineation of the phenotype. Am J Med Genet. 2010;152A:2588–2594. doi: 10.1002/ajmg.a.33600. [DOI] [PubMed] [Google Scholar]
- Pianigiani E, De Aloe G, Andreassi A, et al. Rothmund-Thomson syndrome (Thomson-type) and myelodysplasia. Pediatr Dermatol. 2001;18:422–425. doi: 10.1046/j.1525-1470.2001.01971.x. [DOI] [PubMed] [Google Scholar]
- Porter WM, Hardman CM, Abdalla SH, et al. Haematological disease in siblings with Rothmund-Thomson syndrome. Clin Exp Dermatol. 1999;24:452–454. doi: 10.1046/j.1365-2230.1999.00530.x. [DOI] [PubMed] [Google Scholar]
- Rodgers W, Ancliff P, Ponting CP, et al. Squamous cell carcinoma in a child with Clericuzio-type poikiloderma with neutropenia. Br J Dermatol. 2012;75:543–545. doi: 10.1111/bjd.12016. [DOI] [PubMed] [Google Scholar]
- Mostefai R, Morice-Picard F, Boralevi F, et al. Poikiloderma with neutropenia, Clericuzio type, in a family from Morocco. Am J Med Genet. 2008;146A:2762–2769. doi: 10.1002/ajmg.a.32524. [DOI] [PubMed] [Google Scholar]
- Altunay I, Fisek N, Gokdemir G, et al. Therapy-resistant leg ulcer in a patient with Rothmund-Thomson syndrome. Int Wound J. 2010;7:531–535. doi: 10.1111/j.1742-481X.2010.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LL, Gannavarapu A, Clericuzio CL, et al. Absence of RECQL4 mutations in poikiloderma with neutropenia in Navajo and Non-Navajo patients. Am J Med Genet. 2003;118A:299–301. doi: 10.1002/ajmg.a.10057. [DOI] [PubMed] [Google Scholar]