

Abstract
Deletions involving 17q21–q24 have been identified previously to result in two clinically recognizable contiguous gene deletion syndromes: 17q21.31 and 17q23.1–q23.2 microdeletion syndromes. Although deletions involving 17q22 have been reported in the literature, only four of the eight patients reported were identified by array-comparative genomic hybridization (array-CGH) or flourescent in situ hybridization. Here, we describe five new patients with 1.8–2.5-Mb microdeletions involving 17q22 identified by array-CGH. We also present one patient with a large karyotypically visible deletion involving 17q22, fine-mapped to ∼8.2 Mb using array-CGH. We show that the commonly deleted region in our patients spans 0.24 Mb and two genes; NOG and C17ORF67. The function of C17ORF67 is not known, whereas Noggin, the product of NOG, is essential for correct joint development. In common with the 17q22 patients reported previously, the disease phenotype of our patients includes intellectual disability, attention deficit hyperactivity disorder, conductive hearing loss, visual impairment, low set ears, facial dysmorphology and limb anomalies. All patients displayed NOG-related bone and joint features, including symphalangism and facial dysmorphology. We conclude that these common clinical features indicate a novel clinically recognizable, 17q22 contiguous microdeletion syndrome.
Keywords: 17q22 microdeletion, array-CGH, NOG
Introduction
Deletions involving chromosome 17q21–q24 have been reported to result in two clinically recognizable contiguous gene deletion syndromes: 17q21.31 (MIM#610443) and 17q23.1–q23.2 (MIM#613355) microdeletion syndromes. A third region of interest to us and the subject of this study is 17q22. Eight patients with deletions encompassing 17q22 have been reported previously.1, 2, 3, 4, 5, 6, 7, 8 Four of these were identified using array comparative genomic hybridization (array-CGH) or flourescent in situ hybridization (FISH),4, 6, 7, 8 and of these, two involved the NOG gene.4, 8 There have been also three reported patients with NOG-like features, where NOG was considered not to be involved; however, these were assessed using standard karyotyping with poor resolution of breakpoints.9, 10, 11 To date, all reported patients with deletions of NOG have been associated with symphalangism, proximally placed thumbs and upslanting palpebral fissures. In four patients hypertelorism and posteriorly rotated low set ears were noted.1, 3, 4, 8
The NOG gene consists of a single exon12 and the protein product, Noggin, is important for normal development of bone and joints,13, 14 inactivating bone morphogenetic proteins.15 This is illustrated by the excess bone morphogenetic protein activity and cartilage formation in NOG null mutant mice.15 To date, there are five described syndromes associated with heterozygous NOG mutations: symphalangism in proximal interphalangeal joints (MIM#185800), multiple synostoses syndrome (MIM#186500), stapes ankylosis with broad thumb and toes (MIM#184460), brachydactyly type B2 (MIM#611377) and tarsal–carpal coalition syndrome (MIM#186570). A locus-specific database for NOG variants can be found at https://grenada.lumc.nl/LOVD2/mendelian_genes/home.php. Thus far, a total of 36 human variations in NOG have been reported, leading to the collective term ‘NOG-related symphalangism spectrum disorder' being put forward for the five described syndromes.16, 17
In the current study, we set out to refine genotype–phenotype correlations for patients with 17q22 deletions, investigate evidence for a clinically recognizable 17q22 microdeletion syndrome and to identify contributing genes.
Materials and methods
Patients
Clinical data were collected from five patients for whom a 17q22 microdeletion was identified in DNA samples. They were of Swedish (patient 1), British (patient 2), US (patient 4 and 5) and Brazilian (patient 6) origin. The patients were unrelated except the mother and daughter from the United States. A further British patient (patient 3) with a karyotypically visible deletion of 17q22 was also included in the study. Facial and hand photographs of patients 1–5 are shown in Figures 1 and 2. The study was approved by the ethical committee at Karolinska University Hospital (ethical approval number: 2010/1930–32).
Figure 1.

Facial characteristics of the 17q22 microdeletion patients.
Figure 2.

Symphalangism in the 17q22 microdeletion patients showing as: (a) ankylotic proximal interphalangeal joint in the fifth digit in patient 4 (white arrow). Note also the fusion of the capitate and hamate bones (black arrow). (b) and (c) Stiff proximal interphalangeal joints with reduced dorsal creasing (white arrows) in patient 3 (b) and patient 2(c). (d) Reduced joint space in the fifth digit proximal interphalangeal joint in patient 1 (white arrow). (e) Stiff fifth finger proximal interphalangeal joint with absence of volar crease (white arrow) compared with normal crease in the second digit proximal interphalangeal joint (black arrow).
Cytogenetic studies
For the G-banding analyses, metaphase chromosome spreads were prepared from peripheral blood according to standard procedures. The karyotypes were described according to the International System for Human Cytogenetic Nomenclature ISCN (1999).
Array-CGH and FISH
For all six patients array-CGH was carried out using DNA isolated from peripheral blood. Patient 1 was analyzed on a 180K array from Oxford Gene Technology (Oxford, UK). Patients 2 and 3 were analyzed on 185K arrays from Agilent Technologies (Santa Clara, CA, USA). Patients 4 and 5 were analyzed on 105K arrays from the SignatureChip Oligo Solution, made for Signature Genomics Laboratories (Seattle, WA, USA) by Agilent Technologies as previously described.18 Patient 6 was analyzed on a 4 × 180K array from Agilent Technologies.19
The effective mean resolution of the arrays was ∼16–54 kb based on three to four consecutive oligonucleotides showing a change. Experiments were performed according to the manufacturer's recommendations. Slide scanning was performed using an Agilent Microarray scanner. Data were extracted using Agilent Feature Extraction software (Agilent, Santa Clara, CA, USA). Data from patient 1 were analyzed using the OGT cytosure interpret software (z-score algorithm, OGT, Oxford, UK), whereas data from patient 2, 3 and 6 were analyzed using the Agilent CGH Analytics software, Agilent (z-score setting for patients 2 and 3 and ADM-2 algorithm for patient 6). For patients 4 and 5, metaphase FISH analyses with regional BAC probes were used to confirm deletions detected by array-CGH and test for additional chromosome rearrangements as previously described.20
Results
Cytogenetics
Karyotyping at ∼550-band resolution was performed using peripheral blood lymphocytes, and all patients revealed normal karyotypes except for an apparently balanced pericentric inversion of chromosome 2 (46, XX, inv(2)(p11.2q13)) in the familial patients 4 and 5 and the large deletion encompassing 17q22 (46, XY, del(17q21q23.1)) noted in patient 3. Metaphase comparative genomic hybridization analyses in patients 4 and 5 failed to detect any deletions or duplications at the inversion breakpoint regions, findings consistent with these inversions being balanced.
Array-CGH and FISH
In all patients, array-CGH showed microdeletions involving the NOG gene on 17q22 (Table 1 and Figure 3). In patients 1, 2, 3 and 6 they were de novo. FISH confirmed the 1.9-Mb microdeletion in patients 4 and 5. For patient 6 array-CGH also revealed a de novo duplication of 122 933 bp on chromosome 1, involving ST7L and WNT2B. Apart from the latter, patients 1, 2, 3 and 6 did not show any gene dose imbalances except copy number variants noted already in the Database of Genomic Variants (http://projects.tcag.ca/variation/) with no apparent clinical significance. The deletions involved an 8 184 525-bp region (chr17:48389130-56925730, hg19), with a maximal deletion size of 8 184 525 bp and a minimal size of 1 860 732 bp. The commonly deleted region of 237 304 bp (chr17:54875445-54638141, hg19) encompassed the NOG gene and an open reading frame gene called C17ORF67. Neither deletions nor duplications of this region have been reported in the region in control individuals. The position, size and genes involved in each deletion are shown in Table 1 and Figure 3. Array-CGH and FISH data are shown in Supplementary Figures 1a–f and 2.
Table 1. Position (hg19), size and involved genes and transcripts of the 17q22 microdeletions.
| Puusepp et al8 | Khattab et al6 | Nimma-kayalu et al7 | Shimizu et al4 | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Position of the 17q22 deletion (hg19) | chr17:50845001/50945001–chr17:56845001/56945218 | chr17:55717537–59257880 | chr17:56429075–60181763 | Bac-clones 367C1, 9G4, 649B10, 826B22 | chr17:54329389–56314137 | chr17:53609015–56109938 | chr17:48389130–56573655 | chr17:53014713–54875445 | chr17:53014713–54875445 | chr17:54638141–56925730+ dupchr1:113036203–113159136 |
| Size of the 17q22 deletion | 5.9 Mb | 3.54 Mb | 3.75 Mb | 2.8–3.7 Mb | 1.98 Mb | 2.50 Mb | 8.18 Mb | 1.86 Mb | 1.86 Mb | 2.29 Mb+dup chr1:0.12 Mb |
| Genes involved in deletion | ||||||||||
| XYLT2 | + | |||||||||
| MRPL27 | + | |||||||||
| EME1 | + | |||||||||
| LRRC59 | + | |||||||||
| DQ599569 | + | |||||||||
| ACSF2 | + | |||||||||
| CHAD | + | |||||||||
| ACSF2 | + | |||||||||
| RSAD1 | + | |||||||||
| MYCBPAP | + | |||||||||
| EPN3 | + | |||||||||
| SPATA20 | + | |||||||||
| LOC253962 | + | |||||||||
| CACNA1G | + | |||||||||
| ABCC3 | + | |||||||||
| BC131755 | + | |||||||||
| ANKRD40 | + | |||||||||
| Y_RNA | + | |||||||||
| LUC7L3 | + | |||||||||
| AK090674 | + | |||||||||
| LINC00483 | + | |||||||||
| C17ORF73 | + | |||||||||
| WFIKKN2 | + | |||||||||
| TOB1 | + | |||||||||
| LOC400604 | + | |||||||||
| SPAG9 | + | |||||||||
| NME1 | + | |||||||||
| NME2 | + | |||||||||
| NME1-NME2 | + | |||||||||
| MBTD1 | + | |||||||||
| UTP18 | + | |||||||||
| AK124832 | + | |||||||||
| CA10 | + | |||||||||
| LOC100506650 | + | |||||||||
| KIF2B | + | + | ||||||||
| TOM1L1 | + | + | + | + | + | |||||
| TRNA | + | + | + | + | + | |||||
| COX11 | + | + | + | + | + | |||||
| STXBP4 | + | + | + | + | + | |||||
| HLF | + | + | + | + | + | |||||
| MMD | + | + | + | + | + | |||||
| TMEM100 | + | + | + | + | + | + | ||||
| PCTP | + | + | + | + | + | + | ||||
| ANKFN1 | + | + | + | + | + | + | + | |||
| BC037494 | + | + | + | + | + | + | + | |||
| NOG | + | + | + | + | + | + | + | + | ||
| C17ORF67 | + | + | + | + | + | + | + | + | ||
| DGKE | + | + | + | + | + | + | ||||
| MTVR2 | + | + | + | + | + | + | ||||
| TRIM25 | + | + | + | + | + | + | ||||
| BC114339 | + | + | + | + | + | + | ||||
| MIR3614 | + | + | + | + | + | + | ||||
| COIL | + | + | + | + | + | + | ||||
| SCPEP1 | + | + | + | + | + | + | ||||
| RNF126P1 | + | + | + | + | + | + | ||||
| AKAP1 | + | + | + | + | + | + | ||||
| MSI2 | + | + | + | + | + | + | + | |||
| MRPS23 | + | + | + | + | + | + | ||||
| CUEDC1 | + | + | + | + | + | + | ||||
| VEZF1 | + | + | + | + | + | + | ||||
| AK095112 | + | + | + | + | + | |||||
|
AK12 |
+ |
+ |
|
|
+ |
+ |
|
|
|
+ |
| SFRS1 | + | + | + | + | + | |||||
| DYNLL2 | + | + | + | + | ||||||
| OR4D1 | + | + | + | + | ||||||
| MSX2P1 | + | + | + | + | ||||||
| OR4D2 | + | + | + | + | ||||||
| EPX | + | + | + | + | ||||||
| MKS1 | + | + | + | + | ||||||
| LPO | + | + | + | |||||||
| MPO | + | + | + | |||||||
| BX648763 | + | + | + | |||||||
| BZRAP1 | + | + | + | |||||||
| LOC100506779 | + | + | + | |||||||
| MIR142 | + | + | + | |||||||
| MIR4736 | + | + | + | |||||||
| LOC100506779 | + | + | + | |||||||
| SUPT4H1 | + | + | + | + | ||||||
| RNF43 | + | + | + | + | ||||||
| HSF5 | + | + | + | + | ||||||
| MTMR4 | + | + | + | + | ||||||
| SEPT4 | + | + | + | + | ||||||
| C17orf47 | + | + | + | + | ||||||
| TEX14 | + | + | + | + | ||||||
| RAD51C | + | + | + | + | ||||||
| PPM1E | + | + | + | + | ||||||
Figure 3.
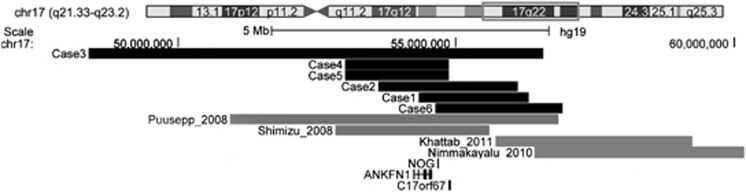
Schematic representation of the size and location of 17q22 microdeletions detected by array-CGH in patients described here (black bars) and previously described patients with similar microdeletions (gray bars).4, 6, 7, 8 Displayed and magnified chromosomal region is boxed in red on chromosome 17 at the top. The numbers below represent the genomic distance (in base pairs) from the 17p telomere according to UCSC Genome Browser Build 37/hg19 (2009). The tracks of the commonly deleted genes ANKFN1, NOG and CORF67 are retrieved from the UCSC Genes track(http://genome.ucsc.edu/).
Clinical description (Table 2)
Table 2. Common clinical characteristics of our and the four previously reported 17q22 microdeletion patients.
| Phenotype | Puusepp et al8 | Khattab et al6 | Nimma-kayalu et al7 | Shimizu et al4 | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Totala |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Head | |||||||||||
| Microcephaly | + | – | + | NA | + | – | + | – | − | + | 5/9 (56%) |
| Narrow long face | NA | NA | NA | NA | − | + | + | − | − | + | 3/6 (50%) |
| Maxillary hypoplasia | + | NA | NA | NA | − | + | + | + | + | NA | 5/6 (83%) |
| Short philtrum | NA | NA | NA | NA | + | + | + | + | + | NA | 5/5 (100%) |
| Thin border of upper lip | + | + | + | NA | + | + | + | + | + | NA | 8/8 (100%) |
| Micrognathia | + | + | NA | NA | + | − | − | + | + | NA | 5/7 (71%) |
| Large bulbous nose | + | NA | NA | NA | + | + | + | + | + | NA | 6/6 (100%) |
| Hypoplastic alae nasi | NA | NA | NA | NA | − | + | + | + | + | NA | 4/5 (80%) |
| Prominent columella | NA | NA | NA | NA | + | + | + | + | + | NA | 4/5 (80%) |
| Large dysplastic ears | NA | NA | NA | NA | + | − | − | − | − | NA | 1/5 (20%) |
| Upslanting palpebral fissures | + | + | NA | NA | + | − | + | + | + | NA | 6/7 (86%) |
| Narrow palpebral fissures | NA | + | NA | NA | + | + | + | + | + | NA | 6/6 (100%) |
| Epicantal folds | + | NA | NA | NA | + | + | NA | + | + | NA | 5/5 (100%) |
| Strabismus | + | NA | NA | NA | + | + | NA | − | − | NA | 3/5 (60%) |
| Hypertelorism | + | + | NA | + | + | − | + | + | + | NA | 7/8 (88%) |
| Blepharophimosis | + | NA | NA | NA | + | + | + | − | − | NA | 4/6 (67%) |
| Absent or hypoplastic teeth | NA | NA | NA | NA | NA | + | NA | + | NA | NA | 2/6 (33%) |
| Chest and spine | |||||||||||
| Narrow thorax | NA | NA | NA | NA | − | − | NA | + | + | NA | 2/4 (50%) |
| Vertebral anomalies | + | − | NA | NA | − | + | + | − | NA | NA | 3/6 (50%) |
| Upper extremity | |||||||||||
| Symphalangism | + | + | − | + | + | + | + | + | + | NA | 8/9 (89%) |
| Brachydactyly | NA | NA | − | NA | + | − | − | + | + | NA | 3/6 (50%) |
| Clinodactyly | NA | NA | + | NA | − | − | + | + | + | NA | 4/6 (67%) |
| Short first metacarpal | NA | NA | − | NA | + | + | − | + | + | NA | 4/6 (67%) |
| Proximally placed thumbs | + | − | − | NA | + | + | NA | + | + | NA | 5/7 (71%) |
| Lower extremity | |||||||||||
| Coxa Valga | NA | NA | NA | NA | − | + | + | NA | NA | NA | 2/3 (67%) |
| Genu Valgum | NA | NA | NA | NA | − | + | + | NA | NA | NA | 2/3 (67%) |
| Broad halluces | NA | NA | NA | NA | − | − | − | + | + | NA | 2/5 (40%) |
| Symphalangism | NA | NA | NA | + | + | + | + | − | − | NA | 4/6 (67%) |
| Urogenital | |||||||||||
| Urogenital malformation | + | NA | NA | + | NA | + | + | + | + | + | 7/7 (100%) |
| General | |||||||||||
| Intellectual disability | + | NA | + | + | + | + | + | + | + | + | 9/9 (100%) |
| Attention-deficit hyperactivity disorder | NA | NA | NA | NA | NA | − | NA | + | + | NA | 2/5 (20%) |
| Conductive hearing loss | + | NA | NA | + | NA | − | + | + | + | NA | 5/7 (71%) |
| Stapes ankylosis | NA | NA | NA | + | NA | − | NA | − | − | NA | 1/4 (25%) |
| Speech delay | NA | NA | NA | NA | NA | + | + | + | + | + | 5/6 (83%) |
| Hyperopia | + | NA | NA | + | + | + | + | − | − | NA | 5/7 (71%) |
| Astigmatism | NA | NA | NA | + | − | + | − | + | − | NA | 3/6 (50%) |
NA, information not available (due to not examined or not reported in article. In some cases not applicable due to gender, age at examination or severe intellectual disability).
The total number of patients with a specific phenotype differs depending on whether presence or absence of the phenotype was specifically mentioned in the reports or examined in our study.
The main shared clinical features for each patient are given in Table 2. Photos of faces and hands are shown in Figures 1 and 2. More detailed descriptions follow below.
Patient 1 was the second child born to healthy unrelated parents. The pregnancy was uneventful and she was delivered in week 41 with birth weight 3120 g (−0.7 standard deviation score (SDS), according to Albertsson Wikland21), birth length 50 cm (0 SDS) and head circumference 33 cm (−1.1 SDS). The head circumference at 1 year of age was 42 cm (−3.2 SDS), hence she became microcephalic. Shortly after birth, dysmorphic facial features were noticed, and ultrasonography revealed aortic hypoplasia and a persistent ductus arteriosus. Examination at 1 year of age showed short, upslanting palpebral fissures, blepharophimosis, hypertelorism, strabismus and epicanthal folds together with a large bulbous nose, high nasal bridge, prominent columella, thin vermilion border of upper lip and a short philtrum. In both upper extremities there was an inability to flex the proximal interphalangeal joints due to symphalangism. The thumbs appeared proximally placed with a short first metacarpal. The forearm had limited rotation on examination despite non-dislocated radial heads. In the lower extremities there was symphalangism of the toes and variable terminal deficiencies, that is, the toes ended at various lengths. The patient had intellectual disability, impaired hearing and hyperopia. At examination she was too young for speech evaluation.
Patient 2 was born at 42 weeks gestation after an uneventful pregnancy to a healthy unrelated couple. She weighed 3270 g (−0.4 SDS) and had a head circumference of 38 cm (1.7 SDS). She had a long, thin face with maxillary hypoplasia and a large bulbous nose with a prominent columella, hypoplastic alae nasi and a high nasal bridge. There was a short philtrum and a thin vermillion border to the upper lip. Later, hypertelorism, blepharophimosis, strabismus, narrow palpebral fissures and epicanthic folds were noted. In addition, she had preauricular sinuses. There were two fused thoracic vertebrae, first and second, causing scoliosis. She had hypogonadotrophic hypogonadism, an absent uterus and a rudimentary vagina. In the hands there was a short first metacarpal with the appearance of proximally placed thumbs and symphalangism in all fingers including the thumbs. The proximal interphalangeal joints in digits 2,3 and 4 had a flexion contracture due to the symphalangism and this was progressively more severe towards the ulnar border of the hands. She had Raynaud phenomenon in the hands and feet. Her shoulders were anteriorly positioned and internally rotated and the forearm had limited rotation on examination, most likely due to dislocated radial heads. The hips were dysplastic, internally rotated and had reduced movement (coxa valga). The knees had a fixed flexion deformity with genu valgum and there was symphalangism of toes 2, 3 and 5. The patient was delayed in speech and had intellectual disability.
Patient 3 is a young adult male, the youngest of three siblings born to unrelated parents. In the first month of life he was dysmorphic and found to have a cytogenetically visible de novo deletion of 17q22. He was slow to feed, microcephalic, had a prominent nose, mild contractures of the fingers, stiff hips, penile chordee and cryptorchidism. He had intellectual disability and his mobility was poor (requiring a wheelchair by age 7 years) due to increasing contractures of his knees, and he developed a seizure disorder without grand mal attacks (well controlled on carbamazepine, later lamotrigine) and asthma. Communication has always been extremely limited. Through his childhood and adolescence his contractures increased at the hips (tendon release left hip performed age 13), knees and ankles, and to a lesser extent in his digits. By age 17 years he had developed a spinal kyphosis with vertebral wedging of T11–12. When assessed at age 17 years of age, his head circumference was 50.5 cm (∼−4.2 SDS according to Nellhaus22), he was essentially wheelchair-bound (though could walk with support) with anteriorly placed shoulders, contractures of his fingers (Figure 2), hips, knees, ankles and toes. He had fair hair, prominent supraorbital ridges, short and slightly upslanting palpebral fissures with telecanthus, mildly dysplastic helices, a large bulbous nose, short philtrum, thin upper lip and prominent lower lip, a rugiose tongue, clefting of uvula, long digits and toes with symphalangism, an appearance similar to ‘pseudoclubbing' and variable terminal deficiencies. Feeding was still problematic as he could not chew effectively, and he had no sphincter control.
Patient 4, a 5-year-old female, the daughter of patient 5, was born after 40 weeks gestation to a primagravida woman whose pregnancy was complicated by breech presentation, requiring a cesarean section. She required 4-day hospitalization in the neonatal intensive care unit for mild respiratory difficulties. Her birth weight was low, 2795 g (−1.4 SDS). Her parents were unrelated. In preschool years, she demonstrated intellectual disability and speech delay, attention deficit hyperactivity disorder, conductive hearing loss and astigmatism. She had a markedly round face with maxillary hypoplasia, a large bulbous nose with prominent columella, hypoplastic alae nasi and mild micrognathia. The eyes had short, upslanting palpebral fissures, hypertelorism and epicanthal folds. There were duplicated renal collecting system, limited range of motion in the neck and a narrow thorax. Her upper extremities revealed carpal fusions, shortening of the metacarpals and phalanges of the fifth digits bilaterally. The thumbs appeared proximally placed, broad and had asymmetric shortening of the metacarpals. The proximal symphalangism on digits 1, 4 and 5 bilaterally was more severe in digit 5, where the proximal interphalangeal joint was ankylotic with clinodactyly. Some of the phalanges were broad and some were hypoplastic with terminal deficiencies. In the lower extremities, there were congenital muscular anomalies in the calves requiring lengthening of the Achilles tendon and short feet with broad and short halluces. She had pain in multiple joints. Mother and daughter were the only individuals within their extended pedigree who showed any evidence of skeletal anomalies or symphalangism.
Patient 5, the mother of patient 4, presented as an early teen with delay of speech, intellectual disability, attention deficit hyperactivity disorder, conductive hearing loss and pain in multiple joints. Her face showed maxillary hypoplasia, micrognathia, a large bulbous nose with prominent columella and hypoplastic alae nasi and an upper lip with thin vermilion border and a short philtrum. The eyes had short upslanting palpebral fissures, epicanthal folds and hypertelorism. There were duplicated renal collecting system, vertebral anomalies, a webbed neck with limited range of motion and a narrow thorax. In the upper extremity there were carpal fusions, broad digits and hypoplastic phalanges (mid and distal phalanx) with variable terminal deficiency. The thumb appeared proximally placed with a short first metacarpal. The fingers showed proximal symphalangism (progressively worse to the ulnar side) and brachydactyly. There was clinodactyly of the fifth finger. The feet were short with broad halluces. There were variable terminal deficiencies in the toes, absence of distal phalanges and hypoplastic toenails. The mother differed from her daughter by demonstrating webbed neck, a partial cleft of the alveolar ridge and a history of vesicourethral reflux.
The duplicated renal collecting systems segregated with the chromosome 2 inversion in multiple maternal family members, including two maternal aunts and a maternal uncle. These other individuals did not have the chromosome 17 deletion.
Patient 6. Unfortunately limited information on the phenotype was available. It was concluded that the patient had delayed speech and intellectual disability. His head was microcephalic with a narrow long face and a high palate. The patient's genitals showed chordee in a small penis and chryptorchidism. In the upper extremity there was camptodactyly.
Discussion
In the current study, we describe evidence for a clinically recognizable 17q22 microdeletion syndrome. The syndrome includes microcephaly, intellectual disability, visual impairment, distinctive facial features such as thin border of upper lip, upslanting palpebral fissures, micrognathia, hypertelorism and NOG-related bone and joint-features such as symphalangism, conductive hearing loss and joint-contractures. It has previously been shown that larger deletions across the region and heterozygous missense and nonsense mutations in NOG cause symphalangism17 All six patients in our study had bone and joint symptoms described in the five OMIM NOG-related symphalangism spectrum disorders. The short and broad phalanges and symphalangism of joints in digits and toes are most likely caused by haploinsufficency of NOG.
Symphalangism, dysmorphic facial features and intellectual disability have previously been reported in four 17q22 microdeletion patients4, 6, 7, 8 (Table 2). In addition, eight patients with similar phenotype and cytogenetically visible deletions between 17q22 and 17q24 have been reported.8 The lack of genomic coordinates for the deletions based on G-band patterning alone makes it difficult to make detailed genotype–phenotype correlations. The 17q22 microdeletions described in this study involve a ∼8.5-Mb region that is rich in segmental duplications (http://projects.tcag.ca/variation/). Segmental duplications comprise ∼5% of the human genome and are known to mediate clinically relevant deletions, duplications, and inversions through non allelic homologous recombination.23 The commonly deleted region of 17q22 identified in this study is ∼0.24 Mb in size and encompasses two genes: the NOG gene and an open reading frame gene called C17ORF67. When we exclude patient 6 in which limited clinical information was available, the commonly deleted region is ∼0.55 Mb, involving NOG, C17ORF67, ANKFN1 and the mRNA-coding sequence BC037494. We did not find any associated phenotypes noted in OMIM, Decipher (except our Patient 6, https://decipher.sanger.ac.uk/patient/2292) or Ensembl for ANKFN1, BC037494 or C17ORF67.
The phenotypic and genotypic differences between our patients are interesting to explore. Patient 1 differed from patients 2, 3, 4 and 5 by aortic hypoplasia and deletion of DYNLL2, OR4D1, OR4D2, MSX2P1 and MKS1. Patient 6 also had a deletion involving these genes but no cardiac abnormality. It is notable that the patient with a 3.54-Mb microdeletion of 17q22–17q23.1, reported by Khattab et al.6 had heart anomalies, pulmonary hypertension and deletion of MKS1 but not NOG. This patient also had symphalangism, which might indicate positional effects of other genes in the region in the development of symphalangism.
Patient 2 is the only patient in our study with hypogonadotrophic hypogonadism and absence of uterus. Interestingly, Shimizu et al.4 also reported a patient with a 17q22 deletion and hypogonadotrophic hypogonadism. When excluding the genomic region of Patient 4 and 5 (mother to patient 4), the breakpoints that the Shimizu et al.4 patient and our patient 2 share includes DGKE, MTVR2, TRIM25, BC114339, MIR3614, COIL, SCPEP1, RNF126P1, AKAP1 and MSI2. To our knowledge, there is no human phenotype related to hypogonadism or absence of uterus reported in PubMed, OMIM or Ensemble for mutations in these genes, but Orimo et al.24 have shown that mice carrying a loss of function mutation in Trim25, also named Efp (oestrogen-responsive finger protein), have underdeveloped uterus, suggesting that Efp could be involved in the normal oestrogen-induced cell proliferation of uterus and the uterine swelling.
Mutations in NOG-related symphalangism spectrum disorders have not resulted in intellectual disability.25, 26, 27, 28, 29 Two previously reported 17q22 microdeletion, patients were intellectually disabled.7, 8 All patients in our study, including patient 6, have intellectual disability and deletions of NOG and C17ORF67. However, all cases harbored deletions of several other dosage-sensitive genes outside the region of overlap, making haploinsufficiency of NOG and C17ORF67 unlikely to be the cause of intellectual disability in our patients.
Our findings support the proposal of Ballif et al.30 that copy number variants of 17q22 and 17q23.1q23.2 represent clinically distinct entities. They do share some symptoms such as hearing loss, intellectual disability and microcephaly. However, there are some major differencies in the bone, joint and facial phenotypes, such as symphalangism and short palpebral fissures. All patients with 17q23.1q23.2 deletions presented with heart defects, which is not the case with the 17q22 microdeletion patients, except for the above mentioned patient 1 with aortic hypoplasia and the patient reported by Khattab et al.6, 30, 31
In the 17q21.31 microdeletion syndrome, patients present with intellectual disability, hypotonia, high palate, slender, long fingers and facial features, including blepharophimosis, upslanting palpebral fissures, epicanthal folds, a bulbous nose and prominent ears.32, 33, 34, 35 In addition, cardiac abnormalities, kidney problems and skeletal anomalies have been described. In our opinion, some of the facial features resemble patients in our study, possibly indicating positional effects of other genes involved in the two syndromes.33, 34
The hand descriptions noted for NOG-related symphalangism spectrum disorders raise a specific point of interest, in particular the proximally placed thumbs reported previously. On clinical examination patients 1, 2, 4 and 5 appeared to have proximally placed thumbs and patient 1 also had a deep first web space. X-rays revealed a short first metacarpal in patients 1, 2 and 4 (Figure 2), but the base of the first metacarpal and the carpometacarpal-joint was not proximally placed. Therefore, the clinical appearance of a proximally placed thumb is due to a short first metacarpal or in some patients a short proximal phalanx and should be reported as such. Cushing36 noticed, as many after him, that the symphalangism progressed in severity from the postaxial side of the hands to the preaxial side. This was also the case in our study for all patients except patient 6. Despite the fact that patient 1 had stiff proximal interphalangeal joints and absent flexion creases at clinical examination, her X-rays at one and a half years of age were almost normal except for narrowing of the joint space of the proximal interphalangeal joint of the fifth digit (Figure 2). There could be many reasons for this. First, an X-ray of the hands at early age is likely inadequate for determining the presence of a proper proximal interphalangeal joint. Second, the gap showing at the proximal interphalangeal joints may be cartilage without joint space and the ankylosis of the proximal interphalangeal joints may progress later during life.37
In conclusion, we have investigated molecularly and clinically, patients with 17q22 microdeletions and present evidence for a clinically recognizable 17q22 microdeletion syndrome. The identification of additional patients with microdeletions on 17q22 should further elucidate the clinical features of this contiguous microdeletion syndrome.
Acknowledgments
We thank all colleagues in pediatric departments who provided the patient samples. This work was supported by grants from the Swedish Research Council, Kronprinsessan Lovisa, Karolinska Institutet, Frimurarna Barnhuset in Stockholm and Linnea and Joseph Carlsson foundation. We would like to acknowledge Dr Regina Regan for performing the array hybridization for patient 2 in UK. SJLK is supported by the NIHR Biomedical Research Centre, Oxford with funding from the Department of Health's NIHR Biomedical Research Centres funding scheme. The views expressed in this publication are those of the authors and not necessarily those of the Department of Health. SJLK is also supported by the Wellcome Trust Core Award Grant [090532/Z/09/Z].
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary Material
References
- Dallapiccola B, Mingarelli R, Digilio C, Obregon MG, Giannotti A. Interstitial deletion del(17) (q21.3q23 or 24.2) syndrome. Clin Genet. 1993;43:54–55. doi: 10.1111/j.1399-0004.1993.tb04452.x. [DOI] [PubMed] [Google Scholar]
- Khalifa MM, MacLeod PM, Duncan AM. Additional case of de novo interstitial deletion del(17)(q21.3q23) and expansion of the phenotype. Clin Genet. 1993;44:258–261. doi: 10.1111/j.1399-0004.1993.tb03893.x. [DOI] [PubMed] [Google Scholar]
- Park JP, Moeschler JB, Berg SZ, Bauer RM, Wursterhill DH, Unique Denovo A. Interstitial Deletion Del(17) (Q21.3q23) in a Phenotypically Abnormal Infant. Clin Genet. 1992;41:54–56. doi: 10.1111/j.1399-0004.1992.tb03631.x. [DOI] [PubMed] [Google Scholar]
- Shimizu R, Mitsui N, Mori Y, et al. Cryptic 17q22 deletion in a boy with a t(10;17)(p15.3;q22) translocation, multiple synostosis syndrome 1, and hypogonadotropic hypogonadism. Am J Med Genet A. 2008;146A:1458–1461. doi: 10.1002/ajmg.a.32319. [DOI] [PubMed] [Google Scholar]
- Marsh AJ, Wellesley D, Burge D, et al. Interstitial deletion of chromosome 17 (del(17)(q22q23.3)) confirms a link with oesophageal atresia. J Med Genet. 2000;37:701–704. doi: 10.1136/jmg.37.9.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khattab M, Xu F, Li P, Bhandari V. A de novo 3.54 Mb deletion of 17q22-q23.1 associated with hydrocephalus: a case report and review of literature. Am J Med Genet A. 2011;155A:3082–3086. doi: 10.1002/ajmg.a.34307. [DOI] [PubMed] [Google Scholar]
- Nimmakayalu M, Major H, Sheffield V, et al. Microdeletion of 17q22q23.2 encompassing TBX2 and TBX4 in a patient with congenital microcephaly, thyroid duct cyst, sensorineural hearing loss, and pulmonary hypertension. Am J Med Genet A. 2011;155A:418–423. doi: 10.1002/ajmg.a.33827. [DOI] [PubMed] [Google Scholar]
- Puusepp H, Zilina O, Teek R, et al. 5.9 Mb microdeletion in chromosome band 17q22-q23.2 associated with tracheo-esophageal fistula and conductive hearing loss. Eur J Med Genet. 2009;52:71–74. doi: 10.1016/j.ejmg.2008.09.006. [DOI] [PubMed] [Google Scholar]
- Levin ML, Shaffer LG, Lewis R, Gresik MV, Lupski JR. Unique de novo interstitial deletion of chromosome 17, del(17) (q23.2q24.3) in a female newborn with multiple congenital anomalies. Am J Med Genet. 1995;55:30–32. doi: 10.1002/ajmg.1320550110. [DOI] [PubMed] [Google Scholar]
- Mickelson EC, Robinson WP, Hrynchak MA, Lewis ME. Novel case of del(17)(q23.1q23.3) further highlights a recognizable phenotype involving deletions of chromosome (17)(q21q24) Am J Med Genet. 1997;71:275–279. doi: 10.1002/(sici)1096-8628(19970822)71:3<275::aid-ajmg5>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Thomas JA, Manchester DK, Prescott KE, Milner R, McGavran L, Cohen MM. Hunter-McAlpine craniosynostosis phenotype associated with skeletal anomalies and interstitial deletion of chromosome 17q. Am J Med Genet. 1996;62:372–375. doi: 10.1002/(SICI)1096-8628(19960424)62:4<372::AID-AJMG9>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Valenzuela DM, Economides AN, Rojas E, et al. Identification of mammalian noggin and its expression in the adult nervous system. J Neurosci. 1995;15:6077–6084. doi: 10.1523/JNEUROSCI.15-09-06077.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Krakow D, Marcelino J, et al. Heterozygous mutations in the gene encoding noggin affect human joint morphogenesis. Nat Genet. 1999;21:302–304. doi: 10.1038/6821. [DOI] [PubMed] [Google Scholar]
- Zimmerman LB, De Jesus-Escobar JM, Harland RM. The Spemann organizer signal noggin binds and inactivates bone morphogenetic protein 4. Cell. 1996;86:599–606. doi: 10.1016/s0092-8674(00)80133-6. [DOI] [PubMed] [Google Scholar]
- Brunet LJ, McMahon JA, McMahon AP, Harland RM. Noggin, cartilage morphogenesis, and joint formation in the mammalian skeleton. Science. 1998;280:1455–1457. doi: 10.1126/science.280.5368.1455. [DOI] [PubMed] [Google Scholar]
- Usami S, Abe S, Nishio S, et al. Mutations in the NOG gene are commonly found in congenital stapes ankylosis with symphalangism, but not in otosclerosis. Clin Genet. 2012;82:514–520. doi: 10.1111/j.1399-0004.2011.01831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potti TA, Petty EM, Lesperance MM. A comprehensive review of reported heritable noggin-associated syndromes and proposed clinical utility of one broadly inclusive diagnostic term: NOG-related-symphalangism spectrum disorder (NOG-SSD) Hum Mutat. 2011;32:877–886. doi: 10.1002/humu.21515. [DOI] [PubMed] [Google Scholar]
- Ballif BC, Theisen A, McDonald-McGinn DM, et al. Identification of a previously unrecognized microdeletion syndrome of 16q11.2q12.2. Clin Genet. 2008;74:469–475. doi: 10.1111/j.1399-0004.2008.01094.x. [DOI] [PubMed] [Google Scholar]
- Zhang ZF, Ruivenkamp C, Staaf J, et al. Detection of submicroscopic constitutional chromosome aberrations in clinical diagnostics: a validation of the practical performance of different array platforms. Eur J Hum Genet. 2008;16:786–792. doi: 10.1038/ejhg.2008.14. [DOI] [PubMed] [Google Scholar]
- Traylor RN, Fan Z, Hudson B, et al. Microdeletion of 6q16.1 encompassing EPHA7 in a child with mild neurological abnormalities and dysmorphic features: case report. Mol Cytogenet. 2009;2:17. doi: 10.1186/1755-8166-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikland KA, Luo ZC, Niklasson A, Karlberg J. Swedish population-based longitudinal reference values from birth to 18 years of age for height, weight and head circumference. Acta Paediatr. 2002;91:739–754. doi: 10.1080/08035250213216. [DOI] [PubMed] [Google Scholar]
- Nellhaus G. Head circumference from birth to eighteen years. Practical composite international and interracial graphs. Pediatrics. 1968;41:106–114. [PubMed] [Google Scholar]
- Sharp AJ, Cheng Z, Eichler EE. Structural variation of the human genome. Annu Rev Genomics Hum Genet. 2006;7:407–442. doi: 10.1146/annurev.genom.7.080505.115618. [DOI] [PubMed] [Google Scholar]
- Orimo A, Inoue S, Minowa O, et al. Underdeveloped uterus and reduced estrogen responsiveness in mice with disruption of the estrogen-responsive finger protein gene, which is a direct target of estrogen receptor alpha. Proc Natl Acad Sci USA. 1999;96:12027–12032. doi: 10.1073/pnas.96.21.12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson K, Seeman P, Sebald E, et al. GDF5 is a second locus for multiple-synostosis syndrome. Am J Hum Genet. 2006;78:708–712. doi: 10.1086/503204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Takahashi I, Komatsu M, et al. Mutations of the NOG gene in individuals with proximal symphalangism and multiple synostosis syndrome. Clin Genet. 2001;60:447–451. doi: 10.1034/j.1399-0004.2001.600607.x. [DOI] [PubMed] [Google Scholar]
- Brown DJ, Kim TB, Petty EM, et al. Autosomal dominant stapes ankylosis with broad thumbs and toes, hyperopia, and skeletal anomalies is caused by heterozygous nonsense and frameshift mutations in NOG, the gene encoding noggin. Am J Hum Genet. 2002;71:618–624. doi: 10.1086/342067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomeer HG, Admiraal RJ, Hoefsloot L, Kunst HP, Cremers CW. Proximal symphalangism, hyperopia, conductive hearing impairment, and the NOG gene: 2 new mutations. Otol Neurotol. 2011;32:632–638. doi: 10.1097/MAO.0b013e318211fada. [DOI] [PubMed] [Google Scholar]
- Weekamp HH, Kremer H, Hoefsloot LH, Kuijpers-Jagtman AM, Cruysberg JR, Cremers CW. Teunissen-Cremers syndrome: a clinical, surgical, and genetic report. Otol Neurotol. 2005;26:38–51. doi: 10.1097/00129492-200501000-00008. [DOI] [PubMed] [Google Scholar]
- Ballif BC, Theisen A, Rosenfeld JA, et al. Identification of a recurrent microdeletion at 17q23.1q23.2 flanked by segmental duplications associated with heart defects and limb abnormalities. Am J Hum Genet. 2010;86:454–461. doi: 10.1016/j.ajhg.2010.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonewolf-Greulich B, Ronan A, Ravn K, et al. Two new cases with microdeletion of 17q23.2 suggest presence of a candidate gene for sensorineural hearing loss within this region. Am J Med Genet A. 2011;155A:2964–2969. doi: 10.1002/ajmg.a.34302. [DOI] [PubMed] [Google Scholar]
- Koolen DA, Kramer JM, Neveling K, et al. Mutations in the chromatin modifier gene KANSL1 cause the 17q21.31 microdeletion syndrome. Nat Genet. 2012;44:639–641. doi: 10.1038/ng.2262. [DOI] [PubMed] [Google Scholar]
- Koolen DA, Sharp AJ, Hurst JA, et al. Clinical and molecular delineation of the 17q21.31 microdeletion syndrome. J Med Genet. 2008;45:710–720. doi: 10.1136/jmg.2008.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koolen DA, Vissers LE, Pfundt R, et al. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet. 2006;38:999–1001. doi: 10.1038/ng1853. [DOI] [PubMed] [Google Scholar]
- Dubourg C, Sanlaville D, Doco-Fenzy M, et al. Clinical and molecular characterization of 17q21.31 microdeletion syndrome in 14 French patients with mental retardation. Eur J Med Genet. 2011;54:144–151. doi: 10.1016/j.ejmg.2010.11.003. [DOI] [PubMed] [Google Scholar]
- Cushing H. Hereditary anchylosis of the proximal phalan-geal joints (symphalangism) Genetics. 1916;1:90–106. doi: 10.1093/genetics/1.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek GH, Lee HJ. Classification and surgical treatment of symphalangism in interphalangeal joints of the hand. Clin Orthop Surg. 2012;4:58–65. doi: 10.4055/cios.2012.4.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.