

Abstract
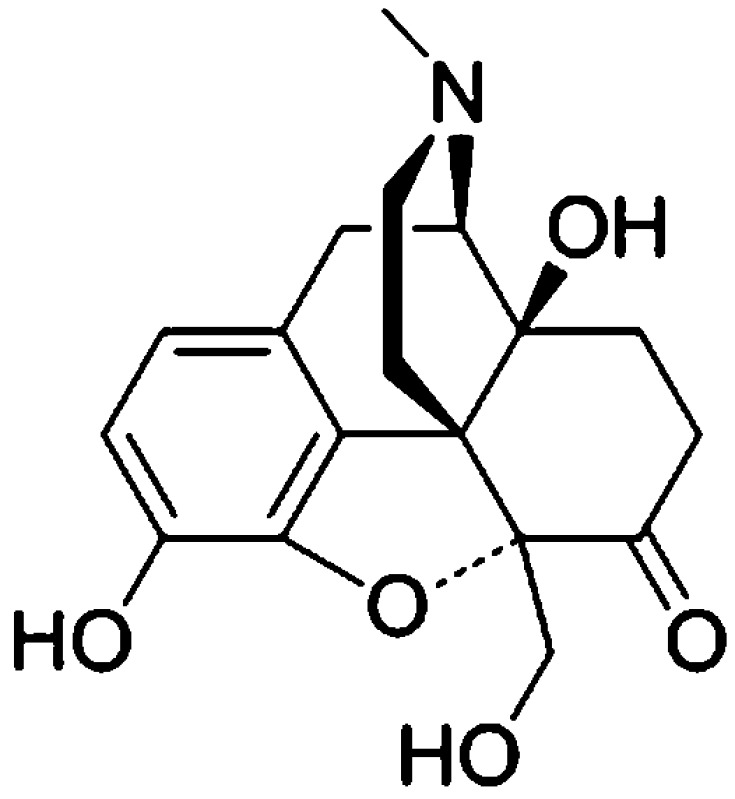
Opioid narcotics are used for the treatment of moderate-to-severe pain and primarily exert their analgesic effects through μ receptors. Although traditional μ agonists can cause undesired side effects, including tolerance, addition of δ antagonists can attenuate said side effects. Herein, we report 4a,9-dihydroxy-7a-(hydroxymethyl)-3-methyl-2,3,4,4a,5,6-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7(7aH)-one (UMB 425) a 5,14-bridged morphinan-based orvinol precursor synthesized from thebaine. Although UMB 425 lacks δ-specific motifs, conformationally sampled pharmacophore models for μ and δ receptors predict it to have efficacy similar to morphine at μ receptors and similar to naltrexone at δ receptors, due to the compound sampling conformations in which the hydroxyl moiety interacts with the receptors similar to orvinols. As predicted, UMB 425 exhibits a mixed μ agonist/δ antagonist profile as determined in receptor binding and [35S]GTPγS functional assays in CHO cells. In vivo studies in mice show that UMB 425 displays potent antinociception in the hot plate and tail-flick assays. The antinociceptive effects of UMB 425 are blocked by naloxone, but not by the κ-selective antagonist norbinaltorphimine. During a 6-day tolerance paradigm, UMB 425 maintains significantly greater antinociception compared to morphine. These studies thus indicate that, even in the absence of δ-specific motifs fused to the C-ring, UMB 425 has mixed μ agonist/δ antagonist properties in vitro that translate to reduced tolerance liabilities in vivo.
Keywords: Antinociception, conformationally sampled pharmacophore, delta antagonist, mu agonist, opioid receptor, tolerance
Opioid analgesics, including morphine, are the traditional standards for individuals suffering from cancer pain, postoperative pain, or pain from other severe trauma.1,2 The prescription of opioids has risen significantly since the early 1980s.3 While opioids may be the standard for treating moderate-to-severe pain, the side effects, including respiratory depression, tolerance, physical/psychological dependence, constipation, sedation, nausea/vomiting, and dizziness, can be problematic.4 These severe side effects often lead to the undertreatment of chronic pain and increase the possibility of death.5 Therefore, there is a pressing need to identify pharmacological agents that maintain potent analgesic properties while eliminating the problematic side effects.
Opioid receptors are G protein-coupled receptors6 located in the central nervous system, peripheral nervous system, and gastrointestinal tract.7 To date, three opioid receptor subtypes have been identified: mu (μ),8 delta (δ),9 and kappa (κ).10 Traditional opioid analgesics exert their pain-relieving properties through μ receptors located within the central nervous system.11 Opioid interactions at the δ receptor have shown synergistic analgesic effects in combination with activation of the μ receptor.12 However, research suggests that the activation of the δ receptor may play a role in the side effect liabilities associated with chronic opioid use, including tolerance.13 In contrast, selective δ antagonists can reduce tolerance when given in conjunction with the traditional μ agonists, including morphine,13 and opioid peptides that display mixed μ agonist/δ antagonist activity have reduced tolerance liabilities compared to traditional opioid analgesics.14
The identification of nonpeptidic opioid analgesics that display dual characteristics of μ agonism/δ antagonism could convey therapeutic advantages compared to peptides, with regards to ease of administration and delayed metabolic breakdown. Several approaches toward this goal have been undertaken, specifically the characterization of bivalent and bifunctional opioid ligands. Portoghese introduced the concept of bivalent ligands, compounds that embody two specific pharmacophores connected via an optimized linker.15 Bivalent ligands containing both μ agonist and δ antagonist motifs were shown to exhibit greater analgesic effects, while also reducing tolerance and physical dependence liabilities.16 The existence of homo- and hetero-oligomeric opioid receptor complexes, including a μ–δ complex,17 suggest that bivalent opioid ligands are a viable therapeutic tool. However, the physiochemical properties profiles of bivalent ligands may prove problematic with regards to an oral absorption formulation.18
Bifunctional ligands possess a single pharmacophore that targets two binding sites with functional activity distinct for each of the respective sites,19 ideally circumventing the potentially problematic characteristics associated with bivalent ligands. Such ligands include structural motifs that are seen in traditional μ agonists as well as δ antagonists. Bifunctional ligands depicting mixed μ agonism/δ antagonism have displayed potent analgesic activity with reduced side effect liabilities, including tolerance.20 However, those developed are traditionally characterized in vivo by intracerebroventricular administration, a method unsuitable for widespread therapeutic use.
The studies herein report the synthesis, modeling and pharmacological characterization of the novel opioid ligand, 4a,9-dihydroxy-7a-(hydroxymethyl)-3-methyl-2,3,4,4a,5,6-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7(7aH)-one (UMB 425). UMB 425 was originally designed as a precursor to a series of bifunctional 5,14-bridged morphinan-based orvinols. The compound has a unique structure, such that it exhibits no δ-specific motifs fused to the C-ring, and the 5′-hydroxymethyl substituent is the only functional group that distinguishes UMB 425 from the chemical structure of oxymorphone. Application of conformationally sampled pharmacophore (CSP) models of μ agonism and δ antagonism,21−23 with the latter updated as part of the present study, predicted mixed μ agonist/δ antagonist effects for UMB 425. Moreover, analysis of the conformations of UMB 425 generated as part of the CSP protocol showed that the 5′-hydroxymethyl moiety can spatially overlap with the hydroxyl group linked to the C19 of orvinols. Since some orvinols, such as buprenorphine, act as mixed μ agonist/δ antagonists,24 it was hypothesized that UMB 425 may also interact with the receptors in a similar way as the orvinols, despite UMB 425’s lack of the classical δ antagonist motif on the C-ring. Indeed, in vitro and in vivo pharmacological characterization of UMB 425 show it to have high affinity and the desired μ agonism and δ antagonism profile in CHO cells, and to have antinociceptive effects with decreased development of tolerance in mice.
Results and Discussion
The development of novel opioid ligands with mixed μ agonist/δ antagonist activity has therapeutic potential for the treatment of pain with a lower side effect liability than commonly marketed opioid analgesics. Toward this goal, a number of peptidic as well as bivalent and bifunctional nonpeptidic ligands have been developed that achieve this desired pharmacological profile, but have inherent problems that limit their potential as therapeutic agents. We report herein on UMB 425, a novel opioid that lacks a typical δ-selective motif, yet exhibits mixed μ agonist/δ antagonist activity, robust antinociceptive effects, and a reduced tolerance liability compared to morphine.
Synthesis
A highly efficient method for the synthesis of UMB 425 is summarized in Scheme 1. Briefly, deprotonation of thebaine (1) followed by in situ reaction of the anion with ethyl chloroformate yielded 2.25 Reduction of the ester (2) with lithium aluminum hydride gave 3.26 By oxidation of 3 with a mixture of formic acid and hydrogen peroxide at 4 °C (70 h), we obtained 4,27 which was reduced by catalytic hydrogenation to obtain olefin reduced diol (5). O-Demethylation of the diol (5) using boron tribromide/chloroform gave UMB 425 at a 64% yield.28
Scheme 1. Chemical Synthesis of UMB 425 from Thebaine.

(a) n-BuLi, ethyl chloroformate, THF, −78 °C, 4 h, 64%; (b) LiAlH4, THF, 0 °C–rt, 2 h, 81%; (c) H2O2, HCOOH, H2SO4, 4 °C, 70 h, 61%; (d) 10% Pd/C, H2, 1:1 ethanol/glacial acetic acid, 4 h, 70%; (e) BBr3, CHCl3, −20 °C, 3 h, 64%.
μ Agonism/δ Agonism Dual-Profile CSP Model
Prediction of the efficacy of UMB 425 applied previously developed μ agonism21 and δ agonism22,23 CSP models, with the latter updated to include a larger number of nonpeptidic opioid δ ligands. CSP updated δ model generation involved the development of multiple individual models based on different pharmacophoric descriptors (Figure S1, Supporting Information); the top five models of the updated δ CSP are listed in Table 1, each with an R2 greater than 0.89. The final CSP model is based on averaging the predicted efficacies from these top five models. From the model, overlap of the aromatic ring (A) to hydrophobic group (B) distance distributions was identified as the most important descriptor. AB distances of compounds showing agonism at δ opioid receptors had greater overlap with those of (±)-4-((α-R*)-α-((2S*,5R*)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-hydroxybenzyl)-N,N-diethyl-benzamide (BW373U86) than antagonists; however, efficacy was explained not solely by the AB distance, but in combination with the relative position of the hydrophobic group with respect to the aromatic ring and basic N. Accordingly, overlap coefficients of angles ANB, BAN, and ABN were identified as important descriptors by the automated variable selection applied during model construction.
Table 1. Top Five δ Receptor Conformationally Sampled Pharmacophore Models That Define the Final Predictive Modela.
| model # | a | X1 | b | X2 | c | R2 | p-value | correlation coefficient |
|---|---|---|---|---|---|---|---|---|
| 1 | 0.465 | AB | 0.514 | BN | 0.028 | 0.962 | 0.00005 | 0.815 |
| 2 | 0.730 | AB | 0.235 | ANB | –0.031 | 0.919 | 0.00054 | 0.519 |
| 3 | 0.760 | AB | 0.187 | BAN | –0.026 | 0.911 | 0.00071 | 0.442 |
| 4 | 0.749 | AB | 0.182 | ABN | –0.037 | 0.893 | 0.00121 | 0.603 |
| 5 | 1.094 | BN | –0.196 | ANB | 0.108 | 0.890 | 0.00133 | 0.797 |
Multiple regression equations, efficacy = aX1 + bX2 + c. N represents the basic nitrogen, A is the aromatic ring and B is the hydrophobic group, as shown in Figure S1 of the Supporting Information. X1 and X2 are overlap integrals with respect to the reference compound, while a and b are coefficients for variables X1 and X2 and c is the y-intercept in the regression equations. R2 is the goodness of fit, p-value is the significance of models, and correlation coefficients between X1 and X2 overlap coefficients.
Calculated efficacies for the training set molecules are shown in Table 2 together with experimental values reported previously.29 The model predicts buprenorphine to be a weak partial agonist at δ receptors due to its resemblance to etorphine, particularly with respect to the AB distances. Oxymorphindole and naltrindole were not differentiated by the model. The only difference between them is the N-substituent (N-methyl for oxymorphindole and N-cyclopropylmethyl for naltrindole) and the present model did not include the N-substituent as a pharmacophoric descriptor because the length of the N-substituent is not as critical for the δ receptor as it is for the μ receptor. However, the weak partial agonism of oxymorphindole seems to be due to the short methyl N-substituent.
Table 2. Comparison between Experimental and Calculated Efficacy Values for Compounds in Training Seta.
| relative %Emax |
||
|---|---|---|
| name | exptl | calcd |
| BW373U86 | 1.00 | 0.96 |
| etorphine | 0.36 | 0.36 |
| SIOM | 0.18 | 0.13 |
| oxymorphindole | 0.12 | 0.04 |
| diprenorphine | 0.08 | 0.01 |
| buprenorphine | 0.00 | 0.13 |
| naltrexone | 0.00 | 0.01 |
| naltrindole | 0.00 | 0.04 |
| (E)-BNTX | 0.00 | 0.07 |
Experimental data, except buprenorphine and naltrexone, is previously reported.29 Buprenorphine and naltrexone were experimentally designated %Emax = 0 values as both are classified as antagonists at the δ receptor.6,24 BW373U86 = (±)-4-((α-R*)-α-((2S*,5R*)-4-Allyl-2,5-dimethyl-1-piperazinyl)-3-hydroxybenzyl)-N,N-diethyl-benzamide, SIOM = 7-spiroindanyloxymorphone, (E)-BNTX = [(E)-benzylidenenaltrexone].
The CSP models for the μ and δ receptor ligands was applied to UMB 425, as well as all ligands being developed as part of our ongoing research program. Predicted efficacy (%Emax) values for UMB 425 were 101 and 1.4 for the μ and δ receptors, respectively. The high efficacy at μ receptors is consistent with the structural similarity of UMB 425 with morphine or oxymorphone, while the low efficacy at δ receptors is consistent with the C-ring substituents of naltrexone.
To better understand the contribution of the 5′-hydroxymethyl to efficacy, additional analysis was performed on conformations of UMB 425 generated during CSP model development. Distances and angle distributions between the basic nitrogen and oxygen in the 5′-hydroxymethyl in UMB 425 or the 19-hydroxyl substituent in the orvinols were calculated and compared. Figure 1 shows N–O distance and N–C9–O angle probability distributions of three orvinols and UMB 425. In Figure 1b, two large distributions are present that are separated by around 1.5 Å, although a small peak is noted in the UMB 425 distribution at 6.5 Å that overlaps with that of the orvinols. The N–C9–O angle indicates the relative position of the hydroxyl group with respect to the plane of the aromatic A-ring. The hydroxyl group of UMB 425 is slightly above the A-ring plane while that of the orvinols is below; however, a small overlap between UMB 425 and the orvinols is observed (Figure 1c). While preliminary, these results indicate that UMB 425 can assume conformations in which its hydroxyl moiety participates in interactions with the receptors that are similar to those occurring with the orvinols. The recent availability of X-ray crystal structures of the μ and δ receptors will allow for future evaluation of the present model in the context of 3D interactions between UMB 425, as well as other ligands, and the receptors.30,31
Figure 1.

(a) Images of UMB 425 and etorphine with the hydroxyl oxygen highlighted in red. Probability distributions of the (b) basic N to oxygen and (c) the basic N–C9-oxygen angle from the simulations used in CSP model development for etorphine (red line), buprenorphine (green dashed line), diprenorphine (blue dashed line), and UMB 425 (purple dashed line).
Opioid Receptor Binding Affinities
Given its chemical structure, it is not surprising that UMB 425 demonstrates greater selectivity for the μ receptor than either the δ receptor or the κ receptor. Table 3 summarizes the binding of UMB 425 and morphine at human μ, δ, and κ opioid receptors stably transfected into and overexpressed in CHO cells. Consistent with previous reports, morphine has higher binding affinity for the μ receptor to that of the δ receptor and the κ receptor (δ/μ = 51; κ/μ = 41). UMB 425 also has high binding affinity for the μ receptor with preference for it compared to both the δ receptor and the κ receptor (δ/μ = 65; κ/μ = 66).
Table 3. In Vitro Pharmacological Profiles of Morphine and UMB 425a.
|
Ki ± SEM
(nM) |
EC50 ± SEM (nM) |
% Emax ± SEM |
pA2 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| μ | δ | κ | μ | δ | κ | μ | δ | κ | δ | |
| morphine | 1.7 ± 0.34 | 87 ± 6.6 | 69 ± 1.3 | 38 ± 4.9 | 316.5 ± 4.9b | 484 ± 213b | 81 ± 2.3 | 103 ± 7b | 62 ± 7b | nd |
| UMB 425 | 3.2 ± 0.14 | 208 ± 18 | 212 ± 21 | 35 ± 3.7 | n/e | n/e | 73 ± 7.3 | nd | nd | (−0.91) |
Receptor binding and [35S]GTPγS functional activity for morphine and UMB 425 are summarized for studies performed in CHO cell membranes stably transfected and overexpressing the human μ, δ, and κ opioid receptors. Competition binding for compounds were performed in triplicate of duplicates and reported as mean Ki values ± SEM. Mean EC50 and %Emax values ± SEM for the [35S]GTPγS functional assays were performed in triplicate of duplicates. pA2 is defined as the negative logarithm of antagonist concentration needed to shift the dose response curve by a factor of 2. A slope of at or near −1 is indicative of competitive antagonism for the drug at the receptor.
Reference (32). n/e = no effect, nd = not determined.
Functional Assays for Agonist and Antagonist Activity
Agonist and antagonist activities for morphine and UMB 425 at each of the human opioid receptor subtypes overexpressed in CHO cells are summarized in Table 3. For the [35S]GTPγS functional assay, UMB 425 displayed comparable partial agonistic capabilities at the μ receptor to that of morphine (EC50 = 35 ± 3.7 and 38 ± 4.9 nM; %Emax = 73 ± 7.3 and 81 ± 2.3 nM for UMB 425 and morphine, respectively). Unlike morphine, which is highly efficacious at the δ receptor (EC50 = 316.5 ± 4.9 nM; %Emax = 103 ± 7),32 UMB 425 displayed no significant agonist activity through the δ or κ receptors, yet it did demonstrate antagonistic activity through the δ receptor indicated by a corresponding pA2 value of 6.12 (−0.91).
Acute Antinociceptive Effects
Subcutaneous injection of morphine or UMB 425 demonstrated antinociceptive effects in mice in a time- and dose-dependent manner for the thermal nociceptive assays (Figure 2). Table 4 summarizes respective ED50 values for morphine and UMB 425 from testing performed 30 min after drug treatment. The potency of the antinociceptive activity of UMB 425 (ED50 = 4.30 and 8.83 mg/kg for the hot plate and tail-flick assays, respectively) was similar to that of morphine (ED50 = 2.73 and 6.85 mg/kg for the hot plate and tail-flick assays, respectively). While not as potent as other opioid compounds previously tested,33 UMB 425 was able to achieve a maximal antinociceptive response at comparable doses to morphine.
Figure 2.

Acute dose–response and time–response curves for s.c. morphine and UMB 425 treatment for the hot plate and tail-flick assays. Male, Swiss-Webster mice were treated with morphine (0.1–20 mg/kg, s.c.) or UMB 425 (0.1–20 mg/kg, s.c.). Latencies were recorded 30 min after drug administration and every 20 min thereafter for 150 or 210 min. (a) Dose–response and time–response curves for s.c. morphine in the hot plate assay. (b) Dose–response and time–response curves for s.c. morphine in the tail-flick assay. (c) Dose–response and time–response curves for s.c. UMB 425 in the hot plate assay. (d) Dose–response and time–response curves for s.c. UMB 425 in the tail-flick assay.
Table 4. ED50 Values for Morphine and UMB 425 in the Acute Treatment Paradigma.
| ED50 (mg/kg) |
||
|---|---|---|
| morphine | UMB 425 | |
| hot plate | 2.73 | 4.30 |
| tail-flick | 6.85 | 8.83 |
Summary of antinociceptive activity of acute morphine (0.1–20 mg/kg, s.c.) and UMB 425 (0.1–20 mg/kg, s.c.) treatment in Swiss Webster mice for the hot plate and tail-flick assays. Respective ED50 values (in mg/kg, s.c.) were obtained at the 30 min time point after drug administration.
Opioid Antagonism
To corroborate opioid-induced antinociception, various antagonist pretreatments were given in conjunction with UMB 425 administration. One-way analysis of variance (ANOVA) indicated differences among mice pretreated with saline, naloxone, and nor-BNI (F(2,12) = 8.88, p < 0.005; F(2,13) = 22.61, p < 0.0001 for the hot plate and tail-flick assays, respectively). UMB 425’s partial agonistic effects through the μ receptor appear primarily responsible for the observed antinociceptive effects seen in vivo as naloxone significantly attenuated UMB 425-mediated antinociception (Figure 3; q = 4.55, p < 0.05; q = 8.48, p < 0.001 for the hot plate and tail-flick assays, respectively; Tukey’s posthoc). In contrast, pretreatment with the κ antagonist nor-BNI failed to significantly attenuate UMB 425-mediated antinociception (Figure 3; q = 0.01, n.s.; q = 1.39, n.s. for the hot plate and tail-flick assays, respectively; Tukey’s posthoc). Consistent with the lack of agonist activity observed in the [35S]GTPγS assay at κ receptors by UMB 425, the κ receptor does not appear to contribute significantly to the antinociceptive effects of UMB 425.
Figure 3.

Antagonism of UMB 425 antinociception using various opioid antagonists. Male, Swiss Webster mice received a pretreatment of the nonselective opioid antagonist, naloxone (1 mg/kg, i.p. t = −30 min) or the κ-selective antagonist, nor-BNI (30 mg/kg, i.p. t = −24 h) prior to a s.c. injection with an ED90 dose of UMB 425 (15 mg/kg). Latencies were determined 30 min after UMB 425 administration. (a) Antagonism of UMB 425 antinociception in the hot plate assay by naloxone, but not nor-BNI. *p < 0.05. (b) Antagonism of UMB 425 antinociception in the tail-flick assay by naloxone, but not nor-BNI. ***p < 0.001.
Tolerance to Antinociceptive Effects
The results of administration of ED90 doses of morphine and UMB 425 to mice twice daily for a period of 5 days, with test latencies determined 30 min after drug administration, are summarized in Figure 4. One-way repeated measures ANOVA demonstrated statistical differences among treatment days for morphine administration in both the hot plate (Figure 4a; F(4,76) = 15.22, p < 0.0001) and tail-flick assays (Figure 4b; F(4,76) = 8.52, p < 0.0001). Dunnett’s posthoc analysis revealed that morphine administration significantly decreased antinociceptive activity on Day 4 and 5 of the tolerance paradigm for both the hot plate (Figure 4a; q = 4.45, p < 0.01; q = 6.52, p < 0.01; respectively) and tail-flick assays (Figure 4b; q = 2.72, p < 0.05; q = 5.12, p < 0.01; respectively). One-way repeated measures ANOVA also demonstrated a statistical difference among treatment days for UMB 425 administration in the hot plate assay (Figure 4a; F(4,84) = 9.32, p < 0.0001) but not for the tail-flick assay (Figure 4b, F(4,84) = 1.53, n.s.). Dunnett’s posthoc analysis revealed that UMB 425 administration significantly decreased antinociceptive activity on Day 4 and 5 of the tolerance paradigm in the hot plate assay (Figure 4a; q = 3.43, p < 0.01; q = 5.11, p < 0.01; respectively).
Figure 4.

Antinociceptive tolerance development for morphine and UMB 425 for the hot plate and tail-flick assays. Male, Swiss Webster mice were given an ED90 dose of morphine (15 mg/kg, s.c.) or UMB 425 (15 mg/kg, s.c.) twice daily for a 5 day period. Latencies were determined 30 min after drug administration. (a) Antinociceptive tolerance development in the hot plate assay. **p < 0.01 vs Morphine Day 1/UMB 425 Day 1; #p < 0.05 vs Morphine Day 4; ###p < 0.001 vs Morphine Day 5. (b) Antinociceptive tolerance development in the tail-flick assay. *p < 0.05, **p < 0.01 vs Morphine Day 1; #p < 0.05 vs Morphine Day 5.
Two-way repeated measures ANOVA revealed statistical differences between morphine and UMB 425 treatment in time points for both the hot plate (Figure 4a; p < 0.0001) and tail-flick assays (Figure 4b; p < 0.0001). Bonferroni’s posthoc analysis demonstrated that UMB 425 maintained statistically greater antinociceptive activity than morphine on Day 4 and 5 for the hot plate assay (Figure 4a; t = 2.86, p < 0.05; t = 4.15, p < 0.001; respectively) and on Day 5 for the tail-flick assay (Figure 4b; t = 2.88, p < 0.05).
Our tolerance paradigm involves thermal nociceptive testing on a daily basis to ensure that a statistical difference in antinociceptive activity is seen between morphine and UMB 425 prior to a dose–response challenge. Table 5 summarizes respective ED50 values for morphine and UMB 425 from the dose–response challenge on Day 6 of the tolerance paradigm. UMB 425 (ED50 = 12.96 and 11.58 mg/kg for the hot plate and tail-flick assays, respectively) produced markedly less tolerance development than morphine (ED50 = 21.31 and 44.11 mg/kg for the hot plate and tail-flick assays, respectively), as evident by the respective rightward shifts in ED50 values (7.8- vs 3.0-fold and 6.4- vs 1.3-fold for morphine vs UMB 425 in the hot plate and tail-flick assays, respectively). Respective ED50 shifts in the morphine-treated tolerance paradigm seen on Day 6 were comparable to tolerance paradigms previously performed using male ICR mice, despite variations regarding drug dosing, route of administration, number of injections per day as well as the time length of the paradigm.34
Table 5. ED50 Values for Morphine and UMB 425 in the Tolerance Treatment Paradigmsa.
| morphine |
UMB
425 |
|||
|---|---|---|---|---|
| ED50 (mg/kg) | tolerance | shift | tolerance | shift |
| hot plate | 21.31 | 7.8 | 12.96 | 3.0 |
| tail-flick | 44.11 | 6.4 | 11.58 | 1.3 |
Summary of antinociceptive activity obtained for the tolerance paradigm for morphine and UMB 425 for the hot plate and tail-flick assays. Mice were treated with the ED90 dose of morphine (15 mg/kg, s.c.) or UMB 425 (15 mg/kg, s.c.) determined in the acute treatment paradigm, twice a day for 5 days. ED50 values for the tolerance paradigm were obtained during a dose–response challenge on Day 6 of treatment, whereby latencies were determined 30 min after drug administration (tolerance columns). The shifts represent fold-shifts in the ED50 determined in the acute vs tolerance treatment paradigms.
The antagonistic potency of UMB 425 through the δ receptor (pA2 = 6.12) is lower than previously highlighted μ agonist/δ antagonist analgesics, notably 11-(4-chlorophenyl)-7-(cyclopropylmethyl)-8a-(3-phenylpropoxy)-6,7,8,8a,9,13b-hexahydro-5H-4,8 methanobenzofuro[3,2-h]pyrido[3,4-g]quinolin-1-ol or “17d” (δ Ke = 0.091 ± 0.01 nM),35 as well as the δ-selective antagonist naltrindole (pA2 = 10.92).32 Yet, UMB 425 demonstrated a significant reduction in tolerance liabilities compared to morphine itself, specifically a 2.6- and 4.9-fold reduction in respective ED50 shifts for the hot plate and tail-flick assays. The recently reported mixed μ agonist/δ antagonist analgesic 17d was found to have a 5.6-fold decrease in respective A50 shifts compared to morphine itself in the warm-water tail-withdrawal assay.35 The respective shift-fold variations between the in vivo studies are thus quite comparable despite the lower affinity and antagonist potency of UMB 425 compared to 17d for the δ receptor in vitro. Thus, equipotence at the μ and δ receptor does not seem required to improve tolerance liabilities of opioid analgesics.
The initial pharmacological effects of UMB 425 seen in vivo are promising, though further testing is needed to assess other potential side effects including respiratory depression, decreased gastrointestinal motility, physical as well as psychological dependence. It is therefore noteworthy that δ antagonists and δ opioid receptor knockout mice have been shown in earlier studies to attenuate the rewarding effects of morphine,36,37 reduce respiratory depression liabilities associated with fentanyl analogues,38 and promote colonic propulsion.39 In addition, mixed μ agonist/δ antagonist analgesics have been shown to reduce physical dependence.34
The molecular mechanisms surrounding opioid-induced tolerance are still not clear, although many efforts have been taken to discern the labyrinth of potential components involved. Morphine, a partial μ agonist, is unable to properly internalize the μ receptor upon activation, whereby desensitization and uncoupling of the G-protein potentially leads to the development of tolerance.40 δ receptor recruitment to the plasma membrane has been shown to increase with extended exposure to μ agonists.41 Furthermore, δ antagonists can provide synergistic effects in combination with μ agonists that enhances μ receptor binding and signaling in cells expressing μ–δ heterodimers, potentially through altered G-protein activation.42 It is conceivable that the μ agonist/δ antagonist interactions through UMB 425 allow for proper internalization, driven by the δ receptor in the hetero-oligomeric complex, and subsequent μ receptor mediated recycling and resensitization thereby leading to reduced tolerance development. In addition, GPCR kinase (GRKs) and β-arrestin regulatory processes have been linked to opioid-induced tolerance.43,44 Concurrent μ agonist/δ antagonist activity may alter phosphorylation and recruitment mechanisms associated with these processes, potentially limiting GPCR desensitization. Follow up studies will be needed to delineate the underlying mechanisms through which UMB 425 reduces the development of tolerance compared to morphine.
Conclusion
In summary, UMB 425 is a novel opioid mixed μ agonist/δ antagonist that possesses antinociceptive effects comparable to morphine with reduced tolerance liabilities. It has nanomolar affinity and efficacy for μ receptors similar to morphine and moderate affinity for δ receptors at which it exhibits antagonistic effects. UMB 425 is structurally similar to traditional μ agonists, and CSP models suggest that its hydroxyl moiety assumes conformations that allow interactions similar to orvinols with mixed μ agonist/δ antagonist effects. Together, the data suggest that traditional δ selective motifs need not be added to a traditional μ agonist pharmacophore to achieve δ antagonism and reduce opioid-induced tolerance.
Methods
Chemistry
Synthesis
All reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO) and used without further purification. NMR spectra were recorded on a Varian Mercury 400 system (400 MHz for 1H and 100 MHz for 13C NMR), using CDCl3 or CD3OD as a solvent. Mass spectra were determined using a Bruker Amazon X Ion Trap spectrometer. Melting points are uncorrected. Purification was performed by column chromatography over Whatman silica gel 60 (230–400 mesh) using dichloromethane and methanol. Reactions were monitored by thin layer chromatography (TLC) using dichloromethane/methanol (92:8). The purity of UMB 425 was determined by high resolution mass spectrometry (HRMS) and high performance liquid chromatography (HPLC) analysis and is >95% (Figure S2 and S3, Supporting Information).
Ethyl 7,9-Dimethoxy-3-methyl-2,3,4,7a-tetrahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinoline-7a-carboxylate (2)
A solution of 1 (1g) in 20 mL of tetrahydrofuran (THF) was placed in a flame-dried round-bottom flask, and 1.92 mL (1.5 equiv) of a 2.5 M solution of n-butyllithium in hexane was added while stirring at −78 °C under nitrogen. The mixture immediately turned deep wine-red and was stirred for 45 min at −78 °C. Ethyl chloroformate (0.36 mL, 1.2 equiv) was added, and the mixture was stirred for 4 h at −78 °C. The color changed to orange-yellow. Saturated NH4Cl (5 mL) was then added per drop, and most part of the solvent was removed under reduced pressure. The residual brown product was dissolved in chloroform, washed with brine solution, dried (Na2SO4), and concentrated. The residue was subjected to column chromatography on silica gel using dichloromethane–methanol (95:5) as the eluent to isolate 0.786 g (64% yield) of 2 as a pale yellow foam. 1H NMR (CDCl3, 400 MHz): δ 6.67 d (1H, J = 8.6 Hz), 6.61 d (1H, J = 8.6 Hz), 5.2 d (1H, J = 6.2 Hz), 5.19 d (1H, J = 6.2 Hz), 4.44–4.33 m (1H), 4.28–4.16 m (1H), 3.85 s (3H), 3.66 d (1H, J = 6.2 Hz), 3.57 s (3H), 3.29 d (1H, J = 17.9 Hz), 2.87–2.74 m (1H), 2.7 d (1H, J = 7.0 Hz), 2.68–2.57 m (1H), 2.45 s (3H), 2.16 td (1H, J = 4.6, 12.5 Hz), 1.64 d (1H, J = 13.3 Hz), 1.30 t (3H, J = 7.0 Hz). 13C NMR (CDCl3, 100 MHz): δ 168.31, 152.64, 143.62, 142.66, 132.30, 130.56, 127.38, 119.86, 113.44, 112.09, 97.05, 95.42, 61.80, 61.26, 56.45, 55.27, 49.09, 45.49, 42.12, 31.01, 30.42, 14.21. MS (ESI): m/z 384.1 (M + 1)+.
(7,9-Dimethoxy-3-methyl-2,3,4,7a-tetrahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7a-yl)methanol (3)
Lithium aluminum hydride (LAH) (0.00496g, 0.13 mmol) was added to a stirred solution of 2 (0.050g, 0.13 mmol) in dry THF (2 mL) at 0 °C and allowed to warm to room temperature. After 2 h, the reaction mixture was quenched with saturated sodium sulfate solution and stirred for 30 min. The reaction mixture was filtered through Celite; the organic layer was separated, concentrated, and purified by silica gel column chromatography (15:85 methanol–dichloromethane) to yield the pure product 3 as a pale yellow solid (0.036 g, 81% yield) with mp 169–171 °C. 1H NMR (CDCl3, 400 MHz): δ 6.62 d (1H, J = 7.8 Hz), 6.58 d (1H, J = 7.8 Hz), 5.57 d (1H, J = 6.2 Hz), 5.19 d (1H, J = 6.2 Hz), 4.26 d (1H, J = 10.9 Hz), 4.07 d (1H, J = 11.7 Hz), 3.79 s (3H), 3.66 d (1H, J = 6.2 Hz), 3.55 s (3H), 3.32 d (1H, J = 17.9 Hz), 2.90–2.79 m (1H), 2.78–2.66 m (2H), 2.53 td (1H, J = 4.6, 12.5 Hz), 2.46 s (3H), 1.8 d (1H, J = 12.5 Hz). 13C NMR (CDCl3, 100 MHz): δ 153.13, 143.21, 142.58, 133.86, 130.59, 126.59, 119.64, 113.08, 112.36, 98.34, 94.0, 61.68, 61.48, 56.08, 55.17, 47.19, 45.27, 41.63, 31.25, 28.32. MS (ESI): m/z 342.2 (M + 1)+.
4a-Hydroxy-7a-(hydroxymethyl)-9-methoxy-3-methyl-2,3,4,4a-tetrahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7(7aH)-one (4)
An ice-cold mixture of 0.4 mL of 0.7% H2SO4, 0.125 mL of 88% HCO2H, and 0.251 mL of 30% H2O2 was added to 0.3 g (0.879 mmol) of 3. The mixture was stirred at 0 °C until it became transparent (∼30 min). The resulting solution was kept for 70 h in a refrigerator (4 °C) and then poured into 3 mL of ice water which was made alkaline by the addition of concentrated ammonia solution. The mixture was extracted with five portions of chloroform, and the organic extracts were combined, dried over sodium sulfate, and evaporated to obtain 0.184 g (61%) of 4 as a white solid with mp 231–233 °C. 1H NMR (CDCl3, 400 MHz): δ 6.69–6.55 m (3H), 6.17 d (1H, J = 9.3 Hz), 4.19 d (1H, J = 10.1 Hz), 4.05 d (1H, J = 10.1 Hz), 3.78 s (3H), 3.22 d (1H, J = 17.9 Hz), 3.18–3.1 m (1H), 2.84–2.53 m (3H), 2.48 s (3H), 2.34–2.22 m (1H), 1.57 d (1H, J = 10.1 Hz). 13C NMR (CDCl3, 100 MHz): δ 198.06, 146.15, 143.18, 142.85, 134.36, 130.72, 124.2, 119.83, 114.77, 91.83, 67.57, 63.46, 60.15, 56.57, 47.6, 45.37, 42.3, 25.56, 22.4. MS (ESI): m/z 344.2 (M + 1)+.
4a-Hydroxy-7a-(hydroxymethyl)-9-methoxy-3-methyl-2,3,4,4a,5,6-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7(7aH)-one (5)
To a solution of 4 (0.13g, 0.379 mmol) in 5 mL of 1:1 ethanol–glacial acetic acid, Pd/C (10%, 15 mg) was added. The mixture was evacuated and filled with H2 gas in a hydrogenation flask and maintained under 40 psi H2 pressure for 4 h. The reaction mixture was filtered through Celite, the solvent was evaporated, and the residue was basified with aqueous ammonia prior to CHCl3 extraction. Organic phases were combined, washed with brine, and dried over anhydrous Na2SO4, and the solvent was evaporated. The residue was subjected to column chromatography (7:93 methanol–dichloromethane) to give 5 as a white foam (0.0915 g, 70%). 1H NMR (CDCl3, 400 MHz): δ 6.7 d (1H, J = 8.6 Hz), 6.65 d (1H, J = 8.6 Hz), 4.16 d (1H, J = 10.9 Hz), 4.01 d (1H, J = 11.7 Hz), 3.86 s (3H), 3.16 d (1H, J = 18.7 Hz), 3.07 td (1H, J = 5.4, 14.8 Hz), 2.96 d (1H, J = 5.4 Hz), 2.67–2.51 m (2H), 2.43 s (3H), 2.40–2.29 m (2H), 2.25–2.15 m (1H), 1.96–1.87 m (1H), 1.74–1.63 m (1H), 1.47 d (1H, J = 12.5 Hz). 13C NMR (CDCl3, 100 MHz): δ 210.47, 144.17, 142.88, 129.60, 124.43, 119.64, 114.49, 96.26, 70.29, 64.20, 61.74, 56.53, 50.58, 44.67, 42.65, 37.31, 31.21, 26.88, 21.91. MS (ESI): m/z 346.1 (M + 1)+.
4a,9-Dihydroxy-7a-(hydroxymethyl)-3-methyl-2,3,4,4a,5,6-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7(7aH)-one (UMB 425)
Compound 5 (0.104g, 0.3 mmol) was dissolved in 2 mL CHCl3 and cooled to −20 °C, followed by the slow addition of BBr3 solution (1 M in CHCl3, 1.5 mL). The mixture was stirred at −20 °C for 3 h, carefully quenched with ice and basified with ammonia solution. The resulting mixture was stirred for 30 min at 0 °C, the aqueous layer was separated and the aqueous phase was extracted with CHCl3 (3 × 10 mL). The organic phases were combined, washed with brine and dried over sodium sulfate and the solvent was evaporated. The residue was subjected to column chromatography (20:80 methanol–dichloromethane) to obtain UMB 425 as an off-white solid (0.0638 g, 64%) with mp 193–195 °C. 1H NMR (CD3OD, 400 MHz): δ 6.6 d (1H, J = 8.1 Hz), 6.56 d (1H, J = 8.1 Hz), 5.46 s (1H), 4.23 d (1H, J = 11.9 Hz), 4.0 d (1H, J = 11.9 Hz), 3.16 d (1H, J = 18.9 Hz), 3.12–3.01 m (1H), 2.97–2.9 m (1H), 2.62–2.49 m (2H), 2.41 s (3H), 2.4–2.33 m (1H), 2.25–2.13 m (2H), 1.91–1.83 m (1H), 1.69–1.59 m (1H), 1.33 d (1H, J = 12.4 Hz). 13C NMR (CD3OD, 100 MHz): δ 212.7, 144.6, 140.7, 130.9, 125.3, 120.8, 119.2, 99.20, 71.94, 65.92, 62.47, 52.62, 46.05, 42.89, 37.32, 33.49, 27.34, 23.01. MS (ESI): m/z 332.1 (M + 1)+.
CSP Modeling Calculations
Quantitative conformationally sampled pharmacophore (CSP) models developed for μ and δ receptor ligands were used for studies herein,21,22 with some modifications to the δ receptor model. Updating of the δ receptor model was performed prior to predictions of efficacy of UMB 425. As this study involved derivates of 4,5-epoxymorphinans, the training set was limited to small nonpeptidic opioids: BW373U86, etorphine, 7-spiroindanyloxymorphone (SIOM), oxymorphindole, diprenorphine, buprenorphine, naltrexone, naltrindole, and (E)-benzylidenenaltrexone [(E)-BNTX]. Figure S1 of the Supporting Information shows the chemical structures of compounds used as the training set and their experimental efficacies measured in earlier reported [35S]GTPγS assays.29 For updating the δ receptor CSP model, the selected ligands were modeled using the CHARMM22/CMAP45−47 and CHARMM General Force Field (CGenFF) (48) with Replica Exchange Molecular Dynamics (REX-MD)49,50 for conformational sampling, as previously described.21,51 Pharmacophoric descriptors were designated for calculations of distances and angles between varying functional groups and are identified as an aromatic ring (A), a basic nitrogen (N), and a hydrophobic group (B) (Figure S1, Supporting Information). BW373U86 was used as the reference compound for model development. Statistical models were trained using both agonists and antagonists to differentiate overlapping patterns between the two classes of compounds as well as develop a model that allows for quantitative estimations of efficacy. Changes with respect to the original δ receptor model include the following: (1) for BW373U86, the center of mass of the two piperazine nitrogen groups was designated the N pharmacophoric descriptor; and (2) 1D overlap coefficients with respect to the reference compound were used to obtain multiple regression models with two independent variables; tests using three independent variables did not lead to significant improvements in the models.
Pharmacology
Animals
Male, Swiss Webster mice (21–30 g, Harlan, Indianapolis, IN; Frederick, MD) were housed in groups of five in polysulfone cages (Techniplast, Philadelphia, PA) with a 12:12-h light/dark cycle with food and water ad libitum. Animals were acclimated 1 week prior to experimental use and randomly assigned to treatment groups. All procedures were performed in accordance with the Institutional Animal Care and Use Committee at the West Virginia University Health Sciences Center.
Chemicals
Dulbecco’s modified Eagle’s medium (DMEM) and Dulbecco’s modified Eagle’s medium:Nutrient Mixture F12 with HEPES 1:1 (DMEM:F12) were purchased from Fisher Scientific (Hanover Park, IL). Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Lawrenceville, GA). Penicillin-streptomycin solution (10,000 units/mL), trypsin-EDTA (0.05% trypsin), and G418 (Geneticin) 50 mg/mL were purchased from Invitrogen (Carlsbad, CA). Morphine sulfate, [d-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO), [D-Pen2,5]-enkephalin (DPDPE), N-methyl-2-phenyl-N-[(5R,7S,8S)-7-(pyrrolidin-1-yl)-1-oxaspiro[4.5]dec-8-yl]acetamide (U69,593), naloxone, norbinaltorphimine (nor-BNI), guanosine 5′-diphosphate sodium salt, polyethyleneimine (PEI), and Trizma preset crystals were obtained from Sigma Aldrich (St. Louis, MO). GTPγS lithium salt was obtained from Tocris Bioscience (Ellisville, MO). [3H]DAMGO, [3H]DPDPE, [3H]U69,593, and [35S]GTPγS were purchased from Perkin-Elmer (Shelton, CT).
Binding Studies
Chinese hamster ovary (CHO) cells stably transfected with and overexpressing the human μ opioid receptor (hMOR-CHO), the human δ opioid receptor (hDOR-CHO), or the human κ opioid receptor (hKOR-CHO) were grown in 150 mm dishes (Fisher Scientific, Hanover Park, IL) in DMEM supplemented with 10% FBS, penicillin-streptomycin, and G418 at 37 °C in a 5% CO2 atmosphere. Specifically, DMEM:F12 (1:1) with HEPES, l-Gln solution was used when preparing hMOR-CHO and DMEM 4.5 g/L glucose was used when preparing hDOR-CHO and hKOR-CHO. At 80–90% confluency, cells were scraped from dishes and centrifuged at 2200 rpm for 12 min at 4 °C. Cell pellets were resuspended in 50 mM Tris buffer, pH 7.7, and homogenized using a polytron, and then spun down twice more at 13 500 rpm for 20 min at 4 °C. Membrane was suspended in 50 mM Tris buffer, pH 7.7, at protein concentrations of about 3 mg/mL for hMOR-CHO and hKOR-CHO and 4 mg/mL for hDOR-CHO. Membranes were aliquoted into polypropylene tubes and frozen at −80 °C for future use. Protein concentration was determined using bicinchoninic acid (BCA) reagent and bovine serum albumin (BSA) protein standard provided by the manufacturer (Pierce, Rockford, IL).
Membranes were incubated with 10–12 concentrations of drug (0.001–100 000 nM) and radiolabeled ligand in 50 mM Tris buffer, pH 7.7, at a final volume of 1 mL. μ receptors were labeled using 1.3 nM [3H]DAMGO. δ receptors were labeled using 1.2 nM [3H]DPDPE. κ receptors were labeled using 1.7 nM [3H]U69,593. Nonspecific binding was determined using nonlabeled equivalents at each receptor subtype: 10 μM DAMGO, 1 μM DPDPE, and 10 μM U69,593. Each concentration was tested in triplicates of duplicates with a total volume of 1 mL for each well. Following a 60 min incubation period, reactions were terminated via rapid vacuum filtration over Perkin-Elmer Unifilter-96, GF/B filters (Fisher Scientific, Hanover Park, IL) that were presoaked in 0.5% polyethyleneimine (PEI) for 30 min. After filtration, filters were washed three times with 1.5 mL of cold 50 mM Tris buffer, pH 7.7, and counted in 40 μL of Perkin-Elmer Microscint 20 (Fisher Scientific, Hanover Park, IL). Mean Ki values ± SEM were determined by performing experiments in triplicates of duplicates and calculated using Kd values obtained from saturation binding assays and the Cheng-Prusoff equation.
[35S]GTPγS Functional Assays
Membrane preparation for [35S]GTPγS binding was similar to the aforementioned opioid binding studies, with the exception that CHO cells expressing hMOR, hDOR, and hKOR were resuspended in a final solution (“Buffer A”) consisting of 100 mM NaCl, 10 mM MgCl2, 20 mM HEPES, pH 7.4. Membranes were incubated with 10–12 concentrations of drug and 0.11 nM [35S]GTPγS in “Buffer A” at a final volume of 1 mL. Exogenous guanosine diphosphate (GDP, 100 μL) was added to “Buffer A” per 96-well plate using a 10 mM stock that was made fresh daily. Basal activity was determined in the presence of exogenous GDP and in the absence of an agonist, while nonspecific binding was determined in the presence of 10 μM unlabeled GTPγS. Following a 60 min incubation period, reactions were terminated via rapid vacuum filtration over Perkin-Elmer Unifilter-96, GF/B filters that were presoaked in a 1% BSA solution for 30 min. After filtration, filters were washed three times with 1.5 mL of cold 50 mM Tris buffer, pH 7.7 and counted in 40 μL/well of Perkin-Elmer Microscint 20. Data are presented as the percentage of agonist stimulation (%Emax) of [35S]GTPγS binding normalized against maximal stimulating concentrations of 10 μM DAMGO, 1 μM DPDPE or 10 μM U69,593. %Emax values were determined using the equation: [(CPMbound – CPMbasal)/(CPMmax – CPMbasal)] × 100. Antagonistic properties at δ receptors were determined by constructing dose–response curves of DPDPE (0.01–1000 nM) in the presence and absence of UMB 425. UMB 425 concentrations were 1× (200 nM), 3× (600 nM), and 10× (2 μM) the estimated Ki value obtained from binding studies. pA2 values are indicative of antagonistic potency at a particular receptor subtype and were determined using a Schild plot, whereby the plot’s x-intercept equals pA2. Corresponding slope values at or near −1 indicate competitive antagonism for the compound at that particular receptor subtype. For compounds displaying %Emax < 50, potential antagonistic properties were determined. Means ± SEM were determined by performing experiments in triplicates of duplicates.
Hot Plate Antinociceptive Testing
Mice were placed within a plastic cylinder (10.8 cm ID) atop a black anodized, aluminum plate (27.9 cm × 26.7 cm × 1.9 cm) uniformly regulated at 53 °C (IITC Life Science Inc., Woodland Hills, CA). The latency to the first sign of excessive shaking, lifting, and/or licking of the hind paws was determined and recorded as the behavioral end point. Two baseline latencies (BL) were recorded prior to drug administration, with an average BL within 8–10 s needed for further testing. Mice were then given subcutaneous injections of either morphine (0.1–20 mg/kg) or UMB 425 (0.1–20 mg/kg), and testing latencies (TL) for nociceptive responses were recorded at various time points thereafter. Previous studies have reported that the higher doses of morphine will induce at or near full antinociceptive activity for thermal nociceptive assays.52 A similar dosing paradigm was used for UMB 425, since in vitro opioid binding and functional data for UMB 425 were comparable to that of morphine. The subcutaneous route of injection was chosen because of its common usage with antinociception regimens. A 30 s cutoff latency (CL) was predetermined so as to not cause tissue damage. Data obtained were reported as % Maximum Possible Effect (%MPE), which is indicative of antinociceptive activity associated with a particular compound. %MPE is determined using the following formula: %MPE = [(TL – BL)/(CL – BL)] × 100.
Tail-Flick Antinociceptive Testing
Mice were placed in restraints (2.5 cm ID × 10.2 cm length), and their tails were placed underneath an overhead halogen light source (IITC Life Science Inc., Woodland Hills, CA), whereby the latency to the first sign of a rapid tail flick was determined and recorded as the behavioral end point. Two BL values were recorded prior to drug administration, with an average BL within 2–4 s needed for further testing. Animals were then administered test compound at dosages reported for hot plate antinociceptive testing, and TL values recorded at various time points thereafter. A 10 s CL was predetermined so as to not cause tissue damage. %MPE values were determined as described above.
Antagonist Studies
To determine the opioid receptors involved in the antinociceptive effects of UMB 425, mice were pretreated with the nonselective opioid antagonist naloxone (1 mg/kg i.p., t = −30 min) or the κ-selective antagonist nor-BNI (30 mg/kg i.p., t = −24 h). Antinociceptive testing was performed 30 min after subcutaneous administration of an ED90 dose of morphine or UMB 425. The selected antagonist dosages and pretreatment time points have been shown to correspond with the intended opioid receptor subtype and peak antagonist effect.53,54
Tolerance Assay
The tolerance regimen was performed using previously published methods, with some modifications.34,52 Mice were administered twice daily (8 a.m. and 8 p.m.) subcutaneous injections of a test compound at respective ED90 doses for a 5 day period. On Day 6, animals were given varying doses of morphine (0.1–20 mg/kg, s.c.) or UMB 425 (0.1–20 mg/kg, s.c.) and antinociceptive activity was determined using both the hot plate and tail-flick assays to determine tolerance development. Respective ED50 values determined during the tolerance assay were then compared to values obtained in the acute treatment paradigm. On Days 1–5, the order of the antinociceptive measurements were counterbalanced so that half the mice were assessed for hot plate latencies in the a.m. and tail-flick latencies in the p.m.; the other half were tested for tail-flick latencies in the a.m. and hot plate latencies in the p.m. On Day 6, animal test latencies were determined, first with the tail-flick assay followed 15 min later by the hot-plate assay.
Statistical Analysis
Graphpad Prism software (San Diego, CA) was used for all statistical analysis. Opioid binding and [35S]GTPγS binding data analysis was performed using a nonlinear regression binding model. Ki, EC50, %Emax, and pA2 values were determined as described above. For in vivo antinociceptive assays, agonist ED50 values were calculated using a nonlinear regression model. For antagonist studies, a one-way analysis of variance (ANOVA) followed by Tukey’s posthoc tests was used to determine significance between groups. For the tolerance assay, repeated measures one-way ANOVA followed by Dunnett’s posthoc tests was used to determine significance between treatment days for test compound treatment. Repeated measures two-way ANOVA and Bonferroni’s posthoc tests were used to determine significance between groups. For all comparisons, p < 0.05 was considered statistically significant.
Acknowledgments
CHO cells stably transfected to express each of the human opioid receptor subtypes (hMOR-CHO, hDOR-CHO, hKOR-CHO cells) were generously provided by Dr. Larry Toll (Torrey Pines Institute for Molecular Studies, Port St. Lucie, FL).
Glossary
Abbreviations
- ANOVA
analysis of variance
- BCA
bicinchoninic acid
- BL
baseline latencies
- BSA
bovine serum albumin
- BW373U86
(±)-4-((α-R*)-α-((2S*,5R*)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-hydroxybenzyl)-N,N-diethyl-benzamide
- CHO
Chinese hamster ovary
- CL
cutoff latency
- CPM
counts per minute
- CSP
conformationally sampled pharmacophore
- DAMGO
[d-Ala2, N-MePhe4, Gly-ol]-enkephalin
- DMEM
Dulbecco’s modified Eagle’s medium
- DPDPE
[D-Pen2,5]-enkephalin
- (E)-BNTX
[(E)-benzylidenenaltrexone]
- FBS
fetal bovine serum
- GDP
guanosine diphosphate
- hDOR
human δ opioid receptor
- hKOR
human κ opioid receptor
- hMOR
human μ opioid receptor
- HPLC
high performance liquid chromatography
- HRMS
high resolution mass spectrometry
- i.p.
intraperitoneal
- LAH
lithium aluminum hydride
- MPE
maximum possible effect
- nor-BNI
norbinaltorphimine
- Pd/C
palladium on carbon
- s.c.
subcutaneous
- SEM
standard error of the mean
- SIOM
7-spiroindanyloxymorphone
- TL
testing latencies
- TLC
thin layer chromatography
- THF
tetrahydrofuran
- U69,953
N-methyl-2-phenyl-N-[(5R,7S,8S)-7-(pyrrolidin-1-yl)-1-oxaspiro[4.5]dec-8-yl]acetamide
- UMB 425
4a,9-dihydroxy-7a-(hydroxymethyl)-3-methyl-2,3,4,4a,5,6-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7(7aH)-one
Supporting Information Available
Compounds included in the δ receptor CSP training set as well as spectroscopic and chromatographic experimental procedures and analysis for UMB 425. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
Participated in research design: J.R.H. and R.R.M. (pharmacology), P.B. and A.C. (synthesis), and A.D.M. (modeling). Conducted experiments: J.R.H. (pharmacology), P.B. (synthesis), and J.S. (modeling). New reagents: P.B. and A.C. Data analysis: J.R.H. and R.R.M. (pharmacology), P.B. and A.C. (synthesis), J.S. and A.D.M. (modeling), and J.W.J and M.A.K. (analytical). Wrote and approved the manuscript: J.R.H., P.B., J.S., J.W.J., M.A.K., A.D.M., A.C., and R.R.M.
This work was supported by a grant from the National Institute on Drug Abuse at the National Institutes of Health (DA013583), a PA-08-190 research supplement from the National Institute on Drug Abuse and the University of Maryland Computer-Aided Drug Design Center.
A portion of the work was presented at the 2012 International Narcotics Research Conference (INRC) held in Kansas City, Missouri and the 2012 Society for Neuroscience annual meeting in New Orleans, Louisiana.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Cherny N. J.; Chang V.; Frager G.; Ingham J. M.; Tiseo P. J.; Popp B.; Portenoy R. K.; Foley K. M. (1995) Opioid pharmacotherapy in the management of cancer pain: a survey of strategies used by pain physicians for the selection of analgesic drugs and routes of administration. Cancer 76, 1283–1293. [DOI] [PubMed] [Google Scholar]
- Walder B.; Schafer M.; Henzi I.; Tramer M. R. (2001) Efficacy and safety of patient-controlled opioid analgesia for acute postoperative pain. A quantitative systematic review. Acta Anaesthesiol. Scand. 45, 795–804. [DOI] [PubMed] [Google Scholar]
- Caudill-Slosberg M. A.; Schwartz L. M.; Woloshin S. (2004) Office visits and analgesic prescriptions for musculoskeletal pain in US: 1980 vs. 2000. Pain 109, 514–519. [DOI] [PubMed] [Google Scholar]
- Benyamin R.; Trescot A. M.; Datta S.; Buenaventura R.; Adlaka R.; Sehgal N.; Glaser S. E.; Vallejo R. (2008) Opioid complications and side effects. Pain Physician 11, S105–120. [PubMed] [Google Scholar]
- Manchikanti K. N.; Manchikanti L.; Damron K. S.; Pampati V.; Fellows B. (2008) Increasing deaths from opioid analgesics in the United States: an evaluation in an interventional pain management practice. J. Opioid Manage. 4, 271–283. [DOI] [PubMed] [Google Scholar]
- Waldhoer M.; Bartlett S. E.; Whistler J. L. (2004) Opioid receptors. Annu. Rev. Biochem. 73, 953–990. [DOI] [PubMed] [Google Scholar]
- Stein C.; Schafer M.; Machelska H. (2003) Attacking pain at its source: new perspectives on opioids. Nat. Med. 9, 1003–1008. [DOI] [PubMed] [Google Scholar]
- Wang J. B.; Johnson P. S.; Persico A. M.; Hawkins A. L.; Griffin C. A.; Uhl G. R. (1994) Human mu opiate receptor. cDNA and genomic clones, pharmacologic characterization and chromosomal assignment. FEBS Lett. 338, 217–222. [DOI] [PubMed] [Google Scholar]
- Evans C. J.; Keith D. E. Jr.; Morrison H.; Magendzo K.; Edwards R. H. (1992) Cloning of a delta opioid receptor by functional expression. Science 258, 1952–1955. [DOI] [PubMed] [Google Scholar]
- Mansson E.; Bare L.; Yang D. (1994) Isolation of a human kappa opioid receptor cDNA from placenta. Biochem. Biophys. Res. Commun. 202, 1431–1437. [DOI] [PubMed] [Google Scholar]
- Matthes H. W.; Maldonado R.; Simonin F.; Valverde O.; Slowe S.; Kitchen I.; Befort K.; Dierich A.; Le Meur M.; Dolle P.; Tzavara E.; Hanoune J.; Roques B. P.; Kieffer B. L. (1996) Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature 383, 819–823. [DOI] [PubMed] [Google Scholar]
- Horan P.; Tallarida R. J.; Haaseth R. C.; Matsunaga T. O.; Hruby V. J.; Porreca F. (1992) Antinociceptive interactions of opioid delta receptor agonists with morphine in mice: supra- and sub-additivity. Life Sci. 50, 1535–1541. [DOI] [PubMed] [Google Scholar]
- Abdelhamid E. E.; Sultana M.; Portoghese P. S.; Takemori A. E. (1991) Selective blockage of delta opioid receptors prevents the development of morphine tolerance and dependence in mice. J. Pharmacol. Exp. Ther. 258, 299–303. [PubMed] [Google Scholar]
- Schiller P. W.; Fundytus M. E.; Merovitz L.; Weltrowska G.; Nguyen T. M.; Lemieux C.; Chung N. N.; Coderre T. J. (1999) The opioid mu agonist/delta antagonist DIPP-NH(2)[Psi] produces a potent analgesic effect, no physical dependence, and less tolerance than morphine in rats. J. Med. Chem. 42, 3520–3526. [DOI] [PubMed] [Google Scholar]
- Portoghese P. S. (1989) Bivalent ligands and the message-address concept in the design of selective opioid receptor antagonists. Trends Pharmacol. Sci. 10, 230–235. [DOI] [PubMed] [Google Scholar]
- Daniels D. J.; Lenard N. R.; Etienne C. L.; Law P. Y.; Roerig S. C.; Portoghese P. S. (2005) Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc. Natl. Acad. Sci. U.S.A. 102, 19208–19213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George S. R.; Fan T.; Xie Z.; Tse R.; Tam V.; Varghese G.; O’Dowd B. F. (2000) Oligomerization of mu- and delta-opioid receptors. Generation of novel functional properties. J. Biol. Chem. 275, 26128–26135. [DOI] [PubMed] [Google Scholar]
- Morphy R.; Rankovic Z. (2005) Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 48, 6523–6543. [DOI] [PubMed] [Google Scholar]
- Schiller P. W. (2010) Bi- or multifunctional opioid peptide drugs. Life Sci. 86, 598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananthan S.; Khare N. K.; Saini S. K.; Seitz L. E.; Bartlett J. L.; Davis P.; Dersch C. M.; Porreca F.; Rothman R. B.; Bilsky E. J. (2004) Identification of opioid ligands possessing mixed micro agonist/delta antagonist activity among pyridomorphinans derived from naloxone, oxymorphone, and hydromorphone [correction of hydropmorphone]. J. Med. Chem. 47, 1400–1412. [DOI] [PubMed] [Google Scholar]
- Shim J.; Coop A.; MacKerell A. D. Jr. (2011) Consensus 3D model of mu-opioid receptor ligand efficacy based on a quantitative Conformationally Sampled Pharmacophore. J. Phys. Chem. B 115, 7487–7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard D.; Coop A.; MacKerell A. D. Jr. (2007) Quantitative conformationally sampled pharmacophore for delta opioid ligands: reevaluation of hydrophobic moieties essential for biological activity. J. Med. Chem. 50, 1799–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard D.; Coop A.; MacKerell A. D. Jr. (2003) 2D conformationally sampled pharmacophore: a ligand-based pharmacophore to differentiate delta opioid agonists from antagonists. J. Am. Chem. Soc. 125, 3101–3107. [DOI] [PubMed] [Google Scholar]
- Lutfy K.; Cowan A. (2004) Buprenorphine: a unique drug with complex pharmacology. Curr. Neuropharmacol. 2, 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates M.; Boden R. M.; Sundararaman P. (1989) Derivatives of the thebaine anion. 2. 5-Methylmorphine, 5-methylcodeine, 5-methylheroin and some related compounds. J. Org. Chem. 54, 972–974. [Google Scholar]
- Woudenberg R. H.; Lie T.-S.; Maat L. (1993) Chemistry of opium alkaloids. 38. Synthesis of rigid morphinans doubly bridged at ring C. J. Org. Chem. 58, 6139–6142. [Google Scholar]
- Lotfy H. R.; Schultz A. G.; Metwally M. A. (2003) Synthesis of 5-methoxymethyl, 5-(2-methoxyethyl), and 5-allyl thebaine, codeinone, and morphinone derivatives. Russ. J. Org. Chem. 39, 1256–1260. [Google Scholar]
- Rice K. C. (1977) A rapid, high-yield conversion of codeine to morphine. J. Med. Chem. 20, 164–165. [DOI] [PubMed] [Google Scholar]
- Clark M. J.; Emmerson P. J.; Mansour A.; Akil H.; Woods J. H.; Portoghese P. S.; Remmers A. E.; Medzihradsky F. (1997) Opioid efficacy in a C6 glioma cell line stably expressing the delta opioid receptor. J. Pharmacol. Exp. Ther. 283, 501–510. [PubMed] [Google Scholar]
- Granier S.; Manglik A.; Kruse A. C.; Kobilka T. S.; Thian F. S.; Weis W. I.; Kobilka B. K. (2012) Structure of the delta-opioid receptor bound to naltrindole. Nature 485, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A.; Kruse A. C.; Kobilka T. S.; Thian F. S.; Mathiesen J. M.; Sunahara R. K.; Pardo L.; Weis W. I.; Kobilka B. K.; Granier S. (2012) Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature 485, 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toll L.; Berzetei-Gurske I. P.; Polgar W. E.; Brandt S. R.; Adapa I. D.; Rodriguez L.; Schwartz R. W.; Haggart D.; O’Brien A.; White A.; Kennedy J. M.; Craymer K.; Farrington L.; Auh J. S. (1998) Standard binding and functional assays related to medications development division testing for potential cocaine and opiate narcotic treatment medications. NIDA Res. Monogr. 178, 440–466. [PubMed] [Google Scholar]
- King M. A.; Su W.; Nielan C. L.; Chang A. H.; Schutz J.; Schmidhammer H.; Pasternak G. W. (2003) 14-Methoxymetopon, a very potent mu-opioid receptor-selective analgesic with an unusual pharmacological profile. Eur. J. Pharmacol. 459, 203–209. [DOI] [PubMed] [Google Scholar]
- Wells J. L.; Bartlett J. L.; Ananthan S.; Bilsky E. J. (2001) In vivo pharmacological characterization of SoRI 9409, a nonpeptidic opioid mu-agonist/delta-antagonist that produces limited antinociceptive tolerance and attenuates morphine physical dependence. J. Pharmacol. Exp. Ther. 297, 597–605. [PubMed] [Google Scholar]
- Ananthan S.; Saini S. K.; Dersch C. M.; Xu H.; McGlinchey N.; Giuvelis D.; Bilsky E. J.; Rothman R. B. (2012) 14-Alkoxy- and 14-acyloxypyridomorphinans: mu agonist/delta antagonist opioid analgesics with diminished tolerance and dependence side effects. J. Med. Chem. 55, 8350–8363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chefer V. I.; Shippenberg T. S. (2009) Augmentation of morphine-induced sensitization but reduction in morphine tolerance and reward in delta-opioid receptor knockout mice. Neuropsychopharmacology 34, 887–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shippenberg T. S.; Chefer V. I.; Thompson A. C. (2009) Delta-opioid receptor antagonists prevent sensitization to the conditioned rewarding effects of morphine. Biol. Psychiatry 65, 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y. F.; McNutt R. W.; Chang K. J. (1998) Delta-opioid ligands reverse alfentanil-induced respiratory depression but not antinociception. J. Pharmacol. Exp. Ther. 287, 815–823. [PubMed] [Google Scholar]
- Foxx-Orenstein A. E.; Jin J. G.; Grider J. R. (1998) 5-HT4 receptor agonists and delta-opioid receptor antagonists act synergistically to stimulate colonic propulsion. Am. J. Physiol. 275, G979–983. [DOI] [PubMed] [Google Scholar]
- Koch T.; Hollt V. (2008) Role of receptor internalization in opioid tolerance and dependence. Pharmacol. Ther. 117, 199–206. [DOI] [PubMed] [Google Scholar]
- Cahill C. M.; Morinville A.; Lee M. C.; Vincent J. P.; Collier B.; Beaudet A. (2001) Prolonged morphine treatment targets delta opioid receptors to neuronal plasma membranes and enhances delta-mediated antinociception. J. Neurosci. 21, 7598–7607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I.; Jordan B. A.; Gupta A.; Trapaidze N.; Nagy V.; Devi L. A. (2000) Heterodimerization of mu and delta opioid receptors: A role in opiate synergy. J. Neurosci. 20, RC110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal K. M.; Bohn L. M. (2005) Mu opioid receptor regulation and opiate responsiveness. AAPS J. 7, E587–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Ferguson S. S.; Barak L. S.; Bodduluri S. R.; Laporte S. A.; Law P. Y.; Caron M. G. (1998) Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proc. Natl. Acad. Sci. U.S.A. 95, 7157–7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R.; Brooks C. L. 3rd; MacKerell A. D. Jr.; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; Caflisch A.; Caves L.; Cui Q.; Dinner A. R.; Feig M.; Fischer S.; Gao J.; Hodoscek M.; Im W.; Kuczera K.; Lazaridis T.; Ma J.; Ovchinnikov V.; Paci E.; Pastor R. W.; Post C. B.; Pu J. Z.; Schaefer M.; Tidor B.; Venable R. M.; Woodcock H. L.; Wu X.; Yang W.; York D. M.; Karplus M. (2009) CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell A. D. Jr.; Bashford D.; Bellott M.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T. K.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E.; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wirkiewicz-Kuczera J.; Yin D.; Karplus M. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- MacKerell A. D. Jr.; Feig M.; Brooks C. L. 3rd. (2004) Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25, 1400–1415. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Hatcher E.; Acharya C.; Kundu S.; Zhong S.; Shim J.; Darian E.; Guvench O.; Lopes P.; Vorobyov I.; MacKerell A. D. Jr. (2010) CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita Y.; Okamoto Y. (1999) Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314, 141–151. [Google Scholar]
- Zhou R.; Berne B. J.; Germain R. (2001) The free energy landscape for beta hairpin folding in explicit water. Proc. Natl. Acad. Sci. U.S.A. 98, 14931–14936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim J.; MacKerell A. D. Jr. (2011) Computational ligand-based rational design: Role of conformational sampling and force fields in model development. MedChemComm 2, 356–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery J. J.; Raymond T. J.; Giuvelis D.; Bidlack J. M.; Polt R.; Bilsky E. J. (2011) In vivo characterization of MMP-2200, a mixed delta/mu opioid agonist, in mice. J. Pharmacol. Exp. Ther. 336, 767–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyman J. S.; Koslo R. J.; Mosberg H. I.; Tallarida R. J.; Porreca F. (1986) Estimation of the affinity of naloxone at supraspinal and spinal opioid receptors in vivo: studies with receptor selective agonists. Life Sci. 39, 1795–1803. [DOI] [PubMed] [Google Scholar]
- Horan P.; Taylor J.; Yamamura H. I.; Porreca F. (1992) Extremely long-lasting antagonistic actions of nor-binaltorphimine (nor-BNI) in the mouse tail-flick test. J. Pharmacol. Exp. Ther. 260, 1237–1243. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.