

Abstract
Parkinson’s disease (PD) is characterized by the progressive degeneration of dopaminergic cells, which implicates a role of dopamine (DA) in the etiology of PD. A possible DA degradation pathway is the Fe(III)-catalyzed oxidation of DA by oxygen, which produces neuronal toxins as side products. We investigated how ATP, an abundant and ubiquitous molecule in cellular milieu, affects the catalytic oxidation reaction of dopamine. For the first time, a unique, highly stable DA–Fe(III)–ATP ternary complex was formed and characterized in vitro. ATP as a ligand shifts the catecholate–Fe(III) ligand metal charge transfer (LMCT) band to a longer wavelength and the redox potentials of both DA and the Fe(III) center in the ternary complex. Remarkably, the additional ligation by ATP was found to significantly reverse the catalytic effect of the Fe(III) center on the DA oxidation. The reversal is attributed to the full occupation of the Fe(III) coordination sites by ATP and DA, which blocks O2 from accessing the Fe(III) center and its further reaction with DA. The biological relevance of this complex is strongly implicated by the identification of the ternary complex in the substantia nigra of rat brain and its attenuation of cytotoxicity of the Fe(III)–DA complex. Since ATP deficiency accompanies PD and neurotoxin 1-methyl-4-phenylpyridinium (MPP+) induced PD, deficiency of ATP and the resultant impairment toward the inhibition of the Fe(III)-catalyzed DA oxidation may contribute to the pathogenesis of PD. Our finding provides new insight into the pathways of DA oxidation and its relationship with synaptic activity.
Keywords: ATP, dopamine, iron, Parkinson’s disease, catalytic oxidation, inhibition
Parkinson’s disease (PD), a leading neurodegenerative disorder, is characterized by the degeneration of dopaminergic neurons in the substantia nigra (SN),1 and the presence of Lewy bodies with abnormal cytoplasmic inclusions in the dying neurons.2 At present, the pathogenesis of PD is still not clear. A line of evidence indicates that the PD-afflicted brains are usually under an extensive and constant oxidative stress.3 Damages of dopamine (DA)-producing neurons in PD-afflicted brain strongly implicate a pivotal role of DA in the etiology of PD, most likely through the production of reactive oxygen species (ROS) or neurotoxins.4
DA is an important neurotransmitter. However, due to its relatively low oxidation potential, it is vulnerable to oxidation. DA oxidation by O2 leads to the production of a range of toxic species,5−10 which in turn contribute to excessive oxidative stress. Therefore, DA is tightly controlled in brain.11,12 It is stored in DA vesicles, and upon exocytosis the released DA molecules are rapidly captured by DA receptor molecules of postsynaptic neurons. Excess DA molecules either reenter the presynaptic neurons via endocytosis or are metabolized through enzymatic and nonenzymatic routes.13,14 It has been proven that a number of DA oxidation intermediates, such as 6-hydroxydopamine (6-OHDA),5 dopachrome (DACHR),9 and dopaminequinone (DAQ),10 are neurotoxic. The kinetics and intermediates/products of DA oxidation are dependent on the DA oxidation pathways.8 Therefore, it is vital to understand what factors influence the oxidation kinetics and pathways.
Iron is a essential metal in almost all living organisms.15 Iron, being highly redox active, is a potential toxin in the free form. Thus, iron is also tightly regulated in brain and usually stored in the redox inactive mineral form encapsulated by intracellular ferritin.16 However, the disruption of iron homeostasis has been linked in most of the neurodegenerative diseases.17 In PD, compared with age-matched disease-free subjects, the total iron concentration in the Parkinsonian SN is significantly increased.17 Interestingly, in vivo biochemical brain imaging has revealed that the increased level of iron is localized in the SN in the ferric form and coordinated to the phenolic oxygen of neuromelanin.17 Since neuromelanin is believed to be produced from oxidation of excess DA, and the loss of dopaminergic neurons in SN is accompanied by an accumulation of ferric ions, the synergy between DA and iron in the etiology of PD has become increasingly evident.18 Furthermore, iron-induced DA neurotoxicity has been confirmed by cell culture experiments, which revealed that cell viability decreases dramatically in the presence of DA and iron.19,20
In vitro, iron has been shown to catalyze the oxidation of DA by oxygen to generate H2O2, neurotoxic intermediates, and neuromelanin (Scheme 1).21−23 We should also mention that the tautomer of DAQ, quinomethide, can produce norepinephrine in the presence of Fe(III) and 3,4-dihydroxybenzaldehyde in the presence of H2O2.7,8 Thus far it is not entirely clear if the Fe(III)-catalyzed DA oxidation occurs in vivo. Any complexes formed between DA and Fe(III) in vivo would render credence to the involvement of rogue iron in DA oxidation/degradation. Particularly in line with this is a recent study showing that in healthy brain iron and DA coexist in the DA vesicles, most likely in the form of the iron–DA complex.24 That such a complex in DA vesicles does not undergo catalytic oxidation of DA (cf. Scheme 1) is probably due to the acidic interior of the vesicles.25 Once the DA vesicles are fused with the synapse, the complex released could interact with oxygen, which is abundant in cytoplasm. Since in vitro studies have shown that the iron-catalyzed DA oxidation is rather rapid, a largely overlooked question is how DA oxidation is prevented/inhibited during the exocytotic process to reduce any DA-induced cytotoxicity.
Scheme 1. Two Proposed Iron-Induced DA Oxidation Pathways.

The iron-catalyzed oxidation of DA by oxygen has been extensively studied in vitro, with the emphasis placed on the production of toxic species (e.g., 6-OHDA, DAQ, and DACHR; cf. Scheme 1). It is well accepted that the catalytic reaction starts with the formation of the Fe(III)–DA complex, an internal charge transfer complex.23 In the complex, the partial internal electron transfer from the ligands causes the iron center to behave as Fe(II) bound to the semiquinone radicals. Such a complex is capable of binding O2 to form a ternary complex within which the electron transfer between oxygen and the ligands (DA) is greatly accelerated.26
Because the formation of the ternary complex among DA, Fe(III), and O2 is the prerequisite for the Fe(III)-catalyzed reaction of DA,26 any ligand that can bind to the DA–Fe(III) complex should at least partially impede the access of O2 to the iron center. As a consequence, the catalytic oxidation kinetics may be significantly retarded and oxidation pathways altered. In cellular milieu, there exist a number of small ligands such as phosphate, citrate, AMP, ADP, and ATP.27 In particular, ATP, as a source of chemical energy essential for organisms, is abundant in cells (1–10 mM).28 Its relevance to PD is manifested by the significant depletion of ATP in neuronal cells of PD-afflicted brain.29 In recent years, many neuronal processes have been shown to occur with ATP as a cotransmitter in cell signaling.30 Interestingly, its interaction with Fe(III) and other important neurotransmitters has not been studied.
In this work, we investigated the formation of a ternary complex among DA, Fe(III), and ATP. Modulations of the redox potentials of DA and Fe(III) by the ATP ligation were studied by electrochemical techniques. Using HPLC, we separated and identified intermediates from the Fe(III)-catalyzed DA oxidation and examined the effect of the ATP ligation on the catalytic reaction kinetics. Our results revealed that the ATP ligation inhibits the catalytic oxidation of DA by blocking the oxygen access to the Fe(III) center. The variation in the kinetics is rationalized by and correlated with the redox potential changes and structural variations. More significantly, we confirmed the existence of this ternary complex in the SN tissues of rat brain. The biological relevance of the ternary complex is also demonstrated by the formation of the complex among DA, ATP, and the abundant iron storage protein ferritin (as the iron source) and the attenuation of cytotoxicity inherent in the Fe(III)–DA complex by ATP. The implication of the findings to the pathogenesis of PD is discussed.
Results
Formation and Characterization of the DA–Fe(III)–ATP Ternary Complex
The Fe(III)–DA complex exhibits a characteristic absorption peak at around 570 nm, which corresponds to the ligand-to-metal charge transfer (LMCT) transition.31 Usually a functional group on the catechol ring or an additional ligand attached to the metal center brings about a shift in the absorption peak.32 To verify the formation of the ternary complex among DA, Fe(III), and ATP, we recorded UV–vis spectra of DA/Fe(III) mixtures with and without ATP (Figure 1). With [DA]/[Fe(III)] ratio of 2/1 and in the absence of ATP, the characteristic absorption peak at 570 nm was observed. It is generally accepted that at neutral pH Fe(III)(DA)2 is the dominant species when Fe(III) is bound by excess DA.33 Decreasing the [DA]/[Fe(III)] ratio to 1/1 causes the characteristic peak height to decrease by ca. 40%, which is indicative of an attenuated LMCT band. The results indicate that at this ratio the composition of the complex is Fe(III)–DA. Interestingly, when a mixture with the [DA]/[Fe(III)]/[ATP] ratio of 2/1/1 was used, the characteristic LMCT peak shifts to 625 nm and the peak height is also decreased by ca. 40% with respect to that of the Fe(III)(DA)2 complex. The shift in the absorption peak suggests that a new ternary complex containing ATP has been formed, while the decrease in the peak height suggests that in the complex only one DA is bound to the Fe(III) center. Formation of the ternary complex is also supported by the observation of an essentially identical LMCT absorption peak at the [DA]/[Fe(III)]/[ATP] ratio of 1:1:1. Increasing [ATP] by more than one equivalent of Fe(III) did not alter the absorption peak. Our results thus confirmed that ATP only replaces one DA molecule in the Fe(III)(DA)2 complex. Given that the second formation constant between Fe(III) and DA is relatively large (pK2, 10.3534 or 13.433), the replacement of DA by ATP suggests that ATP has a strong affinity to the Fe(III)–DA complex. The formation of such a ternary complex is also highly specific to Fe(III), as other excess (2-fold or more) metal cations such as Mg2+ does not affect the formation of the ternary complex (data not shown). Interestingly, EDTA does not form a ternary complex with DA and Fe(III). Instead, EDTA sequesters Fe(III) from the DA–Fe–ATP complex, as indicated by the disappearance of Fe–DA LMCT band (cf. Figure S1, Supporting Information).
Figure 1.
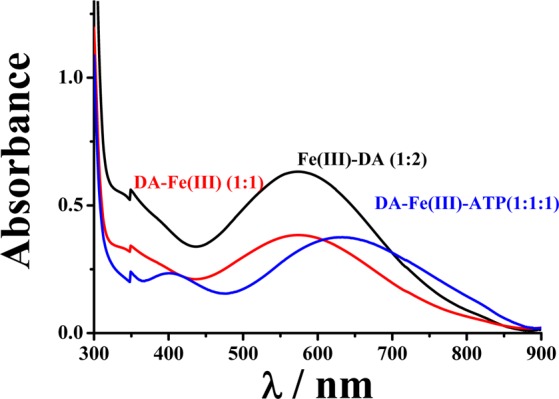
UV–vis spectra of mixtures of DA, Fe(III), and ATP at different concentration ratios: 2:1:0 (black), 1:1:0 (red), and 1:1:1 (blue) in which the Fe(III) concentration was maintained at 250 μM.
To further confirm the formation of the ternary complex, we conducted mass spectrometric measurements of a mixture of DA, Fe(III), and ATP. As shown in Figure 2, at a ratio of 1:1:1, the molecular ion corresponds to the ternary complex. The peak at 713.9942 Da is assigned to the +1 species containing a deprotonated DA, Fe(III), and ATP (i.e., the total mass corresponds to 151.08 (deprotonated DA – 2H+) + 55.93 (Fe) + 507.00 (ATP) or a theoretical mass of 713.99409). The mass peaks at 711.99896 and 714.99774 correlate well with the theoretical molecular weights (711.99877 and 714.99745) of the two major isotopic species of the complex with an error less than 0.5 ppm.
Figure 2.

ES–MS spectrum of the DA–Fe(III)–ATP ternary complex. M denotes the ternary complex.
Formation of the Ternary Complex Shifts the Redox Potentials of Both DA and the Fe(III) Center
A model complex of dioxygenase has been extensively studied previously, in which the covalency of the Fe(III)–catecholate bond is believed to affect its reactivity with oxygen.35 The redox potentials of the iron center and the catechol ligand and the LMCT absorption band have been correlated with the observed reactivity. In our study, the DA–Fe(III)–ATP ternary complex can be regarded as an analogue of the dioxygenase model complex since DA itself is also a catechol. In order to probe how ATP affects the iron-catalyzed oxidation of DA by oxygen, we conducted electrochemical investigations of the DA–Fe(III)–ATP and Fe(III)–DA complexes. Figure 3 shows representative cyclic voltammograms (CVs) of free DA, Fe(III)–DA and DA–Fe(III)–ATP. In the absence of Fe(III), the oxidation of DA to DAQ occurs at around 0.25 V (peak I). As depicted in Scheme 1, DAQ rapidly cyclizes to produce leuco-DACHR. Previous voltammetric studies of DA have assigned the two peaks (II′ and II) to the reduction and oxidation of DACHR and leuco-DACHR, respectively.36−38 In the mixture of DA and Fe(III), the same peaks remain unchanged. However, two shoulder peaks (peaks III and III′) appeared and these two new peaks are more obvious when the voltammetric scan was only conducted between 0.1 and −0.8 V. On the basis that peaks II and II′ were observed only when DA is initially oxidized, peaks III and III′ can be ascribed to the redox reaction of the Fe(III) center. Also noteworthy is that the DA oxidation peak in the Fe(III)–DA complex occurs at 0.37 V, a shift in the anodic (more positive) direction by 0.12 V with respect to the oxidation peak of free DA (i.e., DA is more difficult to oxidize in its complexed form). Notice that the small shoulder peak at 0.2 V can be attributed to the oxidation of DA uncomplexed by Fe(III). The shift in the peak potential is a result of the decrease in the electron density of DA upon partial distribution to the Fe(III) center. Concurrently, the redox potential of Fe(III) is shifted in the opposite direction to ca. −0.28 V. Compared to the redox potential of the Fe(III)/Fe(II) couple in their free forms, 0.575 V vs Ag/AgCl,39 the shift in the negative (cathodic) direction is significant. This indicates a dramatic attenuation in the oxidizing power of the Fe(III) center in the complex (i.e., Fe(III) has become more difficult to reduce). Ligation by additional strong ligands should alter the redox potentials of both the Fe(III) center and catecholate. Indeed, as shown in Figure 3A the additional ligation by ATP shifts the peak potentials of DA and Fe(III) to 0.44 and −0.52 V, respectively. Our voltammetric study therefore renders additional support for the DA–Fe(III)–ATP complex formation. Furthermore, it demonstrates that the redox property of the ternary complex is quite different from that of the Fe(III)–DA complex.
Figure 3.

Cyclic voltammograms of DA (top), Fe(III)–DA (middle), and DA–Fe(III)–ATP (bottom) acquired within the potential window between −0.9 and 0.7 V (A) and between −0.8 and 0.1 V (B). In panel (A), the initial potential was scanned from 0.1 V. The concentrations of DA, Fe(III), and ATP were all 250 μM. The scan rates are 50 mV s–1 in panel (A) and 10 mV s–1 in panel (B). The arrow indicates the initial scan direction. All measurements were performed in a nitrogen-purged glovebox.
Ligation of ATP Dramatically Diminishes the Activity of the Fe(III) Center in Catalyzing the DA Oxidation
DA can be oxidized by oxygen in solution, and in the presence of Fe(III) the oxidation is accelerated.8,26,40 It is interesting to investigate how ATP ligation affects the oxidation kinetics and pathways. Figure 4 displays typical chromatograms of solutions containing DA–Fe(III)–ATP, Fe(III)–DA, or DA that had been incubated at room temperature for 6 h. In all three cases, DACHR (retention time = 3.1 min) was detected, suggesting that DA is oxidized to DAQ, which readily cyclizes to DACHR.37 Also, in all cases, a peak at a retention time of 6.3 min emerged. This peak was verified to be 6-hydroxydopamine (6-OHDA), since it is the same as the peak corresponding to the elution of a 6-OHDA standard out of the chromatographic column. Notice that the 6-OHDA peak is smaller in the DA-only solution, which is understandable since the autoxidation of DA is a slower process.8,26,40 The 6-OHDA peak heights are comparable between the DA/Fe(III) and DA/Fe(III)/ATP mixtures. However, markedly different is the peak (retention time = 7.5 min) of 6-OHDAQ (6-hydroxydopaquinone; cf. Scheme 1), the oxidation product of 6-OHDA. Apparently, ligation of the Fe(III) center by ATP has reduced the amount of 6-OHDAQ produced or the total amount of the neurotoxin 6-OHDA produced in the DA/Fe(III)/ATP mixture is substantially less than that in the DA/Fe(III) mixture. We should also mention that the chromatograms in Figure 4 did not show peaks attributable to norepinephrine and 3,4-hydroxybenzaldehyde. Thus, the pathway through the quinomethide intermediate7 is probably not important under our experimental condition.
Figure 4.

Chromatograms showing the separation of DA from some of its oxidation products in different solutions. The Fe(III)-catalyzed oxidation reactions proceeded for 6 h in the absence and presence of ATP. The initial concentration of DA, Fe(III), and ATP are all 250 μM. Before separation, the sample mixture was diluted 10-fold with 0.1 M HCl.
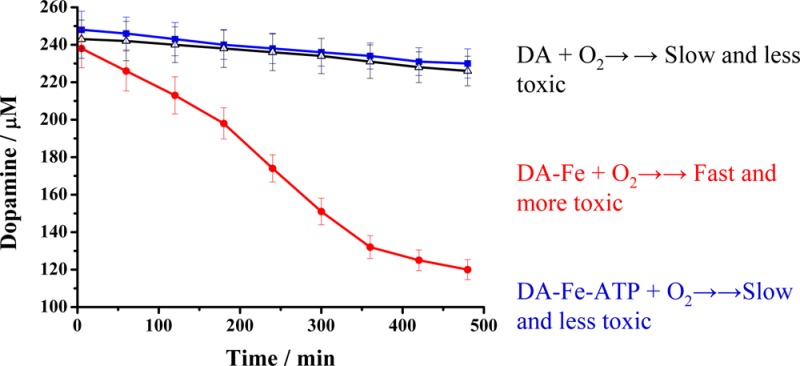
Since most of the DA oxidation products are not stable and their concentration variations do not reflect accurately the catalytic reaction kinetics, we monitored the consumption rate of DA to gauge the catalytic activities of the Fe(III) center in the Fe(III)–DA and DA–Fe(III)–ATP complexes. Figure 5 shows the DA concentration variation with reaction time under different conditions. In the presence of Fe(III), DA concentration decreases considerably faster. The addition of ATP counteracts the Fe(III) catalytic effect, making the DA consumption rate close to that of the DA autoxidation reaction. The initial oxidation rates were calculated to be 0.61 ± 0.05 nM–1 s–1 for DA alone (i.e., DA autoxidation), 3.64 ± 0.33 nM–1 s–1 for the Fe(III)-catalyzed oxidation of DA, and 0.60 ± 0.05 nM–1 s–1 with ATP added. Thus, the ligation of ATP decreases the Fe(III)-catalyzed DA oxidation rate by approximately 6 times. As shown in Scheme 1, the Fe(III)-catalyzed DA oxidation proceeds via formation of the 6-OHDA intermediate or via the DAQ route (though it has also been shown that 6-OHDA can be converted from DAQ).7,8,41 To probe if the ATP ligation affects the oxidation pathway, we plotted the ratio of the 6-OHDAQ/DA peak areas over a span of 7 h (Figure 5B). Clearly the additional ligation by ATP not only slows down the DA oxidation, but also diverts the reaction to the DAQ pathway. Such a diversion dramatically cuts down the amount of the highly neurotoxic 6-OHDA molecules. We also found ADP and AMP bind with DA-Fe(III) much more weakly and exhibit little inhibitive effect on the Fe(III)-catalyzed DA oxidation.
Figure 5.

(A) Variations of DA concentrations under different conditions: DA only (■), in the presence of Fe(III) (●), and in the presence of Fe(III) and ATP (△). (B) Plots of the 6-OHDAQ peak area over that of consumed DA in the presence of 250 μM Fe(III) (○) and 250 μM Fe(III) and 250 μM ATP (●).
Identification of the DA–Fe(III)–ATP Ternary Complex in SN of Rat Brain
Given the presence of ATP, Fe(III), and DA in the SN region and the stability of the ternary complex, we attempted to separate and identify this complex in tissues extracted from SN of rat brain samples. Figure 6 is a representative electrospray high-resolution mass spectrum wherein a relatively large peak at m/z 713.99548 Da was observed, in addition to a few other unidentified species. This peak and two other isotopic peaks shown in the inset are in excellent agreement with the theoretical molecular weights and distribution of isotopic species of the ternary complex. The mass uncertainty is less than 5 ppm (cf. Table 1), suggesting a high degree of accuracy in the peak assignment. Furthermore, spiking into the crude extract the ternary complex preformed in vitro augmented this mass spectrometric peak. In contrast, the extract from other brain tissues (e.g., the stratum region) shows no peaks corresponding to this ternary complex. Based on the relative peak size in the mass spectra between the standard sample and the tissue sample, the concentration of the complex in the SN tissue was estimated to be 65 ± 20 ng/g of wet tissue. We also did not observe the peaks corresponding to the ternary complex when EDTA was introduced to the tissue extract. Thus, EDTA, being an exceedingly strong iron chelator, is capable of disintegrating the ternary complex by forming the iron-EDTA complex. This experiment further confirms the existence of the ternary complex in the SN tissue.
Figure 6.
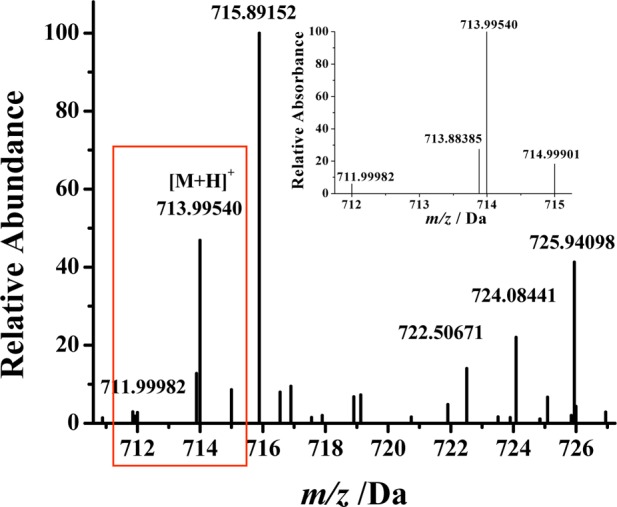
Electrospray high-resolution mass spectrum of tissues extracted from the SN region of rat brain showing the presence of the DA–Fe(III)–ATP ternary complex. The inset is an enlarged spectrum of three major isotopic peaks of the complex.
Table 1. Experimentally Measured and Theoretical Masses of the Three Major Isotopic Peaks of DA–Fe(III)–ATP.
|
m/z |
relative abundance |
|||
|---|---|---|---|---|
| theoretical | detected | error (ppm) | theoretical | detected |
| 711.99877 | 711.99982 | 1.47 | 6.3 | 5.9 |
| 713.99409 | 713.99540 | 1.83 | 100 | 100 |
| 714.99745 | 714.99901 | 2.18 | 19.5 | 18.3 |
Sequestration of Fe(III) by DA and ATP from Ferritin
The successful identification of the ternary complex in the SN tissue demonstrates that iron is biologically available from a donor molecule (e.g., iron-containing proteins such as transferrin and ferritin) to DA and ATP. On the basis that iron overloading is prevalent in PD patients and ferritin is a viable source of iron, we conducted a competitive binding experiment. Shown in Figure 7 is an overlap of the UV spectra of a mixture of DA, ATP, and ferritin at 0 h (blue curve) and 21 h (green curve) of incubation, together with a mixture whose ferritin had been removed after 21 h of incubation (red curve). Notice that no absorption peak is present between 550 and 800 nm at the beginning of the incubation. However, both the red and green curves exhibit an absorption peak at 625 nm that is characteristic of the DA–Fe(III)–ATP ternary complex (cf. black curve in Figure 7). This competitive binding experiment suggests that Fe(III) in the ternary complex is stabilized to an extent similar to that in ferritin and ferritin is likely a source of iron in vivo.
Figure 7.
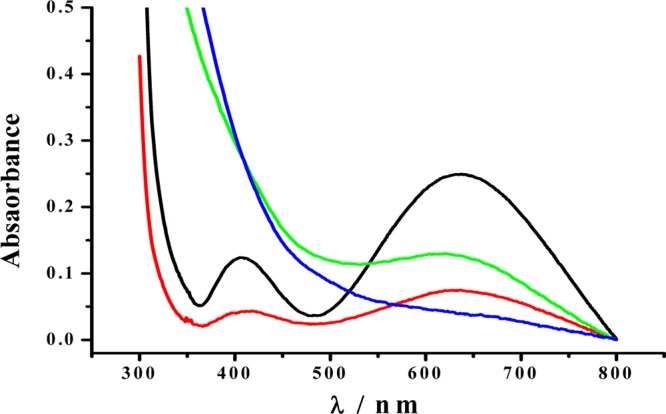
UV–vis spectra of 250 μM DA–Fe(III)–ATP (black curve), a mixture of 250 μM DA, 250 μM ATP, and 0.15 mg/mL ferritin incubated for 0 h (blue), the same ferritin-containing mixture after 21 h of incubation (green), and the same mixture incubated for 21 h followed by removal of ferritin (red).
ATP Ligation Decreases the Cytotoxicity of the Fe(III)–DA Complex
Shown in Figure 8 is the viability values of SH-SY5Y cells in the presence of DA, a mixture of DA and Fe(III), and a mixture of DA, Fe(III), and ATP. The viability value decreased to ca. 86% after exposure to DA for 12 h, a result expected from the cellular toxicity inherent in DA. Interestingly, the cytotoxicity of DA in the presence of Fe(III) is significantly aggravated (viability value further decreased to about 64%), due mainly to the acceleration of DA oxidation by Fe(III). However, inclusion of ATP in the mixture largely abolishes the Fe(III)-induced toxicity (viability value recovered to ca. 78%). The p value for the cell viabilities in the DA/Fe(III) and DA/Fe(III)/ATP mixtures is 0.002, indicating that the difference between the two mixtures is statistically significant. The trend is in line with the results from our kinetic studies of DA oxidation in the presence of these species (vide supra). Therefore, ligation of the Fe(III) center by ATP decreases the cytotoxicity of the Fe(III)–DA complex by suppressing the production of neurotoxic species such as 6-OHDA. The cell viabilities for the Fe–ATP complex and ATP alone are 91% and 94%, respectively. The data indicate that ATP or the Fe–ATP complex has little cytotoxicity to SH-SY5Y cells.
Figure 8.

Viabilities of SH-SY5Y cells in the presence of 250 μM DA, 250 μM DA and 50 μM Fe(III), and a mixture of 250 μM DA, 50 μM Fe(III), and 250 μM ATP after a 12 h incubation. The p value for the cell viabilities of the DA/Fe(III) mixture and the DA/Fe(III)/ATP mixture is 0.002, indicating that the difference between the two mixtures is statistically significant.
Discussion
ATP, Fe(III), and DA form a highly stable ternary complex, and the participation of ATP in the Fe(III) binding inhibits the Fe(III)-catalyzed O2 oxidation of DA. Alteration of the reaction pathway to attain a drastic inhibitory effect is noteworthy from our study. Interestingly, such an inhibitory effect is inconsistent with the prediction from the well documented behaviors of the model complexes of dioxygenase.35,42 It is believed that the increased covalency of the Fe(III)-catecholate bond or the Lewis acidity of Fe(III) increases the catalytic activity of the complex toward the oxidation of catechols involved.35,42 Despite these effects, the catalytic activity of Fe(III) does not increase. A possible explanation of this discrepancy is that the key step in the Fe(III)-catalyzed oxidation of DA is blocked due to the additional coordination by ATP. We propose a mechanism in Figure 9 for the Fe(III)-catalyzed oxidation of DA by O2. Instead of a catalytic mechanism based on an outer-sphere electron transfer reaction, the reaction undergoes an inner sphere electron transfer process. This is analogous to the mechanism of catalytic oxidation of catechols put forth in the literature.35,42 Specifically, Fe(III) rapidly binds DA to form the Fe(III)–DA or Fe(III)–(DA)2 complexes. At neutral pH, Fe(III) ligation by one or two DA molecules is dependent on the DA concentration. In both complexes, the six coordination sites of the Fe(III) center are not completely occupied. Other weak ligands such as H2O molecules can occupy the otherwise empty coordination sites (typically at the distal sites). Consequently, initiation of the DA oxidation begins with the approach of O2 to the empty or weakly occupied coordination sites. The partial electron transfer (distribution) from DA to the Fe(III) center makes the iron center behave similarly to Fe(II). As a result, there is an enhanced propensity for O2 to approach the Fe(III) center ligated by DA. An intermediate of DA–Fe(III)–O2 is formed, and through the metal-centered bridge and the close distance, two electrons from DA can be readily transferred to O2. The intermediate subsequently dissociates into hydrogen peroxide, DAQ, and Fe(III). A side path of the oxidation occurs through the nucleophilic addition of water or hydroxide on the phenol ring of the DA ligand.7 This explains the increased production of 6-OHDA, which is facilitated due probably to the change in the electron density on the DA moiety. ATP is known to be a tetradentate ligand. When ATP is present, formation of the DA–Fe(III)–ATP ternary complex results in the replacement of weak ligands in Fe(III)–DA (e.g., H2O) or Fe(III)–(DA)2 (i.e., the second DA molecule). Consequently, a close shell ternary complex of DA–Fe(III)–ATP is produced. When all the coordination sites are occupied by strong coordination ligands, little space avails O2 in solution. We note that another tetradentate ligand, nitrilotriacetic acid (NTA), which is known to form a close shell ternary complex with Fe(III) and catechols, has been shown to have the same inhibitory effect.43
Figure 9.

Proposed mechanism of the iron-catalyzed DA oxidation by O2.
The fact that DA and ATP together can seize Fe(III) from the iron storage protein ferritin is indicative of the high stability and demonstrates the biological relevance of the ternary complex. Iron overloading has been closely related to aging and an increase in ferritin concentration has been linked to PD progression.17,44 Thus, ferritin is at least a likely source of iron in the formation of this ternary complex. The abundance of this complex may also increase with aging. Even though both SN and stratum contain high concentrations of DA, the fact that the ternary complex was only detected in SN suggests that the bioavailability of iron in SN is likely to be greater than that in the stratum region.45,46
The discovery of the ternary complex in subtantia nigra rather than in stratum of rat brain sheds light on how an oxidation-prone neurotransmitter (e.g., DA and norepinephrine) can be kept in a relatively inactive form in the presence of O2 so that neurotoxic species/intermediates are maintained at a tolerable level or stabilized before the metabolism of excess DA takes effect. Based on our findings and the proposed mechanism (cf. Figure 9), we believe that, at the synaptic cleft, formation of the ATP-containing ternary complex would retard the oxidation of excess DA molecules (uncaptured by the DA receptor or not internalized via endocytosis) by the Fe(III)-catalyzed oxidation. As mentioned in the Introduction, it has been reported that iron is complexed by at least some DA molecules in vesicles.24 Thus, exposure of the complex to O2 in the synaptic cleft could accelerate the degradation of DA and the production of reactive and toxic species. The number of DA molecules bound by iron may increase significantly in aging brains wherein iron overloading is commonplace. Thus, it appears that releasing ATP to produce the DA–Fe(III)–ATP complex might be a process to inhibit the production of a large amount of DA- or norepinephrine-derived toxins. Our kinetic measurements clearly show that, in the presence of ATP, the Fe(III)-catalyzed DA oxidation is decelerated to a rate highly comparable to that of the DA autoxidation. Such retardation of the reaction provides ample time for the clearance of excess DA molecules via enzymatic and nonenzymatic pathways.7 We should mention that releasing ATP as a cotransmitter of norepinephrine (another monoamine neurotransmitter susceptible to the Fe(III)-catalyzed oxidation26) has been found in the locus coeruleus region.30
Our finding also has a significant implication to the pathogenesis of PD. In addition to the common hallmarks of PD,47,48 ATP deficiency is closely linked to the PD symptom.29 In animal models, the connection of ATP deficiency to PD is established by the fact that PD-incurring agent such as the neurotoxin 1-methyl-4-phenylpyridinium (MPP+) leads to the development of PD and ATP deficiency.49,50 The ATP deficiency causes the Fe(III) center to expose the Fe(III)–DA or Fe(III)–(DA)2 complex to O2, which drastically accelerates the DA oxidation. Conversion of DA into its oxidation intermediates and products could compromise the signaling of dopaminergic cells. More adversely, some of the DA oxidation intermediates/products (e.g., 6-OHDA) are highly reactive and neurotoxic. These species can lead to a dramatic loss of the dopaminergic cells.51,52 It is therefore possible that the toxic effect inherent in the iron-catalyzed dopamine oxidation reaction contributes to the loss of dopaminergic cells in PD in a significant way.
Conclusions
We, for the first time, synthesized and characterized in vitro the DA–Fe(III)–ATP ternary complex. Using high-resolution mass spectrometry, we also identified in the extract of substantia nigra of rat brain tissues this ternary complex, which is absent in the stratum region. The spectroscopic and redox properties of the complex are distinctively different from those of the Fe(III)–DA complex. The LMCT band of the ternary complex shifts to a longer wavelength, whereas the redox potential of DA in the complex is positively shifted (i.e., more difficult to oxidize) and that of the Fe(III) center is negatively shifted (i.e., more difficult to reduce). As a consequence of the stabilization of the Fe(III) center by ATP, the catalytic activity of iron is largely annihilated. Retardation of the Fe(III)-catalyzed DA oxidation by O2 helps curb the production of neurotoxic species and oxidative stress associated with iron overloading. Our finding may provide significant insight into how DA, an essential but potentially toxic neurotransmitter, is kept in a relatively unreactive form. Due possibly to the decline in ATP production and iron overloading associated with aging, the catalytic oxidation of DA by O2 is accelerated and the amount of neurotoxins accumulates. This eventually leads to the loss of dopaminergic neurons and contributes to the development of PD.
Methods
Materials
Dopamine hydrochloride and ferritin were obtained from Sigma-Aldrich (St. Louis, MO). Ferric nitrate nonahydrate (Fe(NO3)3·9H2O), sodium hydroxide, trisaminomethane (Tris), NaNO3, acetonitrile, trifluoroacetic acid, 2-[4-(2-hydroxyethyl)-1-piperazine]ethanesulfonic acid (HEPES), and adenosine 5′-triphosphate disodium salt trihydrate (ATP) were of analytical grade and acquired from Thermo Fisher Scientific Inc. (Pittsburgh, PA). SH-SY5Y cells (human neuroblastoma) were purchased from American Type Culture Collection Inc. (Manassas, VA) and fetal bovine serum (FBS) was from Omega (Tarzana, CA). Both Dulbecco’s modified Eagle’s medium (DMEM) and Ham’s F12 were purchased from Mediatech Inc. (Manassas, VA). Ultra filters of 100 kDa molar mass cutoff were obtained from Millipore (Billerica, MA). The stock solution of 10 mM Fe(III) was prepared by dissolving an appropriate amount of Fe(NO3)3·9H2O in 5 mM H2SO4. The stock solution of 10 mM DA was prepared with deionized water. Rat brain samples were kindly provided by Dr. M. Harrington at Huntington Medical Research Institute (Pasadena, CA).
UV–Vis Spectroscopic Measurements
The absorption spectra of the Fe(III)–DA and DA–Fe(III)–ATP complexes were collected in Tris buffer (50 mM, pH 7.2) at 25 ± 1 °C on a Cary 100 Bio UV–vis spectrophotometer (Agilent Technologies, Santa Clara, CA) in a 1 cm quartz spectrophotometer cell (Starna Cells Inc., Atascadero, CA). The absorption peak at 570 nm, characteristic of the phenolate (π) → Fe(III) (dπ*) ligand-to-metal charge transfer (LMCT) transition,8,31 was used to assess the stability of the complexes in the presence of oxygen.
Electrochemical Measurements
A CHI 411 electrochemical workstation (CH Instruments, Austin, TX) was used for the electrochemical measurements. The three-electrode system is composed of a glassy carbon disk electrode with a diameter of 3 mm, a platinum wire auxiliary electrode, and a Ag/AgCl reference electrode. The glassy carbon electrode was polished with diamond pastes down to 3 μm and then alumina slurry down to 0.3 μm (Buehler, Lake Bluff, IL). HEPES buffer (50 mM, pH 7.2) containing 0.1 M NaNO3 was used as the electrolyte solution. The three-electrode cell was housed in a nitrogen-purged glovebox (PlasLab, Lansing, MI). The O2 content in solution, prepared inside this glovebox, was measured to be less than 1 μM by a dissolved oxygen membrane electrode (080515MD, Thermo Fisher Scientific, Pittsburgh, PA).
HPLC Assays
DA and DA oxidation intermediates/products were separated and their concentration variations were monitored at 280 nm by an HPLC equipped with a variable-wavelength UV detector (Waters Delta-600). The analysis was carried out with a C18 column (2.0 mm × 100 mm, 5 μm, Varian Inc.) at a flow rate of 0.2 mL min–1 with a mobile phase containing 0.5 mM sodium octane sulfonate in 5% acetonitrile. The mobile phase pH was adjusted to 3.0 with 6 M ammonia and 150 mM formic acid. The oxidation products were identified by electrospray-mass spectrometry (ES-MS) on a Thermal Fisher LCQ unit (San Jose, CA) and by comparing their retention times with those of standards.8
Extracting the DA–Fe(III)–ATP Ternary Complex from Rat Brain Tissues
Brain tissues (from SN or stratum; pooled from several rat brains to ca. 0.15 g) were homogenized in 1.0 mL of 50 mM ammonium acetate buffer (pH = 7.0) at 4 °C. The final mixture was centrifuged at 15 000 rpm for 10 min at 4 °C. The supernatant was then filtered through a 0.45 μm filter. We further purified the filtrate using HPLC on a reverse phase C18 column (250 mm × 4.6 mm, i.d., 5 μm; Phenomenex Inc., Torrance, CA) at a flow rate of 0.8 mL min–1 with an isocratic mobile phase of 20% acetonitrile and 80% 50 mM ammonium acetate buffer (pH = 7.0).
Mass Spectrometric Measurements
To form the ternary complex, ATP and DA (100 μM each) were dissolved in N2-saturated water solutions with pH adjusted to 7.0 using 1 M NaOH. Aliquots of Fe(NO3)3·9H2O were then added into the DA/ATP solution at a final concentration of 100 μM and the pH was again adjusted to 7.0. A Thermo Fisher Scientific Exactive mass spectrometer (San Jose, CA) was used for high-resolution analysis of the ternary complex. The mixture was diluted 10-fold with acetonitrile before being directly infused at 10 μL s–1 to the MS operated in the positive ion mode. For detecting the ternary complex in rat brain extracts, fractions collected from the HPLC column were freeze-dried and redissolved in 100 μL of acetonitrile/water (7/3, v/v) before being analyzed with MS.
Sequestration of Fe(III) in Ferritin by DA and ATP
DA, ATP, and ferritin stock solutions were prepared with degassed Tris buffer and mixed at different concentration ratios. After purging with N2, UV–vis spectra were recorded at different incubation times. Spectra were also collected from a filtrate collected after passing the mixture through an ultra filter (UFC510024, Milipore) that was under centrifugation at 10 000 rpm for 5 min.
Cell Culture and Cytotoxicity Assay
SH-SY5Y cells were cultured in 45% DMEM/45% Ham’s F12 supplemented with 10% FBS and 1% penicillin/streptomycin. The cultured cells were then transferred to a sterile 96-well plate with approximately 20 000 cells in each well. These cells were acclimatized overnight in the DMEM/F12 medium containing 5% FBS and 1% penicillin/streptomycin in a humidified incubator under 5% CO2 at 37 °C. Solutions of DA, Fe(III)–DA, or DA–Fe(III)–ATP were allowed to interact with the SH-SY5Y cells for 12 h. Viability of cells exposed to each solution was determined with the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT) assay (EMD Inc., Gibbstown, NJ). In brief, MTT was first dissolved in water to 5 mg/mL. Media from the wells in each plate were replaced by the mixture of 10% MTT (5 mg/mL) in media (v/v), and the well contents were subsequently incubated for 4 h. After removal of the MTT-containing media, 150 μL of dimethyl sulfoxide was added into each well to dissolve the formazan precipitate. UV–vis absorption at 490 nm in each well was recorded with a plate reader (Biotek, Winooski, VT). For each solution, MTT assays in five separate wells were conducted, and the final viability, represented as a percentage of the untreated control, is the average from MTT assays conducted on three different days.
Acknowledgments
We thank Dr. Michael Harrington at Huntington Medical Research Institutes and Dr. Alicia Izquierdo at California State University Los Angeles for providing rat brain tissues.
Glossary
Abbreviations
- PD
Parkinson’s disease
- DA
dopamine
- MPP+
1-methyl-4-phenylpyridinium
- MTT
3,[4,5-dimethylthiazol-2-yl]-2,5-diphenyl- tetrazolium bromide
- SN
substantia nigra
- HEPES
2-[4-(2-hydroxyethyl)-1-piperazine]ethanesulfonic acid
- ATP
adenosine 5′-triphosphate
- ES-MS
electrospray-mass spectrometry
- LMCT
ligand metal charge transfer
- 6-OHDA
6-hydroxydopamine
- DACHR
dopachrome
- DAQ
dopaminequinone
Supporting Information Available
Additional figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
D.J. designed the experiments and wrote parts of the paper. S.S. extracted the ternary complex from substantia nigra and performed the mass spectrometric identification. L.Z. conducted the toxicity experiment. L.L. performed the UV–vis, electrochemical, and kinetic studies. B.D. performed sequestration of Fe(III) in ferritin by DA and ATP. B.Z. assisted some of the mass spectrometric measurements. G.Y. developed the separation method. F.Z. oversaw the entire research and revised the paper.
This work was supported by the National Institutes of Health (SC1NS070155-01), a National Science Foundation (NSF) grant (No. 1112105), and the NSF-CREST Program at California State University, Los Angeles (NSF HRD-0931421).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Moore D. J.; West A. B.; Dawson V. L.; Dawson T. M. (2005) Molecular pathophysiology of Parkinson′s disease. Annu. Rev. Neurosci. 28, 57–87. [DOI] [PubMed] [Google Scholar]
- Spillantini M. G.; Schmidt M. L.; Lee V. M.; Trojanowski J. Q.; Jakes R.; Goedert M. (1997) a-Synuclein in Lewy bodies. Nature 388, 839–840. [DOI] [PubMed] [Google Scholar]
- Malkus K. A.; Tsika E.; Ischiropoulos H. (2009) Oxidative modifications, mitochondrial dysfunction, and impaired protein degradation in Parkinson′s disease: how neurons are lost in the Bermuda triangle. Mol. Neurodegener. 4, 24–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y.; Umegaki H.; Wang X.; Abe R.; Roth G. S. (1998) Dopamine induces apoptosis through an oxidation-involved SAPK/JNK activation pathway. J. Biol. Chem. 273, 3756–3764. [DOI] [PubMed] [Google Scholar]
- Irwin I., and Langston J. W. (1995) Endogenous toxins as potential etiologic agents in Parkinson′s disease, pp 153–201, Marcel Dekker, New York. [Google Scholar]
- Graham D. G. (1978) Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol. Pharmacol. 14, 633–643. [PubMed] [Google Scholar]
- Napolitano A.; Pezzella A.; Prota G. (1999) New reaction pathways of dopamine under oxidative stress conditions: nonenzymatic iron-assisted conversion to norepinephrine and the neurotoxins 6-hydroxydopamine and 6,7-dihydroxytetrahydroisoquinoline. Chem. Res. Toxicol. 12, 1090–1097. [DOI] [PubMed] [Google Scholar]
- Pezzella A.; d’Ischia M.; Napolitano A.; Misuraca G.; Prota G. (1997) Iron-mediated generation of the neurotoxin 6-hydroxydopamine quinone by reaction of fatty acid hydroperoxides with dopamine: a possible contributory mechanism for neuronal degeneration in Parkinson′s disease. J. Med. Chem. 40, 2211–2216. [DOI] [PubMed] [Google Scholar]
- Zoccarato F.; Toscano P.; Alexandre A. (2005) Dopamine-derived dopaminochrome promotes H2O2 release at mitochondrial complex I: stimulation by rotenone, control by Ca2+, and relevance to Parkinson disease. J. Biol. Chem. 280, 15587–15594. [DOI] [PubMed] [Google Scholar]
- Sulzer D.; Zecca L. (2000) Intraneuronal dopamine-quinone synthesis: a review. Neurotoxic. Res. 1, 181–195. [DOI] [PubMed] [Google Scholar]
- Kuhar M. J., Minnema K., and Muly E. C. (2006) Catecholamines. In Basic Neurochemistry: Molecular, Cellular, and Medical Aspects (Siegel G. J., Albers R. W., Brady S. T., and Price D. L., Eds.), Elsevier Academic Press, New York. [Google Scholar]
- Clark D. L., Boutros N. N., and Mendez M. F. (2005) The brain and behavior: An introduction to behavioval neuroanatomy, p 255, Cambridge University Press, New York. [Google Scholar]
- Elsworth J. D.; Roth R. H. (1997) Dopamine synthesis, uptake, metabolism, and receptors: relevance to gene therapy of Parkinson′s disease. Exp. Neurol. 144, 4–9. [DOI] [PubMed] [Google Scholar]
- Erickson J. D.; Eiden L. E.; Hoffman B. J. (1992) Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc. Natl. Acad. Sci. U.S.A. 89, 10993–10997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKie A. T.; Barrow D.; Latunde-Dada G. O.; Rolfs A.; Sager G.; Mudaly E.; Mudaly M.; Richardson C.; Barlow D.; Bomford A.; Peters T. J.; Raja K. B.; Shirali S.; Hediger M. A.; Farzaneh F.; Simpson R. J. (2001) An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 291, 1755–1759. [DOI] [PubMed] [Google Scholar]
- Ward R. J., and Crichton R. R. (2006) Iron and Its Role in Neurodegenerative Diseases. In Neurodegenertive Diseases and Metal Ions (Sigel A.; Sigel H.; Sigel R. K. O., Eds.), Vol. 4, John Wiley & Sons Ltd, West Sussex. [Google Scholar]
- Götz M. E.; Double K.; Gerlach M.; Youdim M. B.; Riederer P. (2004) The relevance of iron in the pathogenesis of Parkinson′s disease. Ann. N.Y. Acad. Sci. 1012, 193–208. [DOI] [PubMed] [Google Scholar]
- Golts N.; Snyder H.; Frasier M.; Theisler C.; Choi P.; Wolozin B. (2002) Magnesium inhibits spontaneous and iron-induced aggregation of α-synuclein. J. Biol. Chem. 277, 16116–16123. [DOI] [PubMed] [Google Scholar]
- Paris I.; Martinez-Alvarado P.; Crdenas S.; Perez-Pastene C.; Graumann R.; Fuentes P.; Olea-Azar C.; Caviedes P.; Segura-Aguilar J. (2005) Dopamine-dependent iron toxicity in cells derived from rat hypothalamus. Chem. Res. Toxicol. 18, 415–419. [DOI] [PubMed] [Google Scholar]
- Velez-Pardo C.; Jimenez Del Rio M.; Verschueren H.; Ebinger G.; Vauquelin G. (1997) Dopamine and iron induce apoptosis in PC12 cells. Pharmacol. Toxicol. 80, 76–84. [DOI] [PubMed] [Google Scholar]
- Hermida-Ameijeiras A.; Méndez-Alvarez E.; Sánchez-Iglesias S.; Sanmartín-Suárez C.; Soto-Otero R. (2004) Autoxidation and MAO-mediated metabolism of dopamine as a potential cause of oxidative stress: role of ferrous and ferric ions. Neurochem. Int. 45, 103–116. [DOI] [PubMed] [Google Scholar]
- Linert W.; Herlinger E.; Jameson R. F.; Kienzl E.; Jellinger K.; Youdim M. B. H. (1996) Dopamine, 6-hydroxydopamine, iron, and dioxygen - their mutual interactions and possible implication in the development of Parkinson′ s disease. Biochim. Biophys. Acta 1316, 160–168. [DOI] [PubMed] [Google Scholar]
- Linert W.; Jameson G. N. L. (2000) Redox reactions of neurotransmitters possibly involved in the progression of Parkinson′s Disease. J. Inorg. Biochem. 79, 319–326. [DOI] [PubMed] [Google Scholar]
- Ortega R.; Cloetens P.; Devès G.; Carmona A.; Bohic S. (2007) Iron storage within dopamine neurovesicles revealed by chemical nano-imaging. PLoS One 2, e925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu A.; Wersinger C.; Vernier P. (2004) Does a-synuclein modulate dopaminergic synaptic content and tone at the synapse?. FASEB J. 18, 637–647. [DOI] [PubMed] [Google Scholar]
- Linert W., Jameson G. N. L., Jameson R. F., and Jellinger K. A. (2006) The Chemical Interplay between Catecholamines and Metal Ions in Neurological Diseases. In Metal Ions In Life Sciences (Sigel A., Sigel H., and Sigel R. K. O., Eds.), Vol. 4, pp 281–315, John Wiley & Sons Ltd, West Sussex. [Google Scholar]
- Kakhlon O.; Cabantchik Z. I. (2002) The labile iron pool: characterization, measurement, and participation in cellular processes. Free Radical Biol. Med. 33, 1037–1046. [DOI] [PubMed] [Google Scholar]
- Beis I.; Newsholme E. A. (1975) The contents of adenine nucleotides, phosphagens and some glycolytic intermediates in resting muscles from vertebrates and invertebrates. Biochem. J. 152, 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebadi M., and Pfeiffer R. F. (2005) Parkinson′s Disease, CRC Press, Boca Raton, FL. [Google Scholar]
- Burnstock G. (2007) Physiology and pathophysiology of purinergic neurotransmission. Physiol. Rev. 87, 659–797. [DOI] [PubMed] [Google Scholar]
- Zirong D.; Bhattacharya S.; McCusker J. K.; Hagen P. M.; Hendrickson D. N.; Pierpont C. G. (1992) Studies on bis(catecholato)iron(III) complexes. structure and bonding in members of the Fe(bpy) (Cl4,SQ) (Cl4,Cat)/Fe(bpy) (Cl4Cat)2- redox couple. Inorg. Chem. 31, 870–877. [Google Scholar]
- Arreguin S.; Nelson P.; Padway S.; Shirazi M.; Pierpont C. (2009) Dopamine complexes of iron in the etiology and pathogenesis of Parkinson′s disease. J. Inorg. Biochem. 103, 87–93. [DOI] [PubMed] [Google Scholar]
- Charkoudian L. K.; Franz K. J. (2006) Fe(III)-coordination properties of neuromelanin components: 5,6-dihydroxyindole and 5,6-dihydroxyindole-2-carboxylic Acid. Inorg. Chem. 45, 3657–3664. [DOI] [PubMed] [Google Scholar]
- Abd El Wahed M. G.; Ayad M. (1984) Stability constants of Cu2+, Fe3+, Zr4+ chelates of ampicillin, dopamine and α-methyl L-dopa in aqueous medium. Anal. Lett. 17, 205–216. [Google Scholar]
- Costas M.; Mehn M. P.; Jensen M. P.; Que L. Jr. (2004) Dioxygen activation at mononuclear nonheme iron active sites: enzymes, models, and intermediates. Chem. Rev. 104, 939–986. [DOI] [PubMed] [Google Scholar]
- Hawley M. D.; Tatawawadi S. V.; Piekarski S.; Adarns R. N. (1967) Electrochemical studies of the oxidation pathways of catecholamines. J. Am. Chem. Soc. 89, 447–450. [DOI] [PubMed] [Google Scholar]
- Young T. E.; Babbitt B. W. (1983) Electrochemical study of the oxidation of a-methyldopamine, a-methylnoradrenaline, and dopamine. J. Org. Chem. 48, 562–566. [Google Scholar]
- Chen S.-M.; Peng K.-T. (2003) The electrochemical properties of dopamine, epinephrine, norepinephrine, and their electrocatalytic reactions on cobalt(II) hexacyanoferrate films. J. Electroanal. Chem. 547, 179–189. [Google Scholar]
- Bard A. J. and Faulkner L. R. (2001) Electrochemical Methods. Fundamentals and Applications, John Wiley and Sons, New York. [Google Scholar]
- Shen X.-M.; Dryhurst G. (1998) Iron- and manganese-catalyzed autoxidation of dopamine in the presence of L-Cysteine: possible insights into iron- and manganese-mediated dopaminergic neurotoxicity. Chem. Res. Toxicol. 11, 824–837. [DOI] [PubMed] [Google Scholar]
- Napolitano A.; Pezzella A.; Prota G. (1999) 6,7-Dihydroxy-l,2,3,4-tetrahydroisoquinoline formation by iron mediated dopamine oxidation: a novel route to endogenous neurotoxins under oxidative stress conditions. Tetrahedron lett. 40, 2833–2836. [Google Scholar]
- Abu-Omar M. M.; Loaiza A.; Hontzeas N. (2005) Reaction mechanisms of mononuclear non-heme iron oxygenases. Chem. Rev. 105, 2227–2252. [DOI] [PubMed] [Google Scholar]
- Cox D. D.; Que L. Jr. (1988) Functional models for catechol 1,2-dioxygenase: the role of the iron(III) center. J. Am. Chem. Soc. 110, 8085–8092. [Google Scholar]
- Snyder A. M.; Connor J. R. (2009) Iron, the substantia nigra and related neurological disorders. Biochim. Biophys. Acta 1790, 606–614. [DOI] [PubMed] [Google Scholar]
- Hill J. M. (1988) The distribution of iron in the brain. In Brain Iron: Neurochemistry and Behavioural Aspects (Youdim M. B. H., Ed.), pp 1–24, Taylor and Francis, London. [Google Scholar]
- Erikson K. M.; Pinero D. J.; Connor J. R.; Beard J. L. (1997) Regional brain iron, ferritin and transferrin concentrations during iron deficiency and iron repletion in developing rats. J. Nutr. 127, 2030–2038. [DOI] [PubMed] [Google Scholar]
- Singleton A. B.; Farrer M.; Johnson J.; Singleton A.; Hague S.; Kachergus J.; Hulihan M.; Peuralinna T.; Dutra A.; Nussbaum R.; Lincoln S.; Crawley A.; Hanson M.; Maraganore D.; Adler C.; Cookson M. R.; Muenter M.; Baptista M.; Miller D.; Blancato J.; Hardy J.; Gwinn-Hardy K. (2003) α-Synuclein Locus Triplication Causes Parkinson′s Disease. Science 302, 841. [DOI] [PubMed] [Google Scholar]
- Spillantini M. G.; Crowther R. A.; Jakes R.; Hasegawa M.; Goedert M. (1998) α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson′s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. U.S.A. 95, 6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K.; Bindokas V. P.; Marks J. D.; Wright D. A.; Frim D. M.; Miller R. J.; Kang U. J. (2000) The selective toxicity of 1-methyl-4-phenylpyridinium to dopaminergic neurons: the role of mitochondrial complex I and reactive oxygen species revisited. Mol. Pharmacol. 58, 271–278. [DOI] [PubMed] [Google Scholar]
- Mizuno Y.; Suzuki K.; Sone N. (1990) Inhibition of ATP synthesis by 1-methyl-4-phenylpyridinium ion (MPP+) in mouse brain in vitro and in vivo. Adv. Neurol. 53, 197–200. [PubMed] [Google Scholar]
- Dooley C. T.; Li L.; Misler J. A.; Thompson J. H. (2012) Toxicity of 6-hydroxydopamine: live cell imaging of cytoplasmic redox flux. Cell Biol. Toxicol. 28, 89–101. [DOI] [PubMed] [Google Scholar]
- Hanrott K.; Gudmunsen L.; O’Neill M. J.; Wonnacott S. (2006) 6-Hydroxydopamine-induced apoptosis is mediated via extracellular auto-oxidation and caspase 3-dependent activation of protein kinase Cδ. J. Biol. Chem. 281, 5373–5382. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.