

Abstract
Monoacylglycerol lipase (MAGL) is a principal metabolic enzyme responsible for hydrolyzing the endogenous cannabinoid (endocannabinoid) 2-arachidonoylglycerol (2-AG). Selective inhibitors of MAGL offer valuable probes to further understand the enzyme’s function in biological systems and may lead to drugs for treating a variety of diseases, including psychiatric disorders, neuroinflammation, and pain. N-Hydroxysuccinimidyl (NHS) carbamates have recently been identified as a promising class of serine hydrolase inhibitors that shows minimal cross-reactivity with other proteins in the proteome. Here, we explore NHS carbamates more broadly and demonstrate their potential as inhibitors of endocannabinoid hydrolases and additional enzymes from the serine hydrolase class. We extensively characterize an NHS carbamate 1a (MJN110) as a potent, selective, and in-vivo-active MAGL inhibitor. Finally, we demonstrate that MJN110 alleviates mechanical allodynia in a rat model of diabetic neuropathy, marking NHS carbamates as a promising class of MAGL inhibitors.
Keywords: activity-based protein profiling, 2-arachidonoyl glycerol, carbamate, endocannabinoid, inhibitor, click chemistry, monoacylglycerol lipase
The serine hydrolases monoacylglycerol lipase (MAGL) and fatty acid amide hydrolase (FAAH) are responsible for degrading 2-arachidonoyl glycerol (2-AG) and N-arachidonoyl ethanolamine (anandamide or AEA), respectively, which are endogenous agonists for the cannabinoid receptors, CB1 and CB2.1 The endocannabinoid system modulates a diverse set of physiological processes, including pain, inflammation, sleep, and appetite, and drugs that augment cannabinoid signaling may have therapeutic value.2 Δ9-Tetrahydrocannabinol (THC) and other direct CB1 agonists, for instance, possess medicinally beneficial properties; however, their adverse effects on motor control and cognition have encouraged the pursuit of alternative strategies to enhance endocannabinoid function. One potentially attractive strategy is to inhibit FAAH or MAGL, which elevates brain AEA and 2-AG levels, respectively, and has been shown to produce a subset of the behavioral effects observed with direct CB1 agonists.3 As such, endocannabinoid hydrolase inhibitors have the potential to reduce pain and (neuro)inflammatory responses,3b,3d,4 as well as improve neuropsychiatric function,3a,5 without substantial motor or cognitive impairment.
While FAAH inhibitors have been described for more than a decade6 and include compounds, such as PF-04457845,7 that have advanced to clinical trials, selective chemical probes for MAGL have only recently been reported.2b,8 Genetic and pharmacologic disruption of MAGL has implicated this enzyme in numerous (patho)physiological processes, such as pain,3c,3d,4a,9 neuropsychiatric disorders,5b,10 cancer,11 and neurodegeneration.4b,4c,12 In the nervous system and select peripheral tissues, MAGL not only regulates 2-AG, but also arachidonic acid (AA) and AA-derived eicosanoids.4b,4c,12 These data underscore the importance of developing selective and in-vivo-active MAGL inhibitors as probes for studying MAGL function and as potential therapeutic agents for a range of human disorders.
Our initial efforts to develop irreversible inhibitors for MAGL led to the discovery of JZL184, an O-p-nitrophenyl (PNP) carbamate, which displayed good potency, selectivity, and in vivo activity.3c However, even though JZL184 exhibits >100-fold selectivity for MAGL over FAAH, chronic administration of this compound leads to partial blockade of FAAH, as well as multiple peripheral carboxylesterases (CESs).13 JZL184’s cross-reactivity with FAAH is problematic given that dual inhibition of MAGL and FAAH has been shown to induce catalepsy and drug-dependence in mice,14 highlighting the importance of selectively targeting one but not both arms of the endocannabinoid system to avoid “CB1 agonist-like” activity. We recently overcame this problem by developing an O-hexafluoroisopropyl (HFIP) carbamate analogue of JZL184, KML29, which maintained potency for MAGL and showed negligible cross-reactivity with FAAH and exceptional selectivity across the serine hydrolase class.13b Indeed, the only detectable off-target for KML29 in the mammalian brain was the alternative 2-AG hydrolase ABHD6,15 which was inhibited with ∼300-fold lower potency than MAGL.13b Key to the success of this work was finding an adequate surrogate for the PNP leaving group, which would sufficiently activate the carbamate for attack by the active-site serine nucleophile of MAGL. Additionally, we posited that the bioisoteric nature of the HFIP group, which resembles the glycerol headgroup of 2-AG, also contributed to KML29’s remarkable selectivity for MAGL relative to other serine hydrolases.13b Using these principles, we wondered if N-hydroxysuccinimidyl (NHS) carbamates could also serve as MAGL inhibitors given their reactivity and the isosterism of the NHS and glycerol groups. This hypothesis was recently tested and confirmed by the development of MJN110 (1a), an NHS analogue of JZL184/KML29, which proved to be a potent and selective MAGL inhibitor.16
Here, we expand our proteome-wide reactivity profiling of NHS carbamates to give a full account of the discovery of this class of serine hydrolase inhibitors and describe an extensive characterization of MJN110 activity in vivo. We show that MJN110 possesses distinct advantages over previously developed MAGL inhibitors for pharmacological applications, including excellent in vivo activity and selectivity compatible with acute or chronic dosing in rodents. Furthermore, because of its ease of synthesis and increased potency in vivo, MJN110 is suitable for large-scale studies where inhibitor availability was previously limiting. In this regard, we found that MJN110 displays strong antihyperalgesic activity in a rat model of diabetic neuropathy, matching that of pregabalin and the FAAH inhibitor URB597. Together, these data mark NHS carbamates as a promising class of MAGL inhibitors with the potential to more broadly target enzymes from the serine hydrolase family.
Results and Discussion
Discovery of NHS Carbamates as Serine Hydrolase Inhibitors
Carbamates are a privileged chemotype for irreversible serine hydrolase inhibition that rely on the enhanced nucleophilicity of the active-site serine residue to displace the carbamate’s oxy-substituent, which is typically tuned to favor both the initial nucleophilic attack and its subsequent departure as an oxyanion, resulting in a catalytically dead, carbamylated enzyme adduct (Figure 1A). While O-aryl carbamates are a well-recognized chemotype that have been employed to develop inhibitors for several serine hydrolases,3a,3c,17 most likely due to their synthetic accessibility and easily tuned reactivity, departing from this general motif can offer significant advantages, including improving potency and selectivity in vivo13b,18 (Figure 1B). There are only a handful of examples, however, of potent and selective carbamate serine hydrolase inhibitors featuring alternative (i.e., non-O-aryl) leaving groups, inspiring us to evaluate potentially underutilized carbamate chemotypes. Recognizing the importance of leaving group-ability as a component of carbamate reactivity, we were particularly intrigued by the possibility that NHS carbamates could possess the requisite properties to selectively target serine hydrolases. While NHS esters, carbonates, and secondary carbamates have enjoyed widespread use in synthetic applications due to their attenuated, yet highly tractable reactivity relative to their chloride or anhydride counterparts, tertiary NHS carbamates have remained rather poorly investigated. Interestingly, unlike reactions with NHS esters, carbonates, and secondary carbamates, amine and alkoxy nucleophiles preferentially react with the succinimidyl amide (B) over the carbamate carbonyl (A) of tertiary N,N-dialkyl NHS carbamates (Figure 1C),19 exemplifying the marked stability of their carbamate group and its potential for use in biological systems.
Figure 1.
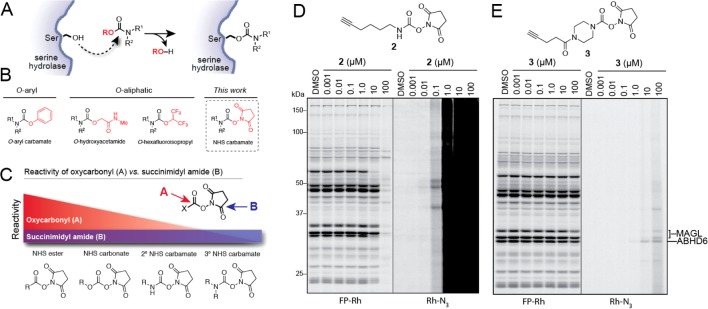
Discovery of NHS carbamates as serine hydrolase inhibitors. (A) Mechanism of serine hydrolase (SH) inhibition by carbamates. (B) General structures of carbamate SH inhibitor classes, including O-aryl and O-aliphatic carbamates, leading to consideration of NHS carbamates as a distinct chemotype for SH inhibition. (C) Model of chemical reactivity toward amine and alkoxy/hydroxy nucleophiles for oxycarbonyl [A] and succinimidyl amide [B] positions in various NHS activated functional groups showing relative stability of tertiary NHS carbamates. Diagnostic clickable probes 2 (D) and 3 (E) evaluate serine hydrolase inhibition and proteome-wide reactivity of secondary and tertiary NHS carbamates, respectively.
To test the general reactivity of NHS carbamates toward the proteome, we treated mouse brain homogenates with two diagnostic clickable NHS carbamates, one secondary (2) and the other tertiary (3) (Figure 1D, E). Each probe bore an alkyne so that labeled proteins could be visualized by copper-catalyzed azide–alkyne cycloaddition (CuAAC or click chemistry (CC)20) conjugation to an azide reporter tag (i.e., rhodamine azide [Rh–N3]).21 Whereas competitive ABPP with the serine hydrolase-directed probe fluorophosphonate-rhodamine (FP-Rh) would reveal the serine hydrolase targets of each NHS carbamate,22 CC-ABPP would provide a full portrait of their protein reactivity across the entire proteome.3b Consistent with its expected heightened reactivity, the secondary NHS carbamate 2 showed a high degree of nonspecific labeling that overwhelmed the signal for its serine hydrolase targets observed by competitive ABPP (Figure 1D). The tertiary NHS carbamate 3, on the other hand, exhibited minimal inhibition of any serine hydrolases or reactivity with other proteins up to 10 μM at which point MAGL and ABHD6 appeared to be the primary targets (Figure 1E). Together these data suggested that while secondary NHS carbamates show indiscriminate reactivity across the entire proteome, the more tempered reactivity of tertiary NHS carbamates may enable selective targeting of serine hydrolase enzymes.
While NHS carbamate 3 did not exhibit significant activity toward any serine hydrolase in the mouse brain proteome, we posited that modifying the piperazine scaffold could improve potency toward specific enzymes. We therefore next synthesized a focused library of NHS carbamates and screened its members at 10 μM against mouse brain serine hydrolases using gel-based competitive ABPP (Figure 2 and Supporting Information Figure S1). Screening this library revealed that many NHS carbamates targeted MAGL, ABHD6, or both (Figure 2B and Supporting Information Figure S1B). Additionally, FAAH and LYPLA1/2 were also inhibited by several members of the NHS carbamate library, albeit without selectivity over MAGL or ABHD6. Interestingly, pyrrolidine 28 and azetidine 29 showed complete selectivity for APEH over other serine hydrolases at 10 μM, indicating that appropriate structural modifications to the NHS carbamate can direct this compound class away from endocannabinoid hydrolases and toward other functionally unrelated serine hydrolases. While taking particular interest in inhibiting MAGL due to its important role in endocannabinoid signaling, we note that recent studies of ABHD615b,23 and LYPLA1/224 indicate that these targets of NHS carbamates also regulate nervous system function and other disease states.
Figure 2.

Evaluation of NHS carbamates as serine hydrolase inhibitors. (A) Structures of representative members of NHS carbamate library. (B) In vitro evaluation of NHS carbamates as serine hydrolase inhibitors in mouse brain (membrane) proteomes showing inhibition of multiple serine hydrolases including MAGL, FAAH, ABHD6, LYPLA1/2, and APEH (enzyme identities were assigned based on gel migration patterns consistent with previous studies3b,3c,29). See Figure S1 for full compound list and ABPP gel profiles.
Evaluation of MJN110 as an Endocannabinoid Hydrolase Inhibitor
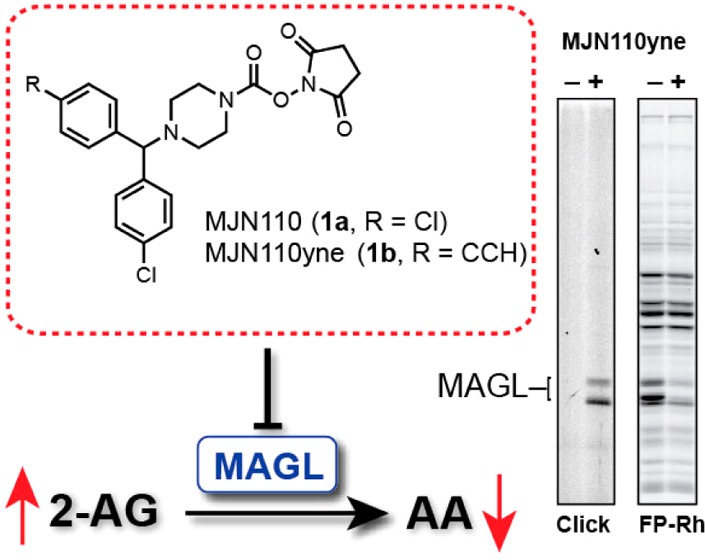
In a comparative analysis of various carbamate chemotypes,16 we recently found that MJN110 (1a) and its clickable analogue MJN110yne (1b, Figure 4C) displayed remarkable potency and selectivity for MAGL and ABHD6, suggesting that this NHS carbamate could serve as a useful pharmacological tool for the functional characterization of MAGL. Here, we expand upon this work and more fully characterize the utility of MJN110 as an endocannabinoid hydrolase inhibitor in vivo. It should be noted that the rationale for choosing the N-benzhydryl piperazine system was guided by numerous studies3c,25 showing that this scaffold enhances carbamate and urea reactivity toward MAGL and improves selectivity over FAAH. While some modifications of this scaffold were investigated, such as dibromobenzhydryl 4 (Figure S1A), MJN110 proved to be superior with regards to potency and selectivity and was thus chosen for more detailed analysis.
Figure 4.

Evaluation of serine hydrolase inhibition and proteome-wide reactivity of MJN110 upon chronic administration. (A) Competitive ABPP gel profiles of brain and liver isolated from mice treated with either vehicle or MJN110 (0.25 or 1.0 mg·kg–1, 6 d, p.o.). (B) Brain 2-AG and AEA levels measured in mice chronically treated with either vehicle or the indicated dose of MJN110. Data are presented as means ± SEM (n = 3 mice per group). *p < 0.05; **p < 0.01; ***p < 0.001 for vehicle-treated versus MJN110-treated mice. (C) Structure of MJN110yne (1b), a clickable analog of MJN110 (1a). (D) Competitive and click-chemistry ABPP profiles of brain and liver from mice treated with MJN110yne (1.0 mg·kg–1, 6 d, p.o.). MJN110yne’s off-target in liver is indicated by an arrow.
As shown previously, we first confirmed by competitive ABPP that MJN110 potently inhibited MAGL and to a lesser extent ABHD6, with excellent selectivity over FAAH and other serine hydrolases in the mouse brain proteome, with LYPLA1/2 being the only other off-targets observed at 10 μM or above (Figure S2).16 These ABPP data were verified by measuring 2-AG and AEA hydrolysis in mouse brain homogenates, which showed that MJN110 potently inhibited 2-AG hydrolysis (IC50 = 2.1 nM) with no effect on AEA hydrolysis up to 50 μM (Figure S2).
Even though tertiary NHS carbamates have been shown to preferentially react with nucleophiles at the succinimidyl amide bond rather than the carbamate carbonyl, we reasoned that MJN110 most likely inhibited MAGL through a carbamylation mechanism, which would arise from optimal positioning of the carbamate near the enzyme’s serine nucleophile. To test this hypothesis, we incubated human recombinant MAGL with either MJN110 or DMSO, proteolyzed each sample with trypsin, and analyzed the tryptic peptides by LC-MS/MS (Figure S3A). From this analysis, we were able to detect a significant reduction in the unmodified active-site peptide (Figure S3B), whereas the mass for the serine-carbamylated active site peptide was observed in only the MJN110-treated sample (Figure S3C). We also searched for the acyl-enzyme adduct that would arise from succinimidyl amide attack by the active-site serine, but were unable to detect this inhibitor-modified peptide species (Figure S3D). These data suggest that the principle mode of MAGL inhibition by MJN110 is via carbamylation of the enzyme’s active-site serine nucleophile, which mirrors the mechanism of other carbamate inhibitors of MAGL.13a,13b
In Vivo Characterization of MJN110 in Mice
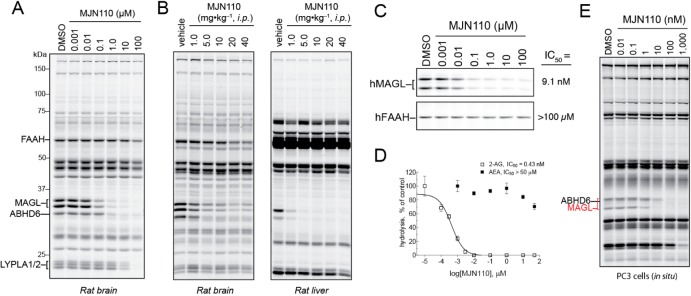
We next evaluated the activity of MJN110 in vivo. We orally administered MJN110 to mice at doses ranging from 0.25 to 5.0 mg·kg–1, and, after 4 h, animals were sacrificed and their tissues harvested for analysis. Dose-dependent inhibition of MAGL was detected by gel-based competitive ABPP with observable inhibition seen at doses as low as 0.5 mg·kg–1 and maximal inhibition detected at 5.0 mg·kg–1 (Figure 3A). Gel-based ABPP of liver proteomes revealed partial MAGL blockade at 0.25 mg·kg–1 and full inhibition by 1.0 mg·kg–1. MJN110 also inhibited MAGL in vivo when administered intraperitoneally, with maximal inhibition observed at 1.0 mg·kg–1 in the brain and 0.25 mg·kg–1 in the liver (Figure 3B). With regard to selectivity, ABHD6 was the sole off-target detected in both brain and liver by gel-based competitive ABPP. We further validated MAGL inhibition by measuring brain levels of 2-AG, AA, and N-acyl ethanolamine (NAE) from mice treated orally with MJN110. Consistent with our gel-based ABPP data showing MAGL inhibition, we observed a dose-dependent increase in 2-AG ranging from 2-fold increases at 0.5 mg·kg–1 (p.o.) MJN110 and 10-fold increases at 5.0 mg·kg–1 (p.o.) MJN110 (Figure 3C). These results indicate that MJN110 can be used to achieve partial inhibition of MAGL, which is noteworthy considering that complete, but not partial, blockade of MAGL leads to drug tolerance through CB1 receptor desensitization.4a,10b,13c A complementary, dose-dependent reduction in brain AA was observed in MJN110-treated mice, while no such changes were observed in brain levels of NAEs, which are substrates of FAAH (Figure 3C).
Figure 3.

In vivo evaluation of MJN110 as a MAGL inhibitor. Competitive ABPP analysis in brain and liver isolated from mice treated with 0.25–5.0 mg·kg–1 MJN110 by oral (A) or intraperitoneal (B) administration. (C) Brain lipid levels from mice treated with indicated doses of MJN110 showing dose-dependent elevation of 2-AG, reduction of AA and no detectable changes in NAEs. Data are presented as means ± SEM (n = 3 mice per group). *p < 0.05; **p < 0.01; ***p < 0.001 for vehicle-treated versus MJN110-treated mice. (D) In vivo time-course analysis of MJN110-mediated MAGL inhibition following a single 1.0 mg·kg–1 (p.o.) dose.
We next evaluated the extent of target inhibition and recovery at various time points following a single dose of MJN110 (1.0 mg·kg–1, p.o.) (Figure 3D). Maximal inhibition of MAGL (∼70%) was observed at 1 h and was sustained until 12 h postadministration. After 72 h, MAGL activity was almost completely recovered. Notably, we did not observe inhibition of any other serine hydrolase across the 72 h time-course analysis. Encouraged by these data, we evaluated MJN110 activity and selectivity following chronic administration by treating mice with either vehicle or MJN110 (0.25 or 1.0 mg·kg–1, p.o.) once per day for 6 days. Four hours following treatment on the sixth day, animals were sacrificed and brain and peripheral tissue proteomes analyzed by competitive ABPP with FP-Rh. At both tested doses, chronic administration of MJN110 produced selective inactivation of MAGL with no detectable cross-reactivity against other serine hydrolases in the brain and liver (Figure 4A), including ABHD6. Chronic MJN110 treatment at 0.25 and 1.0 mg·kg–1 also elevated brain 2-AG levels by two- and 10-fold, respectively, without any significant changes in AEA (Figure 4B). Interestingly, we observed greater blockade of brain MAGL with this chronic dosing regimen compared to single, acute dosing at 1.0 mg·kg–1 (compare Figure 3A, C to Figure 4A, B). Considering that MAGL activity is not completely recovered by 24 h after acute dosing with MJN110 (Figure 3D), we interpret the enhanced MAGL inhibition observed following chronic dosing as being due to serial depletion of active MAGL in the brain, which reduces the demand for MJN110 to achieve complete inhibition after each successive dose. Also consistent with this model is the finding that chronic but not acute dosing with 0.25 mg·kg–1 MJN110 produces a substantial reduction in MAGL activity (>50%, Figure 4A; compare to Figure 3A) and increase in brain 2-AG (>2-fold; Figure 4B; compare to Figure 3C).
We finally queried whether chronic dosing might lead to modification of additional proteins within or outside of the serine hydrolase class by using the clickable analogue MJN110yne (1b, Figure 4C). We treated mice with vehicle or MJN110yne (1.0 mg·kg–1, p.o.) once per day for 6 days, and, 4 h after dosing on the final day of administration, sacrificed the animals and analyzed their brain and liver proteomes by competitive ABPP and CC-ABPP. In both tissues, MJN110yne produced near complete blockade of MAGL with no other detectable serine hydrolase off targets as measured by competitive ABPP (Figure 4D). CC-ABPP confirmed MJN110yne’s complete selectivity for MAGL in mouse brain, as no other detectable protein adducts were observed in this organ. In liver, CC-ABPP revealed that MJN110yne reacted with MAGL and a single additional 65k Da protein that is likely a carboxylesterase, a subclass of serine hydrolases that are known to exhibit broad substrate and inhibitor interactions profiles.26 That we did not observe evidence of inhibition of any liver CES enzymes in our competitive ABPP studies (Figure 4D) indicates the MJN110yne-CES reaction may correspond to a low target engagement event (or a cross-reactivity with a low abundance protein that is obscured in competitive ABPP gels by more highly expressed comigrating serine hydrolases). These data, taken together, indicate that MJN110 maintains excellent selectivity for MAGL when administered chronically to mice.
MJN110 Inhibits Rat MAGL in Vitro and in Vivo
Having previously observed that JZL184 exhibits reduced potency for rat MAGL (IC50 ∼ 260 nM versus 5–10 nM for mouse and human MAGL),13a we wondered if MJN110 would prove suitable for studying this MAGL variant. Competitive ABPP and substrate analyses revealed that MJN110 shows good potency for inhibiting MAGL (IC50 < 100 nM) and to a lesser extent ABHD6 in rat brain proteomes, while not affecting FAAH activity (IC50 > 10 μM) (Figures 5A and S2C). As was observed in mouse brain, LYPLA1/2 were the only other detectable off-targets in rat brain, being inhibited at 10 μM or greater MJN110 (Figure 5A). We next evaluated in vivo activity by administering increasing doses of MJN110 (1.0–40 mg·kg–1, i.p., 4 h) to Wistar rats and analyzing their brain and liver proteomes by competitive ABPP. In both tissues, MAGL inhibition could be detected at doses as low as 1.0 mg·kg–1 with maximal inhibition being reached at 5.0 mg·kg–1 (Figure 5B). ABHD6 inhibition was also observed in both tissues, but little or no cross-reactivity was detected with other serine hydrolases (Figure 5B). These data, taken together, indicate that MJN110 performs well as an inhibitor of rat MAGL both in vitro and in vivo.
Figure 5.
Evaluation of MJN110 for inhibition of rat and human serine hydrolases. (A) In vitro competitive ABPP profiles of MJN110 in rat brain (membrane) proteomes. (B) Competitive ABPP profiles showing serine hydrolase activities in brain and liver proteomes isolated from rats treated with either vehicle or MJN110 (1–40 mg·kg–1, i.p.) for 4 h. (C) Inhibition of recombinant hMAGL and hFAAH by MJN110 as measured by gel-based competitive ABPP with FP-Rh. (D) MJN110-mediated inhibition of recombinant hMAGL and hFAAH activity as measured by hydrolysis of 2-AG and AEA, respectively. (E) Competitive ABPP profiles of human-derived PC3 cells following in situ treatment with MJN110 for 4 h.
MJN110 Selectively Inhibits Human MAGL in Vitro and in Situ
MAGL inhibitors are of potential therapeutic interest;2b,2c we therefore asked whether MJN110 could selectively inhibit human MAGL (hMAGL). Here, it is worth emphasizing the importance of achieving selectivity over FAAH, considering that dual inhibition of MAGL and FAAH has been shown to promote CB1-dependent psychotropic and addictive behaviors in rodents.14 Using lysates from HEK293T cells transiently transfected with hMAGL or hFAAH cDNAs, we determined by competitive ABPP that MJN110 inactivates hMAGL with an IC50 of 9.1 nM, while showing no inhibitory activity against hFAAH over the entire concentration range (0.001–100 μM) (Figure 5C). MJN110 was also found to selectively inhibit hMAGL using a 2-AG substrate hydrolysis assay (IC50 = 0.4 nM) and, again, showed negligible cross-reactivity with hFAAH using an AEA substrate hydrolysis assay (IC50 > 50 μM) (Figure 5D). Finally, we evaluated MJN110 activity against hMAGL in the context of living cells. We treated PC3 cells, a human prostate cancer cell line that expresses both MAGL and ABHD6, with MJN110 (0.01–1000 nM, 4 h) and then harvested cells and analyzed their proteomes by gel-based competitive ABPP with FP-Rh. From this analysis, we identified hMAGL as the primary serine hydrolase target of MJN110 with an IC50 of ∼1 nM and 10- and 100-fold selectivity windows over ABHD6 and LYPLA1/2, respectively (Figure 5E).
MJN110 Alleviates Mechanical Allodynia in a Rat Model of Diabetic Neuropathy
Much of the evidence supporting MAGL’s relevance as a therapeutic target has been obtained using mice by virtue of the availability of genetic models and limitations of previously developed MAGL inhibitors, such as JZL184 (see above). Since MJN110 exhibited good MAGL inhibitory activity in rats, we investigated the effects of this compound in a rat model of diabetic neuropathy.
Neuropathy is a common complication of diabetes, presenting in 30% of patients as tingling and burning sensations, spontaneous pain, and allodynia. In the present study, we use a combination of high fat diet (HFD) and streptozotocin (STZ)-induced pancreatic β-cell death to model the sequelae of Type II diabetes. Mechanistically, these coupled paradigms produce overt hyperglycemia through a combination of diet-induced insulin resistance and insulin reduction through a depletion of β-cells.
The HFD/STZ rats used in this study had significantly (P < 0.001) increased plasma glucose (23.96 ± 0.29 mM) at day 3, compared to matched basal values (8.81 ± 0.09 mM), with similar values at day 31 (Figure 6A). HFD/STZ rats also had significantly (P < 0.001) decreased plasma insulin (0.56 ± 0.05 ng/mL) at day 3 compared to basal levels (1.42 ± 0.09 ng/mL), and this value further decreased further by day 31 (0.29 ± 0.02 ng/mL) (Figure 6B). At day 15, HFD/STZ rats exhibited lowered mechanical withdrawal thresholds to stimulation of the hindpaw, indicative of mechanical allodynia (Figure 6C). At 37 days following model induction, the mechanical withdrawal threshold of the hindpaws was reduced to 6.5 ± 0.6 g from basal values of 13.5 ± 0.4 g (P < 0.001), at which point the antiallodynic effects of MJN110 (5.0 mg·kg–1, i.p.) were measured. In addition to administration of MJN110, both the FAAH inhibitor URB597 (0.3 mg·kg–1, i.p.) and pregabalin (10 mg·kg–1, p.o.) robustly attenuated neuropathic pain behavior whereas vehicle treatment had no effect. At 1 and 3 h following administration of each drug or vehicle, von Frey testing was used to quantify changes in mechanical withdrawal thresholds of the hindpaw. MJN110 significantly increased mechanical withdrawal thresholds in HFD/STZ rats, an effect which was comparable to the antiallodynic effects of URB597 and pregabalin at 1 and 3 h postadministration. These results show that MJN110 attenuated established mechanical allodynia in a rat model of diabetic neuropathy, and thus provides preliminary evidence that MAGL inhibitors may have therapeutic potential for treating chronic pain caused by diabetes.
Figure 6.

Evaluation of antiallodynic effects of MJN110 in rats using the HFD/STZ model of diabetic neuropathy. (A) Fasting plasma glucose in HFD/STZ treated rats (n = 37). All data represent mean ± SEM, analysis was by a repeated measures ANOVA with Dunnett’s multiple comparison posthoc test: ***p < 0.001, compared to baseline (Day −5). (B) Fasting plasma insulin in HFD/STZ treated rats (n = 37). All data represent mean ± SEM, analysis was by Friedman test with Dunn’s posthoc test: *** p < 0.001, compared to baseline (Day −5). (C) Mechanical withdrawal thresholds of the hindpaw in HFD/STZ (45 mg·kg–1) treated rats (n = 36). All data represent mean ± SEM, analysis was by Friedman test with Dunn’s posthoc test: **p < 0.01, ***p < 0.001, compared to baseline (Day −2). (D) Effects of MJN110 (5.0 mg·kg–1, i.p.), URB597 (0.3 mg·kg–1, i.p.) and pregabalin (10 mg·kg–1, p.o.) on mechanical withdrawal thresholds in HFD/STZ treated rats (day 37 post model induction) at 1 and 3 h following drug administration. Data are mechanical withdrawal thresholds expressed as a percentage of baseline values, analysis was with Friedman test with Dunn’s posthoc test: *p < 0.05; **p < 0.01.
Conclusions
MAGL serves as an important metabolic hub that both terminates 2-AG-mediated signaling and controls the release of the AA to be used in the production of proinflammatory eicosanoids in the brain and select peripheral tissues. Inhibitors of MAGL have exhibited beneficial effects in a variety of preclinical models of pain, inflammation, and nervous system disorders, in several cases at doses where few, if any cannabinoid-related psychotropic or motor defects are observed. Several selective and in-vivo-active inhibitors of MAGL have been developed to date, including the O-aryl and O-HFIP carbamates JZL1843c and KML29,13b respectively. Nonetheless, the pursuit of additional classes of MAGL inhibitors is warranted, especially compounds that might exhibit superior cross-species in vivo activity.
As part of our general interest in developing novel chemotypes for the inhibition of serine hydrolases, we recently discovered that MJN110, an NHS carbamate analogue of the MAGL inhibitor JW651 showed attractive features as a MAGL inhibitor.16 In this study, we expand upon this preliminary finding and show that tertiary, but not secondary, NHS carbamates have excellent potential as serine hydrolase inhibitors, exhibiting selectivity for several members of this enzyme class and across the mammalian proteome. We report that MJN110, which can be easily prepared in one synthetic step, exhibits superior potency and comparable selectivity to previously developed MAGL inhibitors, such as KML29, in both acute and chronic dosing regimens, and is, furthermore, well-suited for pharmacological applications in mice and rats. Indeed, we report here that MJN110 reverses established mechanical allodynia in a rat model of diabetic neuropathy, pointing to a potential clinical application for MAGL inhibitors in the treatment of chronic pain caused by diabetes.
Methods
Chemistry
All chemicals were obtained from commercial suppliers and were used without further purification. Anhydrous solvents were obtained by passing solvents through activated alumina columns. Merck s·ilica gel TLC plates (0.25 mm, 60 F254) were used to monitor reactions. Flash chromatography was performed using SiliaFlash F60 silica gel (40–63 μm, 60 Å). NMR spectra were recorded at room temperature on Bruker DRX-500, Varian Inova-400, or Bruker DRX-600 (5 mm DCH Cryoprobe) instruments. Chemical shifts are recorded in ppm relative to tetramethylsilane (TMS) with peaks being reported as follows: chemical shift, multiplicity (s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, m = multiplet, bm = broad multiplet), coupling constant (Hz). High-resolution mass spectra (HRMS) were obtained on an Agilent LC/MSD TOF mass spectrometer by electrospray ionization–time-of-flight (ESI-TOF). All final compounds were determined to be ≥95% pure by HPLC analysis. MJN110 (1a) and MJN110yne (1b) were synthesized according to previously reported methods.16
General Method for Synthesis of NHS Carbamates
To a stirring solution of N,N′-disuccinimidyl carbonate (130 mg, 0.50 mmol, 1.0 equiv) and N-methylmorpholine (0.16 mL, 1.5 mmol, 3.0 equiv) in dry CH2Cl2 (5.0 mL) was added primary or secondary amines (0.50 mmol, 1.0 equiv). The reaction mixture was stirred at room temperature for 12 h. A stream of nitrogen was passed over the reaction mixture to remove the solvent and to the remaining residue was added EtOAc (20 mL). The resulting precipitate was filtered off, and the filtrate was concentrated and purified by SiO2 flash chromatography (EtOAc/hexanes) to give the pure NHS carbamate.
2,5-Dioxopyrrolidin-1-yl Hex-5-yn-1-ylcarbamate (2)
The title compound was synthesized according to the general method for preparation of NHS carbamates from 1-amino-5-hexyne (80 mg, 0.82 mmol), DSC (210 mg, 0.82 mmol), and NMM (0.27 mL, 2.5 mmol). Purification of the crude product by flash chromatography (80% EtOAc) provided the title compound (98 mg, 50%) as a white solid: 1H NMR (600 MHz, CDCl3) δ 5.67–5.63 (m, 1H), 3.27 (q, J = 6.6 Hz, 2H), 2.81 (s, 4H), 2.22 (td, J = 6.8, 2.6 Hz, 2H), 1.95 (t, J = 2.6 Hz, 1H), 1.71–1.66 (m, 2H) 1.60–1.53 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 170.90, 152.27, 84.59, 69.79, 42.36, 29.29, 26.31, 26.13, 18.83. HRMS (ESI-TOF+) m/z calcd for C11H14N2O4 [M+H]+: 239.1032; found, 239.1024.
2,5-Dioxopyrrolidin-1-yl 4-(Pent-4-ynoyl)piperazine-1-carboxylate (3)
To a stirring solution of 4-pentynoic acid (75 mg, 0.76 mmol, 1.0 equiv), N-Boc-piperazine (156 mg, 0.84 mmol, 1.1 equiv) and NMM (0.097 mL, 0.84 mmol, 1.1 equiv) in dry CH2Cl2 (10 mL) was added EDCI (161 mg, 0.84 mmol, 1.1 equiv). After stirring at room temperature for 4 h, the reaction was quenched with a saturated solution of NH4Cl (50 mL) and extracted with CH2Cl2 (50 mL, 3×). The combined organic layers were washed once with brine (50 mL), dried over anhydrous MgSO4, and concentrated to provide tert-butyl 4-(pent-4-ynoyl)piperazine-1-carboxylate (187 mg, 92%) which was used without further purification. To a stirring solution of tert-butyl 4-(pent-4-ynoyl)piperazine-1-carboxylate (187 mg, 0.70 mmol) in CH2Cl2 (4.0 mL) was added TFA (1.0 mL). After 1 h, the reaction mixture was concentrated under a stream of N2. The residue was redissolved in CH2Cl2 (5.0 mL) and concentrated under reduced pressure to remove residual TFA providing a crude colorless oil which was used without further purification. The title compound was synthesized according to the general method for preparation of NHS carbamates from the deprotected piperazine (123 mg, 0.44 mmol), DSC (110 mg, 0.44 mmol), and NMM (0.15 mL, 1.3 mmol). Purification of the crude product by flash chromatography (50% EtOAc/hexanes) provided the title compound (82 mg, 61%) as a white solid: 1H NMR (600 MHz, CDCl3) δ 3.77–3.48 (m, 8H), 2.82 (s, 4H), 2.56 (dd, J = 13.8, 5.9 Hz, 4H), 1.98 (s, 1H). 13C NMR (150 MHz, CDCl3) δ 170.43, 151.29, 151.12, 83.99, 69.89, 45.66, 45.57, 41.80, 32.92, 26.34, 15.32. HRMS (ESI-TOF+) m/z calcd for C14H17N3O5 [M+H]+: 308.1246; found, 308.1239.
2,5-Dioxopyrrolidin-1-yl 4-(4-Methoxyphenyl)piperazine-1-carboxylate (6)
The title compound was synthesized according to the general method for preparation of NHS carbamates from 1-(4-methoxyphenyl)piperazine (100 mg, 0.52 mmol), DSC (130 mg, 0.52 mmol), and NMM (0.17 mL, 1.6 mmol). Purification of the crude product by flash chromatography (70% EtOAc/hexanes) provided the title compound (150 mg, 87%) as a colorless oil: 1H NMR (600 MHz, CDCl3) δ 6.90 (d, J = 8.5 Hz, 2H), 6.84 (d, J = 8.6 Hz, 2H), 3.78 (bm, 2H) 3.76 (s, 3H), 3.67 (bm, 2H), 3.10 (bm, 4H), 2.81 (bm, 4H). 13C NMR (150 MHz, CDCl3) δ 170.60, 155.43, 151.19, 145.96, 120.09, 115.37, 56.39, 51.50, 46.01, 45.57, 26.36. HRMS (ESI-TOF+) m/z calcd for C16H19N3O5 [M+H]+: 334.1403; found, 334.1388.
2,5-Dioxopyrrolidin-1-yl 4-(3-Phenoxybenzyl)piperazine-1-carboxylate (8)
The title compound was synthesized according to the general method for preparation of NHS carbamates from 1-(3-phenoxybenzyl)piperazine (112 mg, 0.42 mmol), DSC (110 mg, 0.42 mmol), and NMM (0.14 mL, 1.3 mmol). Purification of the crude product by flash chromatography (50% EtOAc/hexanes) provided the title compound (140 mg, 82%) as a colorless oil: 1H NMR (600 MHz, CDCl3) δ 7.31 (t, J = 7.87 Hz, 2H), 7.24 (d, J = 7.60 Hz, 1H), 7.08 (t, J = 7.39 Hz, 1H), 7.02 (d, J = 7.56 Hz, 1H), 6.99–6.97 (m, 3H), 6.87 (dd, J = 8.24, 2.41 Hz, 1H), 3.61 (bs, 2H), 3.49 (s, 4H), 2.78 (s, 4H) 2.47 (bs, 4H). 13C NMR (150 MHz, CDCl3) δ 170.60, 155.43, 151.19, 145.96, 120.09, 115.37, 56.39, 51.50, 46.01, 45.57, 26.36; HRMS (ESI-TOF+) m/z calcd for C22H23N3O5 [M+H]+: 410.1716; found, 410.1720.
2,5-Dioxopyrrolidin-1-yl 4-(4-Chlorophenethyl)piperidine-1-carboxylate (9)
The title compound was synthesized according to the general method for preparation of NHS carbamates from 4-[2-(chloro-phenyl)-ethyl-piperidine (130 mg, 0.51 mmol), DSC (130 mg, 0.51 mmol), and NMM (0.17 mL, 1.5 mmol). Purification of the crude product by flash chromatography (30% EtOAc/hexanes) provided the title compound (130 mg, 70%) as a colorless oil: 1H NMR (600 MHz, CDCl3) δ 7.23 (d, J = 7.6 Hz, 2H), 7.08 (d, J = 7.6 Hz, 2H), 4.18 (d, J = 13.2 Hz, 1H), 4.07 (d, J = 13.0 Hz, 1H), 2.97 (t, J = 13.1 Hz, 1H), 2.85 (d, J = 13.1 Hz, 1H), 2.79 (s, 4H), 2.59 (t, J = 8.4 Hz, 2H), 1.76 (d, J = 12.9 Hz, 2H), 1.56 (dd, J = 7.4 Hz, 2H), 1.47 (t, J = 9.7 Hz, 1H), 1.35–1.22 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 170.78, 151.19, 141.41, 132.35, 130.48, 129.33, 46.49, 45.68, 38.66, 35.76, 33.01, 32.56, 32.23, 26.36. HRMS (ESI-TOF+) m/z calcd for C18H21ClN2O4 [M+H]+: 365.1268; found, 365.1264.
2,5-Dioxopyrrolidin-1-yl Benzyl(ethyl)carbamate (10)
The title compound was synthesized according to the general method for preparation of NHS carbamates from N-ethylbenzylamine (110 mg, 0.81 mmol), DSC (210 mg, 0.81 mmol), and NMM (0.27 mL, 2.4 mmol). Purification of the crude product by flash chromatography (50% EtOAc/hexanes) provided the title compound (210 mg, 94%) as a colorless oil: 1H NMR (600 MHz, CDCl3) δ 7.41–7.26 (m, 10H), 4.60 (s, 2H), 4.51 (s, 2H), 3.38 (q, J = 7.1 Hz, 2H), 3.31 (q, J = 7.2 Hz, 2H), 2.80 (s, 8H), 1.21 (t, J = 7.1 Hz, 3H), 1.11 (t, J = 7.2 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ 170.71, 152.98, 151.92, 137.00, 136.94, 129.62, 128.73, 128.70, 128.52, 52.51, 51.05, 44.05, 42.77, 26.37, 14.10, 13.27. HRMS (ESI-TOF+) m/z calcd for C14H16N2O4 [M+Na]+: 299.1002; found, 299.1006.
2,5-Dioxopyrrolidin-1-yl Methyl(3-(pyridin-4-yl)benzyl)carbamate (13)
The title compound was synthesized according to the general method for preparation of NHS carbamates from N-methyl-N-(3-pyridin-4-ylbenzyl)amine (110 mg, 0.55 mmol), DSC (141 mg, 0.55 mmol), and NMM (0.18 mL, 1.7 mmol). Purification of the crude product by flash chromatography (100% EtOAc) provided the title compound (140 mg, 75%) as a colorless oil: 1H NMR (600 MHz, CDCl3) δ 8.66 (s, 4H), 7.72 (s, 1H), 7.64–7.45 (m, 9H), 7.42 (d, J = 7.7 Hz, 1H). 7.33 (d, J = 7.6 Hz, 1H), 4.66 (s, 2H), 4.57 (s, 2H), 3.05 (s, 3H), 2.94 (s, 3H), 2.84 (s, 8H). 13C NMR (150 MHz, CDCl3) δ 170.58, 153.08, 152.19, 151.16, 148.55, 148.47, 139.74, 139.67, 137.49, 137.43, 130.54, 130.49, 129.27, 127.50, 127.46, 127.09, 126.97, 122.54, 122.52, 54.73, 53.50, 36.22, 34.91, 26.38. HRMS (ESI-TOF+) m/z calcd for C18H17N3O4 [M+H]+: 340.1297; found, 340.1289.
2,5-Dioxopyrrolidin-1-yl Methyl(3-(5-methyl-1,2,4-oxadiazol-3-yl)benzyl)carbamate (15)
The title compound was synthesized according to the general method for preparation of NHS carbamates from N-methyl-N-[3-(5-methyl-1,2,4-oxadiazol-3-yl)benzyl]amine (70 mg, 0.34 mmol), DSC (88 mg, 0.34 mmol), and NMM (0.11 mL, 1.0 mmol). Purification of the crude product by flash chromatography (70% EtOAc/hexanes) provided the title compound (110 mg, 93%) as a colorless oil: 1H NMR (600 MHz, CDCl3) δ 8.03–7.99 (m, 3H), 7.94 (s, 1H), 7.63 (d, J = 7.6 Hz, 1H), 7.53 (t, J = 7.7 Hz, 1H), 7.50 (t, J = 7.7 Hz, 1H), 7.44 (d, J = 7.7 Hz, 1H), 4.66 (s, 2H), 4.57 (s, 2H), 3.04 (s, 3H), 2.94 (s, 3H), 2.84 (s, 8H), 2.66 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 177.54, 177.53, 170.55, 170.53, 168.90, 168.89, 153,02, 152.18, 137.28, 137.36, 131.31, 131.23, 130.62, 130.57, 128.11, 127.92, 127.85, 127.82, 127.66, 54.59, 53.40, 36.17, 34.77, 26.37, 13.28. HRMS (ESI-TOF+) m/z calcd for C16H16N4O5 [M+H]+: 345.1199; found, 345.1186.
2,5-Dioxopyrrolidin-1-yl Methyl(phenethyl)carbamate (16)
The title compound was synthesized according to the general method for preparation of NHS carbamates from N-methyl-phenethylamine (140 mg, 1.0 mmol), DSC (270 mg, 1.0 mmol), and NMM (0.34 mL, 3.1 mmol). Purification of the crude product by flash chromatography (50% EtOAc/hexanes) provided the title compound (260 mg, 90%) as a colorless oil: 1H NMR (600 MHz, CDCl3) δ 7.33–7.26 (m, 4H), 7.26–7.19 (m, 6H), 3.58 (t, J = 7.6 Hz, 1H), 3.50 (t, J = 7.6 Hz, 1H), 3.01 (t, J = 7.5 Hz, 2H), 2.96 (s, 3H), 2.88 (t, J = 7.5 Hz, 2H), 2.81 (s, 3H), 2.78 (s, 4H), 2.77 (s, 4H). 13C NMR (150 MHz, CDCl3) δ 170.80, 170.79, 152.20, 152.16, 139.08, 139.02, 129.71, 129.68, 129.54, 129.52, 127.48, 127.45, 53.47, 52.12, 37.68, 35.93, 35.25, 34.34, 26.37, 26.34. HRMS (ESI-TOF+) m/z calcd for C14H16N2O4 [M+H]+: 277.1188; found. 277.1184.
2,5-Dioxopyrrolidin-1-yl Piperidine-1-carboxylate (18)
The title compound was synthesized according to the general method for preparation of NHS carbamates from piperidine (150 mg, 1.7 mmol), DSC (450 mg, 1.7 mmol), and NMM (0.57 mL, 5.2 mmol). Purification of the crude product by flash chromatography (50% EtOAc/hexanes) provided the title compound (310 mg, 79%) as a white solid: 1H NMR (600 MHz, CDCl3) δ 3.56 (s, 2H), 3.44 (s, 2H), 2.79 (s, 4H), 1.62 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 170.78, 151.24, 47.18, 46.44, 26.35, 26.31, 26.02, 24.74. HRMS (ESI-TOF+) m/z calcd for C10H14N2O4 [M+H]+: 227.1032; found, 227.1028.
2,5-Dioxopyrrolidin-1-yl 4-Benzylpiperidine-1-carboxylate (22)
The title compound was synthesized according to the general method for preparation of NHS carbamates from 4-benzylpiperidine (690 mg, 3.9 mmol), DSC (1.0 g, 3.9 mmol), and NMM (1.3 mL, 12 mmol). Purification of the crude product by flash chromatography (60% EtOAc/hexanes) provided the title compound (1.1 g, 89%) as a white solid: 1H NMR (600 MHz, CDCl3) δ 7.28 (t, J = 7.4 Hz, 3H), 7.22–7.18 (m, 1H), 7.13 (d, J = 7.5 Hz, 2H), 4.18 (d, J = 13.1 Hz, 1H), 4.08 (d, J = 13.1 Hz, 1H), 2.94 (t, J = 12.9 Hz, 1H), 2.86–2.79 (m, 5H), 2.56 (d, J = 6.9 Hz, 2H), 1.77–1.67 (m, 3H), 1.41–1.22 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 170.73, 151.17, 140.59, 129.92, 129.20, 126.97, 46.56, 45.73, 43.68, 38.56, 32.46, 32.21, 26.35. HRMS (ESI-TOF+) m/z calcd for C17H20N2O4 [M+Na]+: 339.1315; found, 339.1318.
2,5-Dioxopyrrolidin-1-yl Pyrrolidine-1-carboxylate (28)
The title compound was synthesized according to the general method for preparation of NHS carbamates from pyrrolidine (330 mg, 4.7 mmol), DSC (1.2 g, 4.7 mmol), and NMM (1.5 mL, 14 mmol). Purification of the crude product by flash chromatography (50% EtOAc/hexanes) provided the title compound (720 mg, 72%) as a white solid: 1H NMR (600 MHz, CDCl3) δ 3.56 (t, J = 6.8 Hz, 2H), 3.45 (t, J = 6.8 Hz, 2H), 2.81 (s, 4H), 1.96 (p, J = 6.6 Hz, 2H), 1.90 (p, J = 6.6 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 170.85, 150.41, 48.33, 46.91, 26.67, 26.34, 25.48. HRMS (ESI-TOF+) m/z calcd for C9H12N2O4 [M+H]+: 213.0875; found, 213.0868.
2,5-Dioxopyrrolidin-1-yl Azetidine-1-carboxylate (29)
The title compound was synthesized according to the general method for preparation of NHS carbamates from azetidine (150 mg, 2.6 mmol), DSC (670 mg, 2.6 mmol), and NMM (0.87 mL, 7.9 mmol). Purification of the crude product by flash chromatography (50% EtOAc/hexanes) provided the title compound (340 mg, 65%) as a white solid: 1H NMR (600 MHz, CDCl3) δ 4.30–4.26 (m, 2H), 4.15–4.10 (m, 2H), 2.79 (s, 4H), 2.36 (p, J = 7.8 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 170.70, 150.99, 51.43, 50.83, 26.32, 17.26. HRMS (ESI-TOF+) m/z calcd for C8H10N2O4 [M+H]+: 199.0719; found, 199.0710.
Biology
In Vitro Competitive Activity-Based Protein Profiling
Proteomes (50 μL, 1.0 mg/mL total protein concentration) were preincubated with either DMSO or 1–100 000 nM concentrations of inhibitors at 37 °C. After 30 min, FP-Rh (1.0 μL, 50 μM in DMSO) was added and the mixture was incubated for another 30 min at room temperature. Reactions were quenched with SDS loading buffer (17 μL - 4×) and run on SDS-PAGE. Following gel imaging, serine hydrolase activity was determined by measuring fluorescent intensity of gel bands corresponding to MAGL, ABHD6, and FAAH using ImageJ 1.43u software.
Enzyme Activity Assays
MAGL and FAAH substrate hydrolysis measurements were determined according to the previously reported LC-MS-based assay.14 Activities assays were performed on 25 μg of mouse or rat brain proteomes in 200 μL of DPBS. In contrast, 1.0 μg of hMAGL/hFAAH-transfected HEK293T proteomes in 200 μL of DPBS was used for measuring the human enzyme activity.
Click Chemistry-ABPP
Brain membrane and liver membrane proteomes from either naïve (in vitro) or inhibitor-treated (in vivo) mice were diluted to 1.0 mg/mL prior to use. Tissues were harvested and prepared for analysis according to previously reported methods.13bNote: It is beneficial to remove the soluble fraction from liver proteomes due to its adverse effects on the clickreaction. Using previously developed methods,27 Rh–N3 was conjugated to each alkyne probe for in-gel analysis. Briefly, CuSO4 (1.0 μL/reaction, 50 mM in H2O), TBTA (3.0 μL/reaction, 1.7 mM in DMSO/t-BuOH [1:4]), TCEP (1.0 μL/reaction, 50 mM in H2O [freshly prepared]), and Rh–N3 (1.0 μL/reaction, 1.25 mM in DMSO) were premixed. This click reagent mixture (6.0 μL total volume) was immediately added to each proteome (50 μL, 1.0 mg/mL protein concentration), and the reaction was stirred by briefly vortexing. After 1 h at room temperature, reactions were diluted with 4× SDS loading buffer (17 μL) and resolved by SDS-PAGE.
In Vivo Administration of Carbamate Probes for Gel-Based ABPP and Lipid Metabolite Analyses
Carbamate inhibitors were administered to C57Bl/6J mice or Wistar rats in a vehicle of either saline/emulphor/ethanol (18:1:1) for intraperitoneal injections or PEG300 (Fluka) for administration by oral gavage. After the indicated dosing regimens, the mice were anesthetized using isoflurane (and rats by CO2) and sacrificed by cervical dislocation (mice) or decapitation (rats) and tissues were harvested and flash frozen in liquid N2. Tissue proteomes were prepared for competitive and CC-ABPP using the same protocol which has been previously described.13b The studies were performed with the approval of the Institutional Animal Care and Use Committee at The Scripps Research Institute in accordance with the Guide for the Care and Use of Laboratory Animals.
Measurement of Brain Lipids
Brain lipid levels were determined according to previously reported methods.14
Recombinant Expression of Human MAGL and FAAH in HEK293T Cells
hMAGL and hFAAH were expressed in HEK293T cells according to previously reported methods.15a Cell lysates were diluted with mock proteomes for use in competitive ABPP experiments.
Diabetic Neuropathy Studies
Animals
All experiments were carried out in accordance with the UK Home Office Animals (Scientific Procedures) Act 1986. Thirty seven male Sprague–Dawley rats (200–250g), obtained from Charles River (Margate, Kent), were individually housed on a normal light cycle (lights on: 07:00–19:00) with free access to a high fat diet, 60% fat by caloric content (D12492 diet, from Research Diets, New Brunswick, NJ), and water at all times. Food and water intake, and body weight were monitored twice weekly. After 3 weeks of consumption of the HFD, rats received an intraperitoneal (i.p.) injection of STZ (45 mg·kg–1).
Assessment of Mechanical Withdrawal Thresholds
von Frey monofilaments (Semmes-Weinstein monofilaments of bending forces 1, 1.4, 2, 4, 6, 8, 10, and 15 g) were used to quantify mechanical withdrawal thresholds as previously described;28 a lowering of these thresholds is indicative of mechanical allodynia. von Frey monofilaments were applied to the plantar surface of both hindpaws for a 3 s period. Once a withdrawal reflex was established, the paw was tested again with the next descending von Frey monofilament until no response was elicited. The lowest weight of monofilament which elicited a withdrawal reflex was noted as the paw withdrawal threshold (PWT). The week prior to administration of drugs, the rats were divided into groups based on their von Frey behavioral data (n = 9 for pregabalin, n = 8 for URB597 and MJN110, n = 6 for saline, and n = 5 for 1:1:18 vehicle).
Blood Sampling
After a 4 h fast, 100 μL of blood was taken from the lateral tail vein by vein stab and collected into lithium heparinized tubes (Sarstedt Microvette CB300), and plasma was separated by centrifugation (2400g for 5 min at 4 °C) to produce a single aliquot of plasma (approximately 50 μL) which was frozen (−80 °C) and subsequently assayed for glucose (Thermoelectron infinity glucose reagent) and insulin (Mercodia rat insulin ELISA).
Assessment of Drug Effect on Pain Behavior
von Frey testing was carried out 30 min before administration of the drug (baseline), and at 1 and 3 h after drug administration. MJN110 (5.0 mg·kg–1) and URB597 (0.3 mg·kg–1) were administered via i.p. injection, whereas pregabalin (10 mg·kg–1) was administered orally, all at a volume of 1 mL·kg–1.
Drugs
STZ (batch/lot number 019K1022) was purchased from Sigma Aldrich (Poole, U.K.) and dissolved in 0.05 M citric acid pH 4.5. URB597 was purchased from Sigma Aldrich (Poole, U.K.), pregabalin was kindly provided by Pfizer (Cambridge, U.K.), and MJN110 was synthesized according to the above procedures. MJN110 and URB597 were dissolved in a vehicle of EtOH, Emulphor-620, and saline in a ratio of 1:1:18. Pregabalin was dissolved in saline.
Data and Statistical Analysis
Statistical analysis of changes in mechanical withdrawal thresholds, and plasma insulin was by Friedman test with Dunn’s posthoc test. Analysis of plasma glucose was by a repeated measures ANOVA with Dunnett’s multiple comparison posthoc test. Effect of drugs on mechanical withdrawal thresholds was by a Kruskal–Wallis test with Dunn’s posthoc test. In all analyses, a p value of less than 0.05 was considered statistically significant.
Acknowledgments
Insulin and glucose tests were provided by Renasci, and pregabalin was provided by Pfizer.
Supporting Information Available
Synthetic procedures and 1H NMR, 13C NMR, and HRMS data for NHS carbamates 1–30. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
∥ M.J.N. and A.B.C., III contributed equally to this work.
This work was supported by the National Institutes of Health Grants DA017259, DA033760, DA032933, and DA032541 (to M.J.N.), the Skaggs Institute for Chemical Biology and the Biotechnology and Biological Sciences Research Council (F.B.). This work was supported in part by a grant from Abide Therapeutics.
The authors declare the following competing financial interest(s): B.F.C. is a founder and member of the scientific advisory board of Abide Therapeutics, a company that is interested in developing serine hydrolase inhibitors as therapeutic agents..
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Ahn K.; McKinney M. K.; Cravatt B. F. (2008) Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem. Rev. 108(5), 1687–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Di Marzo V. (2009) The endocannabinoid system: Its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacol. Res. 60(2), 77–84. [DOI] [PubMed] [Google Scholar]; c Fowler C. J. (2008) “The tools of the trade”—an overview of the pharmacology of the endocannabinoid system. Curr. Pharm. Des. 14(23), 2254–2265. [DOI] [PubMed] [Google Scholar]
- a Di Marzo V. (2008) Targeting the endocannabinoid system: to enhance or reduce?. Nat. Rev. Drug. Discovery 7(5), 438–455. [DOI] [PubMed] [Google Scholar]; b Mulvihill M. M.; Nomura D. K. (2013) Therapeutic potential of monoacylglycerol lipase inhibitors. Life Sci. 92(8–9), 492–7. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Fowler C. J. (2012) Monoacylglycerol lipase - a target for drug development?. Br. J. Pharmacol. 166(5), 1568–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kathuria S.; Gaetani S.; Fegley D.; Valino F.; Duranti A.; Tontini A.; Mor M.; Tarzia G.; La Rana G.; Calignano A.; Giustino A.; Tattoli M.; Palmery M.; Cuomo V.; Piomelli D. (2003) Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 9(1), 76–81. [DOI] [PubMed] [Google Scholar]; b Ahn K.; Johnson D. S.; Mileni M.; Beidler D.; Long J. Z.; McKinney M. K.; Weerapana E.; Sadagopan N.; Liimatta M.; Smith S. E.; Lazerwith S.; Stiff C.; Kamtekar S.; Bhattacharya K.; Zhang Y.; Swaney S.; Van Becelaere K.; Stevens R. C.; Cravatt B. F. (2009) Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem. Biol. 16(4), 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Long J. Z.; Li W.; Booker L.; Burston J. J.; Kinsey S. G.; Schlosburg J. E.; Pavon F. J.; Serrano A. M.; Selley D. E.; Parsons L. H.; Lichtman A. H.; Cravatt B. F. (2009) Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol. 5(1), 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Kinsey S. G.; Long J. Z.; O’Neal S. T.; Abdullah R. A.; Poklis J. L.; Boger D. L.; Cravatt B. F.; Lichtman A. H. (2009) Blockade of Endocannabinoid-Degrading Enzymes Attenuates Neuropathic Pain. J. Pharmacol. Exp. Ther. 330(3), 902–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kinsey S. G.; Wise L. E.; Ramesh D.; Abdullah R.; Selley D. E.; Cravatt B. F.; Lichtman A. H. (2013) Repeated Low Dose Administration of the Monoacylglycerol Lipase Inhibitor JZL184 Retains CB1 Receptor Mediated Antinociceptive and Gastroprotective Effects. J. Pharmacol. Exp. Ther. 345, 492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Piro J. R.; Benjamin D. I.; Duerr J. M.; Pi Y.; Gonzales C.; Wood K. M.; Schwartz J. W.; Nomura D. K.; Samad T. A. (2012) A Dysregulated Endocannabinoid-Eicosanoid Network Supports Pathogenesis in a Mouse Model of Alzheimer’s Disease. Cell Rep. 1(6), 617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Chen R.; Zhang J.; Wu Y.; Wang D.; Feng G.; Tang Y.-P.; Teng Z.; Chen C. (2012) Monoacylglycerol Lipase Is a Therapeutic Target for Alzheimer’s Disease. Cell Rep. 2(5), 1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Woodhams S. G.; Wong A.; Barrett D. A.; Bennett A. J.; Chapman V.; Alexander S. P. (2012) Spinal administration of the monoacylglycerol lipase inhibitor JZL184 produces robust inhibitory effects on nociceptive processing and the development of central sensitization in the rat. Br. J. Pharmacol. 167(8), 1609–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gobbi G.; Bambico F. R.; Mangieri R.; Bortolato M.; Campolongo P.; Solinas M.; Cassano T.; Morgese M. G.; Debonnel G.; Duranti A.; Tontini A.; Tarzia G.; Mor M.; Trezza V.; Goldberg S. R.; Cuomo V.; Piomelli D. (2005) Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 102(51), 18620–18625. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kinsey S. G.; O’Neal S. T.; Long J. Z.; Cravatt B. F.; Lichtman A. H. (2011) Inhibition of endocannabinoid catabolic enzymes elicits anxiolytic-like effects in the marble burying assay. Pharmacol., Biochem. Behav. 98(1), 21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Otrubova K.; Ezzili C.; Boger D. L. (2011) The discovery and development of inhibitors of fatty acid amide hydrolase (FAAH). Bioorg. Med. Chem. Lett. 21(16), 4674–4685. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gaetani S.; Cuomo V.; Piomelli D. (2003) Anandamide hydrolysis: a new target for anti-anxiety drugs?. Trends Mol. Med. 9(11), 474–478. [DOI] [PubMed] [Google Scholar]
- Ahn K.; Smith S. E.; Liimatta M. B.; Beidler D.; Sadagopan N.; Dudley D. T.; Young T.; Wren P.; Zhang Y.; Swaney S.; Van Becelaere K.; Blankman J. L. L.; Nomura D. K. K.; Bhattachar S. N.; Stiff C.; Nomanbhoy T. K.; Weerapana E.; Johnson D. S.; Cravatt B. F. (2011) Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective FAAH inhibitor that reduces inflammatory and noninflammatory pain. J. Pharmacol. Exp. Ther. 338, 114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman J. L.; Cravatt B. F. (2013) Chemical probes of endocannabinoid metabolism. Pharmacol. Rev. 65(2), 849–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J.; Guijarro A.; Piomelli D.; Hohmann A. G. (2011) Peripheral antinociceptive effects of inhibitors of monoacylglycerol lipase in a rat model of inflammatory pain. Br. J. Pharmacol. 163(7), 1464–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sciolino N. R.; Zhou W.; Hohmann A. G. (2011) Enhancement of endocannabinoid signaling with JZL184, an inhibitor of the 2-arachidonoylglycerol hydrolyzing enzyme monoacylglycerol lipase, produces anxiolytic effects under conditions of high environmental aversiveness in rats. Pharmacol. Res. 64(3), 226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Busquets-Garcia A.; Puighermanal E.; Pastor A.; de la Torre R.; Maldonado R.; Ozaita A. s. (2011) Differential Role of Anandamide and 2-Arachidonoylglycerol in Memory and Anxiety-like Responses. Biol. Psychiatry 70(5), 479–486. [DOI] [PubMed] [Google Scholar]
- a Nomura D. K.; Lombardi D. P.; Chang J. W.; Niessen S.; Ward A. M.; Long J. Z.; Hoover H. H.; Cravatt B. F. (2011) Monoacylglycerol Lipase Exerts Dual Control over Endocannabinoid and Fatty Acid Pathways to Support Prostate Cancer. Chem. Biol. 18(7), 846–856. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nomura D. K.; Long J. Z.; Niessen S.; Hoover H. S.; Ng S. W.; Cravatt B. F. (2010) Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 140(1), 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ye L.; Zhang B.; Seviour E. G.; Tao K. X.; Liu X. H.; Ling Y.; Chen J. Y.; Wang G. B. (2011) Monoacylglycerol lipase (MAGL) knockdown inhibits tumor cells growth in colorectal cancer. Cancer Lett. 307(1), 6–17. [DOI] [PubMed] [Google Scholar]
- Nomura D. K.; Morrison B. E.; Blankman J. L.; Long J. Z.; Kinsey S. G.; Marcondes M. C. G.; Ward A. M.; Hahn Y. K.; Lichtman A. H.; Conti B.; Cravatt B. F. (2011) Endocannabinoid Hydrolysis Generates Brain Prostaglandins That Promote Neuroinflammation. Science 334(6057), 809–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Long J. Z.; Nomura D. K.; Cravatt B. F. (2009) Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem. Biol. 16(7), 744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chang J. W.; Niphakis M. J.; Lum K. M.; Cognetta A. B.; Wang C.; Matthews M. L.; Niessen S.; Buczynski M. W.; Parsons L. H.; Cravatt B. F. (2012) Highly Selective Inhibitors of Monoacylglycerol Lipase Bearing a Reactive Group that Is Bioisosteric with Endocannabinoid Substrates. Chem. Biol. 19(5), 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Schlosburg J. E.; Blankman J. L.; Long J. Z.; Nomura D. K.; Pan B.; Kinsey S. G.; Nguyen P. T.; Ramesh D.; Booker L.; Burston J. J.; Thomas E. A.; Selley D. E.; Sim-Selley L. J.; Liu Q. S.; Lichtman A. H.; Cravatt B. F. (2010) Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat. Neurosci. 13(9), 1113–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J. Z.; Nomura D. K.; Vann R. E.; Walentiny D. M.; Booker L.; Jin X.; Burston J. J.; Sim-Selley L. J.; Lichtman A. H.; Wiley J. L.; Cravatt B. F. (2009) Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc. Natl. Acad. Sci. U.S.A. 106(48), 20270–20275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Blankman J. L.; Simon G. M.; Cravatt B. F. (2007) A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 14(12), 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Marrs W. R.; Blankman J. L.; Horne E. A.; Thomazeau A.; Lin Y. H.; Coy J.; Bodor A. L.; Muccioli G. G.; Hu S. S.; Woodruff G.; Fung S.; Lafourcade M.; Alexander J. P.; Long J. Z.; Li W.; Xu C.; Moller T.; Mackie K.; Manzoni O. J.; Cravatt B. F.; Stella N. (2010) The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat. Neurosci. 13(8), 951–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J. W.; Cognetta A. B.; Niphakis M. J.; Cravatt B. F. (2013) Proteome-wide reactivity profiling identifies diverse carbamate chemotypes tuned for serine hydrolase inhibition. ACS Chem. Biol. 10.1021/cb400261h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bar-On P.; Millard C. B.; Harel M.; Dvir H.; Enz A.; Sussman J. L.; Silman I. (2002) Kinetic and structural studies on the interaction of cholinesterases with the anti-Alzheimer drug rivastigmine. Biochemistry 41(11), 3555–3564. [DOI] [PubMed] [Google Scholar]; b Chang J. W.; Moellering R. E.; Cravatt B. F. (2012) An activity-based imaging probe for the integral membrane hydrolase KIAA1363. Angew. Chem., Int. Ed. 51(4), 966–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niphakis M. J.; Johnson D. S.; Ballard T. E.; Stiff C.; Cravatt B. F. (2012) O-hydroxyacetamide carbamates as a highly potent and selective class of endocannabinoid hydrolase inhibitors. ACS Chem. Neurosci. 3(5), 418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Vasilevich N. I.; Sachinvala N. D.; Maskos K.; Coy D. H. (2002) Selective conversion of O-succinimidyl carbamates to N-(O-carbamoyl)-succinmonoamides and ureas. Tetrahedron Lett. 43(18), 3443–3445. [Google Scholar]; b Vasilevich N. I.; Coy D. H. (2002) Conversion of O-succinimidyl carbamates to N-(O-carbamoyl)-succinmonoamides and ureas: effects of N-substituents and reaction conditions on the reaction pathway. Tetrahedron Lett. 43(37), 6649–6652. [Google Scholar]
- Rostovtsev V. V.; Green J. G.; Fokin V. V.; Sharpless K. B. (2002) A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem., Int. Ed. 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- a Speers A. E.; Adam G. C.; Cravatt B. F. (2003) Activity-based protein profiling in vivo using a copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 125(16), 4686–4687. [DOI] [PubMed] [Google Scholar]; b Speers A. E.; Cravatt B. F. (2004) Profiling enzyme activities in vivo using click chemistry methods. Chem. Biol. 11(4), 535–546. [DOI] [PubMed] [Google Scholar]
- Leung D.; Hardouin C.; Boger D. L.; Cravatt B. F. (2003) Discovering potent and selective reversible inhibitors of enzymes in complex proteomes. Nat. Biotechnol. 21, 687–691. [DOI] [PubMed] [Google Scholar]
- Tchantchou F.; Zhang Y. (2013) Selective inhibition of alpha/beta-hydrolase domain 6 attenuates neurodegeneration, alleviates blood brain barrier breakdown and improves functional recovery in a mouse model of traumatic brain injury. J. Neurotrauma (30), 565–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dekker F. J.; Rocks O.; Vartak N.; Menninger S.; Hedberg C.; Balamurugan R.; Wetzel S.; Renner S.; Gerauer M.; Scholermann B.; Rusch M.; Kramer J. W.; Rauh D.; Coates G. W.; Brunsveld L.; Bastiaens P. I.; Waldmann H. (2010) Small-molecule inhibition of APT1 affects Ras localization and signaling. Nat. Chem. Biol. 6(6), 449–456. [DOI] [PubMed] [Google Scholar]; b Rusch M.; Zimmermann T. J.; Burger M.; Dekker F. J.; Gormer K.; Triola G.; Brockmeyer A.; Janning P.; Bottcher T.; Sieber S. A.; Vetter I. R.; Hedberg C.; Waldmann H. (2011) Identification of acyl protein thioesterases 1 and 2 as the cellular targets of the Ras-signaling modulators palmostatin B and M. Angew. Chem., Int. Ed. 50(42), 9838–9842. [DOI] [PubMed] [Google Scholar]; c Siegel G.; Obernosterer G.; Fiore R.; Oehmen M.; Bicker S.; Christensen M.; Khudayberdiev S.; Leuschner P. F.; Busch C. J.; Kane C.; Hubel K.; Dekker F.; Hedberg C.; Rengarajan B.; Drepper C.; Waldmann H.; Kauppinen S.; Greenberg M. E.; Draguhn A.; Rehmsmeier M.; Martinez J.; Schratt G. M. (2009) A functional screen implicates microRNA-138-dependent regulation of the depalmitoylation enzyme APT1 in dendritic spine morphogenesis. Nat. Cell Biol. 11(6), 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Long J. Z.; Jin X.; Adibekian A.; Li W.; Cravatt B. F. (2010) Characterization of Tunable Piperidine and Piperazine Carbamates as Inhibitors of Endocannabinoid Hydrolases. J. Med. Chem. 53(4), 1830–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bertrand T.; Augé F.; Houtmann J.; Rak A.; Vallée F.; Mikol V.; Berne P. F.; Michot N.; Cheuret D.; Hoornaert C.; Mathieu M. (2010) Structural Basis for Human Monoglyceride Lipase Inhibition. J. Mol. Biol. 396(3), 663–673. [DOI] [PubMed] [Google Scholar]; c Aaltonen N.; Savinainen J. R.; Ribas C. R.; Ronkko J.; Kuusisto A.; Korhonen J.; Navia-Paldanius D.; Hayrinen J.; Takabe P.; Kasnanen H.; Pantsar T.; Laitinen T.; Lehtonen M.; Pasonen-Seppanen S.; Poso A.; Nevalainen T.; Laitinen J. T. (2013) Piperazine and piperidine triazole ureas as ultrapotent and highly selective inhibitors of monoacylglycerol lipase. Chem. Biol. 20(3), 379–390. [DOI] [PubMed] [Google Scholar]; d Morera L.; Labar G.; Ortar G.; Lambert D. M. (2012) Development and characterization of endocannabinoid hydrolase FAAH and MAGL inhibitors bearing a benzotriazol-1-yl carboxamide scaffold. Bioorgan. Med. Chem. 20(21), 6260–6275. [DOI] [PubMed] [Google Scholar]
- Bachovchin D. A.; Ji T.; Li W.; Simon G. M.; Blankman J. L.; Adibekian A.; Hoover H.; Niessen S.; Cravatt B. F. (2010) Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening. Proc. Natl. Acad. Sci. U.S.A. 107(49), 20941–20946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander J. P.; Cravatt B. F. (2005) Mechanism of carbamate inactivation of FAAH: Implications for the design of covalent inhibitors and in vivo functional probes for enzymes. Chem. Biol. 12(11), 1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guasti L.; Richardson D.; Jhaveri M.; Eldeeb K.; Barrett D.; Elphick M. R.; Alexander S. P.; Kendall D.; Michael G. J.; Chapman V. (2009) Minocycline treatment inhibits microglial activation and alters spinal levels of endocannabinoids in a rat model of neuropathic pain. Mol. Pain 5, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Adibekian A.; Martin B. R.; Wang C.; Hsu K. L.; Bachovchin D. A.; Niessen S.; Hoover H.; Cravatt B. F. (2011) Click-generated triazole ureas as ultrapotent in vivo-active serine hydrolase inhibitors. Nat. Chem. Biol. 7(7), 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Adibekian A.; Martin B. R.; Chang J. W.; Hsu K. L.; Tsuboi K.; Bachovchin D. A.; Speers A. E.; Brown S. J.; Spicer T.; Fernandez-Vega V.; Ferguson J.; Hodder P. S.; Rosen H.; Cravatt B. F. (2012) Confirming target engagement for reversible inhibitors in vivo by kinetically tuned activity-based probes. J. Am. Chem. Soc. 134(25), 10345–10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.