

Abstract
Thrombelastography (TEG) is a method that is used to conduct global assays which monitor fibrin formation and fibrinolysis and platelet aggregation in whole blood. The purpose of this study was to use a well-characterized tissue factor (Tf) reagent and contact pathway inhibitor (corn trypsin inhibitor, CTI) to develop a reproducible thrombelastography assay. In this study, blood was collected from 5 male subjects (3 times). Clot formation was initiated in whole blood with 5 pM Tf in the presence of CTI and fibrinolysis was induced by adding tissue-plasminogen activator (tPA). Changes in visco-elasticity were then monitored by TEG. In quality control assays, our Tf reagent, when used at 5 pM, induced coagulation in whole blood in 3.93±0.23 minutes and in plasma in 5.12±0.23 minutes (n=3). In TEG assays, tPA significantly decreased clot strength (maximum amplitude, MA) in all individuals but had no effect on clot time (R time). The intra-assay variability (CVa<10%) for R time, angle and MA suggests that these parameters reliably describe the dynamics of fibrin formation and degradation in whole blood. Our Tf reagent reproducibly induces coagulation, making it an ideal tool to quantify the processes that contribute to mechanical clot strength in whole blood.
Keywords: Thrombelastography, fibrin, PAI-1, fibrinolysis, tPA
Introduction
Blood coagulation can be initiated via two discrete pathways: the tissue factor (Tf) [1] and the contact pathway [2]. Since deficiencies in the Tf pathway are associated with a severe bleeding diathesis, the Tf pathway is considered to be the physiologically relevant pathway in vivo [3]. By contrast, the contact pathway (kallikrein-factor (f)XII-kininogen) is not considered physiologically relevant in humans because deficiency of clotting fXII [4], prekallikrein or high molecular weight kiniogen are not associated with thrombosis or bleeding. Upon vessel injury, Tf is exposed to the blood and acts as a cofactor for fVIIa which initiates coagulation by activating clotting fIX [5] and fX [6] which then contributes to thrombin generation and subsequent clot formation.
Viscometers (e.g. thrombelastograph) can be used to conduct global assays which monitor fibrin formation and degradation (fibrinolysis) and platelet aggregation in whole blood [7]. The viscosity and elasticity of the blood clot is measured mechanically over time and consequently, quantitative measures describing the physical properties of the clot can be determined during the course of clot formation and fibrinolysis. Currently, thrombelastography assays are initiated with proprietary Tf reagents (commercial products) containing Tf, procoagulant lipids and calcium ion [7]. These Tf reagents are extremely useful in clinical settings where they have been used in thrombelastography based assays for early detection of coagulopathies [8].
A well characterized Tf reagent is essential for quantitative biochemistry since it allows visco-elastic measures to be related directly to the specific activity of the Tf activator. Additionally, whole blood and plasma assays are confounded by activation of the contact pathway. The contact pathway is activated when fXII comes into contact with a negatively charged surface such as plastic or glass [9], thus routine blood collection methods promote procoagulant reactions via a pathway that is artificially activated and not physiologically relevant. In addition to a well characterized Tf reagent, a specific contact pathway inhibitor is required to quantitatively assess Tf-dependent coagulant processes in blood and plasma.
The purpose of the current study was to use a well-characterized Tf reagent and corn trypsin inhibitor (CTI), a specific inhibitor of activated fXII, to develop a reproducible thrombelastography assay. By developing such an assay, we are able to relate changes in visco-elastic properties to a specific Tf stimulus which is not influenced by artificial contact pathway activation.
Subjects, Materials and Methods
Subjects
5 healthy males (S1 – S5) between the ages of 29 and 32 were recruited and advised according to a protocol approved by the Institutional Review Board at the University of Vermont Human Studies Committee. Informed written consent was obtained from all subjects prior to blood collection. Blood from each subject was drawn on 3 occasions spanning a 4 week period. The blood was not drawn into an anticoagulant but was immediately added to an aliquot of CTI (0.1 mg/mL final concentration) to prevent contact pathway activation. All assays were started within 5 minutes of blood collection. Subjects were not restricted in any way and were not on any medications at the time of blood collection. To control for diurnal fluctuations in protein levels blood was always drawn between 8:30 and 9:00 am.
Materials
HEPES, 1-palmitoyl-2-oleoyl-phosphatidyl serine (PS), 1-palmitoyl-2-oleoyl-phosphatidyl choline (PC) were purchased from Sigma Chemical Co. (St. Louis, MO). Recombinant Tf was provided by Drs. Lundblad and Liu (Hyland division, Baxter Healthcare Corp, Duarte, CA) and was relipidated in PCPS (75% PC:25% PS) vesicles by a previously described protocol [10;11]. Corn trypsin inhibitor (CTI) was prepared as previously described [12]. D-phenylalanyl-L-prolyl-L-arginine chloromethyl ketone (FPRck), the anti-human thrombin antibody (Cat. # AHT-5020) and SN-17c (D-Phe-Pro-Arg-6-amino-1-naphthalenesulfonamide-C4H9•2 HCI) was obtained from Haematologic Technologies Inc (Essex Junction, VT) and H-D-Val-Phe-Lys-chloromethyl ketone was purchased from EMD Biosciences (Rockland, MA). Single chain tPA was isolated from tPA which was a gift from Genentech, Inc. (San Francisco, CA) using methods described by Butenas et al. [13]. Greiner Vacuette 3.2% citrate tubes were purchased from Fisher Scientific (Pittsburg, PA), and the Zymutest PAI-1 assay was purchased from Aniara (Mason, OH). HEPES buffered saline (HBS) was used in all experiments unless noted otherwise.
Methods
Cell counts
Phlebotomy blood was drawn into citrate tubes and subjected to complete cell counts using the PoHC-100 automated hematology analyzer (Mundelein, IL).
Tissue factor quality control
The concentration of Tf in our relipidated Tf reagent was determined using an activity assay [14]. Briefly, a standard curve is generated by measuring the rate of SN-17c hydrolysis by the fVIIa-Tf complex. The relipidated Tf reagent is incubated with 2 nM fVIIa and the rate of SN-17c hydrolysis is compared to the standard curve and used to determine the Tf activity in the Tf reagent. Since not all Tf is properly oriented after relipidation, the active Tf concentration determined using this assay is always a fraction of the input Tf concentration. Typicallly, 40 – 50% of Tf activity is measured in our Tf reagent. Our Tf reagent is also quality controlled using blood and plasma clotting assays. Tf is diluted and used to initiate coagulation in either plasma or whole blood. Briefly, Tf is added to either plasma or whole blood at final concentrations ranging from 2 – 10 pM. Plasma assays are conducted with 280 μL of citrated human pooled plasma (10+ donors). The plasma is re-calcified to a final concentration of 15 mM calcium ion. After a 3 minute incubation period, coagulation is initiated by the addition of 2 μM PCPS and Tf (2 - 10 pM). The clot time in plasma-based assays are determined using a STart coagulation analyzer (Diagnostica Stago, Asnières, France). The whole blood clotting assay is performed essentially as previously described [15;16]. Blood is collected and 1 mL is immediately added to tubes containing Tf (2 – 10 pM) and CTI (0.1 mg/mL). The tubes are then set to rock in a temperature controlled chamber (37°C) until the clot time. The clot time is determined visually by 2 independent observers.
Thrombelastography assays
Thrombelastography was conducted using a TEG Haemoscope 5000 (Haemonetics, Braintree MA). Prior to blood collection, TEG cups were spotted with 5 μL each of 360 pM Tf (5 pM final) and 0, 72, 108 or 144 nM tPA (0, 1, 1.5 and 2 nM final). Venous blood was obtained by antecubital phlebotomy and immediately added to 100 μL of CTI to achieve a final concentration of 0.1 mg/mL. The CTI-blood was mixed by inversion (2×) prior to addition of 350 μL to each TEG cup. TEG experiments were performed in duplicate for each blood sample (5 subjects × 3 draws).
Thrombin-antithrombin, PAI-1 and fibrinogen assays
At the end of TEG experiments containing no tPA (3 hours), the cups were ejected and the clot and whole blood was added to an equal volume of an inhibitor cocktail containing 50 μM FPRcK, 20 mM benzamidine, 20 μM EDTA and 50 μM VFKck. The samples were gently mixed, centrifuged at 2000 x g and the cell and fibrin depleted plasma was stored at −80°C and subsequently assayed for α-thrombin-antithrombin (TAT) complexes using an in-house immunoassay (manuscript in preparation). PAI-1 plasma levels were determined for each plasma sample (5 subjects × 3 draws) using the Zymutest PAI-1 antigen ELISA. Fibrinogen levels were determined using the Clauss method at the clinical laboratory of Fletcher Allen Healthcare (Burlington, VT).
Statistics
Between group comparisons were conducted using ANOVA and Tukey HSD test. Data were presented as mean ± standard deviation. The coefficient of variance for the assay (CVa) was calculated as previously described [17]. For all statistical analysis p<0.05 was considered significant.
Results
Cell counts
All subjects had clinically normal cell counts. Platelet counts did not differ significantly between subjects (range: 210.4 ± 25.6 × 103/μL (S3) to 239.6 ± 7.4 × 103/μL (S5). S3 had the lowest platelet (PLT) count but this did not reach the level of statistical significance. Leukocyte counts ranged from 5.0 ± 0.8 × 103/μL (S3) to 8.7 ± 0.5 × 103/μL (S1). S1 had a significantly higher leukocyte count than all other subjects (all p<0.05) which was primarily a result of a higher neutrophil count. Erythrocytes and hematocrit were similar among subjects. The cell counts are summarized in Table 1. LYM and MXD represent lymphocyte and mixed white cell count, respectively. The data represent the mean ±SD, n=3 blood draws.
Table 1.
Complete cell count. The complete blood count consisted of counts of erythrocytes (RBC), hematocrit (HCT), platelets (PLT) and leukocytes (WBC). WBC were further divided into neutrophils (NEUT) lymphocytes (LYM) and mixed white cell count (MXD). The data represent the mean ±SD, n=3 blood draws.
| S1 | S2 | S3 | S4 | S5 | |
|---|---|---|---|---|---|
|
| |||||
| Complete Cell Count | |||||
| RBC (×106/uL) | 5.5 ± 0.2 | 5.0 ± 0.1 | 4.6 ± 0.0 | 5.1 ± 0.1 | 5.0 ± 0.1 |
| HCT (%) | 43.9 ± 1.2 | 44.2 ± 0.3 | 42.3 ± 0.4 | 43.2 ± 1.1 | 42.6 ± 0.8 |
| PLT (×103/uL) | 237.0 ± 15.1 | 235.2 ± 16.3 | 210.4 ± 25.6 | 227.0 ± 10.1 | 239.6 ± 7.4 |
| WBC (×103/uL) | 8.7 ± 0.5 | 6.6 ± 0.7 | 5.0 ± 0.8 | 5.4 ± 0.3 | 5.4 ± 1.0 |
| LYM (×103/uL) | 2.5 ± 0.3 | 2.2 ± 0.2 | 2.0 ± 0.1 | 1.5 ± 0.1 | 1.7 ± 0.1 |
| MXD (×103/uL) | 0.6 ± 0.1 | 0.7 ± 0.1 | 0.5 ± 0.1 | 0.6 ± 0.1 | 0.5 ± 0.1 |
| NEUT (×103/uL) | 5.7 ± 0.1 | 3.7 ± 0.5 | 2.5 ± 0.7 | 3.3 ± 0.3 | 3.3 ± 0.9 |
Tissue factor quality control
The clot time in plasma or whole blood is related to the concentration of the Tf stimulus. Figure 1 shows representative clot times in whole blood (panel A) or plasma (panel B) when coagulation is initiated with 2 – 10 pM Tf. In our TEG assays, we use a 5 pM Tf stimulus. Figure 1 shows that the clot time in both blood and plasma is most sensitive to Tf when present at concentrations ranging from ∼ 3 – 6 pM. For our assays we chose a concentration of 5pM Tf, which was in this range. This concentration has previously been used in our other empirical models to yield a clot time that provides detection and evaluation of the dynamics of the phases of thrombin generation [16] along with platelet activation [18] and fibrin formation [19]. When we use a 5pM Tf stimulus, data from 3 independent Tf preparations yielded plasma and whole blood clot times of 5.12 ± 0.23 and 3.93 ± 0.23 minutes, respectively (Figure 1, panel C).
Figure 1.

Tissue factor quality control. Whole blood (panel A) and plasma (panel B) clot times increase dose dependently as the Tf concentration is decreased from 10 pM to 2 pM. Symbols (○, ●, ▼) represent data generated from 3 independent preparations of Tf. At a nominal concentration of 5 pM Tf which corresponds to the concentration used in out TEG assay, the whole blood and plasma clot times are 3.93 ± 0.23 and 5.12 ± 0.23 minutes, respectively (panel C).
Individual thrombelastography
The R time, or time to initial viscosity varied slightly among subjects [range: 6.35 ± 1.0 (S5) to 9.65 ± 1.23 minutes (S1)]. The R time for S1 was significantly higher when compared to S3 and S5 (p<0.05). In most subjects there was a trend toward an increased R time when tPA was added, however, this increase was not statistically significant. The angle (degrees, °), which approximates the kinetics of clot formation, ranged from 49.48 ± 6.07 ° (S4) to 59.18 ± 2.25 ° (S3) degrees in the absence of tPA but no differences were detected when subjects were compared. In addition, the angle was not significantly affected by the inclusion of tPA. The maximum amplitude (MA), a measure of clot strength, in the absence of tPA ranged from 53.77 ± 5.17 (S2) to 66.02 ± 1.90 mm (S1). In the absence of tPA, S1 had a significantly higher MA when compared to S2 and S4 (p<0.01) which could be explained by elevated fibrinogen levels (Table 3). The addition of tPA (1 – 2 nM) resulted in a concentration dependant decrease in the MA in S2 – S5. In the presence of 2 nM tPA the MA was significantly reduced in S2 – S5 (all p<0.05) compared to the MA in the absence of tPA. With increased tPA concentrations, fibrinolysis also increased. All subjects showed significantly increased fibrinolysis in the presence of 2 nM tPA compared to no tPA or 1 nM tPA (p<0.05). The values for R, angle, MA and LY60 are summarized for each subject in Table 2 and the clot-lysis profiles used to calculate TEG parameters are presented in Figure 2.
Table 3.
Thrombin-antithrombin (TAT), plasma PAI-1 and fibrinogen (Fg) levels. The data represent the mean ±SD, n=3 blood draws.
| TAT (nM) | PAI-1 (nM) | Fg (mg/mL) | |
|---|---|---|---|
| S1 | 119 ± 29 | 2.20 ± 0.42 | 3.50 ± 1.04 |
| S2 | 139 ± 26 | 0.29 ± 0.08 | 2.27 ± 0.18 |
| S3 | 99 ± 16 | 0.11 ± 0.03 | 2.39 ± 0.16 |
| S4 | 197 ± 28 | 0.61 ± 0.21 | 2.13 ± 0.09 |
| S5 | 158 ± 24 | 0.25 ± 0.03 | 2.92 ± 0.82 |
Table 2.
TEG parameters. R time, angle, MA and LY60 are summarized for all 5 subjects. The data represent the mean ±SD, n=3 blood draws.
| S1 | S2 | S3 | S4 | S5 | CVa (%) | |
|---|---|---|---|---|---|---|
|
| ||||||
| Clot Time: R Time (minutes) | ||||||
| No tPA | 9.65 ± 1.23 | 7.38 ± 0.63 | 6.37 ± 0.72 | 7.30 ± 1.10 | 6.35 ± 1.00 | 6 |
| 1nM tPA | 9.80 ± 1.55 | 7.63 ± 0.93 | 6.32 ± 0.48 | 7.88 ± 1.25 | 6.60 ± 0.73 | 7 |
| 1.5nM tPA | 9.88 ± 1.09 | 7.60 ± 0.44 | 6.63 ± 0.47 | 8.37 ± 1.05 | 6.82 ± 1.17 | 6 |
| 2nM tPA | 10.02 ± 1.49 | 7.83 ± 1.01 | 6.88 ± 0.60 | 7.90 ± 0.67 | 6.58 ± 0.68 | 7 |
|
| ||||||
| Kinetics of Clot Formation: Angle (degrees) | ||||||
|
| ||||||
| No tPA | 55.67 ± 3.48 | 53.73 ± 2.66 | 59.18 ± 2.25 | 49.48 ± 6.07 | 56.23 ± 4.24 | 7 |
| 1nM tPA | 53.12 ± 6.13 | 43.78 ± 8.70 | 60.38 ± 2.83 | 46.15 ± 12.81 | 54.17 ± 7.38 | 9 |
| 1.5nM tPA | 49.72 ± 9.51 | 47.75 ± 11.40 | 59.33 ± 1.18 | 44.10 ± 6.76 | 53.80 ± 11.72 | 4 |
| 2nM tPA | 51.13 ± 7.69 | 49.77 ± 4.63 | 54.88 ± 8.78 | 42.87 ± 11.23 | 54.02 ± 8.42 | 9 |
|
| ||||||
| Clot Strength: Maximum Amplitude (mm) | ||||||
|
| ||||||
| No tPA | 66.02 ± 1.90 | 53.77 ± 5.17 | 62.07 ± 1.40 | 53.93 ± 2.90 | 58.07 ± 4.05 | 3 |
| 1nM tPA | 66.55 ± 2.20 | 48.58 ± 10.37 | 58.98 ± 1.35 | 51.62 ± 5.36 | 53.98 ± 2.40 | 5 |
| 1.5nM tPA | 62.88 ± 4.88 | 45.18 ± 3.43 | 53.55 ± 1.69 | 46.25 ± 6.62 | 52.80 ± 6.15 | 6 |
| 2nM tPA | 63.22 ± 4.43 | 41.70 ± 1.67 | 48.90 ± 5.33 | 39.93 ± 4.73 | 48.88 ± 3.94 | 9 |
|
| ||||||
| Fibrinolysis: LY60 (%) | ||||||
|
| ||||||
| No tPA | 0.07 ± 0.12 | 2.28 ± 1.08 | 4.13 ± 0.84 | 3.07 ± 0.88 | 4.10 ± 1.16 | - |
| 1nM tPA | 0.20 ± 0.09 | 14.92 ± 10.08 | 17.23 ± 0.73 | 5.53 ± 2.67 | 8.65 ± 3.21 | 25 |
| 1.5nM tPA | 0.95 ± 0.52 | 43.73 ± 4.86 | 46.98 ± 4.62 | 18.68 ± 11.15 | 21.52 ± 7.90 | 31 |
| 2nM tPA | 2.52 ± 1.46 | 58.58 ± 4.10 | 54.53 ± 6.67 | 47.05 ± 12.77 | 36.47 ± 8.80 | 16 |
Figure 2.
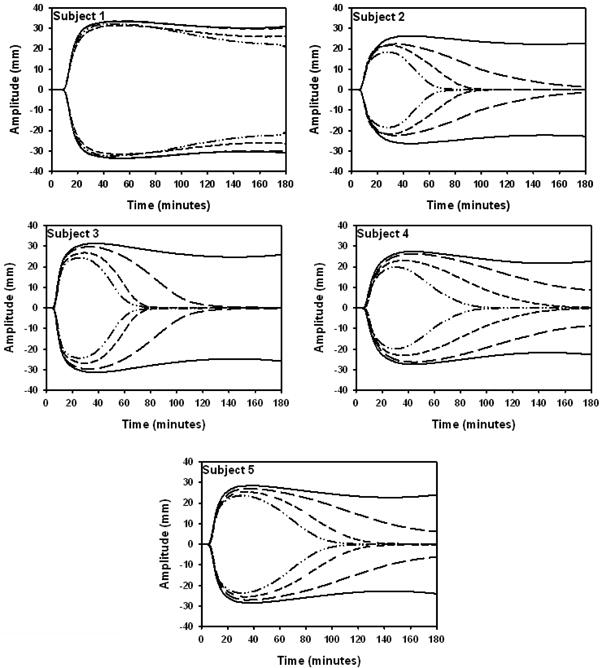
TEG tracings show the individualized response to tPA. Clot-lysis profiles are depicted for all 5 subjects in the absence of tPA (solid line) and with 1 (long dash), 1.5 (short dash) or 2 nM tPA (dash dot). The data represent the mean ±SD, n=3 blood draws (each experiment performed in duplicate).
PAI-1 and TAT
Plasma PAI-1 levels were similar in S2 – S5 (range: 0.11 ± 0.03 to 0.61 ± 0.21 nM) but significantly higher in S1 (2.2 ± 0.42 nM) compared to all other subjects (p<0.01) (Table 3). We observed that plasmin cleaved TAT in a manner that increases its immunogenicity in our TAT assay (data not shown). In the absence of tPA, subjects generated between 99 ± 16 and 197 ± 28 nM TAT over the course of the 3 hour TEG experiment. S4 generated significantly more TAT in the absence of tPA compared to S1 (p<0.05) and S3 (p<0.01).
Discussion
In this study, we developed a thrombelastography-based global assay to measure visco-elastic changes in blood which are due to fibrin formation and platelet activation. Our assay is unique in that it uses a well characterized Tf reagent [14;16;20;21] and CTI to block contact pathway coagulation. Using this system, we show that parameters which describe the dynamics of fibrin formation in whole blood, (R time, angle and MA) have a low intra-assay variability (CVa < 10%). Additionally, thrombin generation and blood cell counts are relatively consistent for a given individual over the 3 week period when blood was collected. Finally, we show that our assay can be used to monitor tPA initiated fibrinolysis in whole blood.
Our data, along with other reports [7] show that the fibrinolysis parameter LY60 has high intra-assay variability (CVa: 16 – 31%) thus making it an unreliable measures of fibrinolysis. With this system, we have identified a novel means to assess fibrinolytic potential in whole blood. Our data show, that there is a dose-dependent decrease in the MA when tPA is present and the MA is highly reproducible (CVa < 10%) at all concentrations of tPA. Our data suggest that the MA has potential to be used instead of the LY60 as a reliable measure of an individual's fibrinolytic potential.
The primary concern when conducting assays with tPA added prior to clot formation is that its inclusion might augment normal hemostasis. Since tPA is highly fibrin specific [22], present at relatively low concentrations and minimally affects the R time then it is not a major concern with this particular assay. Inducing fibrinolysis with low concentrations of tPA makes the fibrinolytic potential particularly susceptible to PAI-1 levels. We were able to show that PAI-1 levels were consistent in a given individual over time, but varied greatly between individuals. In this small study, PAI-1 levels ranged from 0.11 ± 0.03 to 2.20 ± 0.42 nM and the subject with the highest concentration of PAI-1 showed significantly impaired fibrinolysis. Individuals with diabetes [23] or the elderly [24] often have much higher PAI-1 levels than observed here. For these reasons, great care must be taken in choosing an appropriate concentration of tPA which reflects fibrinolytic potential in individuals with low or high PAI-1 without altering the dynamics of thrombin generation and clot formation. The timing of plasminogen activator release from endothelial cells relative to clot formation in vivo is unclear, but presumed to follow endothelial activation by thrombin [25-27]. Consequently, it is unlikely that tPA or urokinase type-PA would be present at significant concentrations prior to clot formation during normal hemostasis.
Our assay has a number of distinct advantages over other similar TEG assays. The primary advantage of our assay is that we are able to relate the visco-elasticity of the emerging blood clot to a very precise concentration of Tf containing a known concentration of procoagulant phospholipids. Our data show that the clot time induced in whole blood or plasma with our Tf reagent is highly reproducible (3.93 ± 0.23 and 5.12 ± 0.23 minutes, respectively for a 5 pM Tf stimulus). Another advantage of our assay is that it incorporates the fXIIa inhibitor, CTI. Since both the Tf pathway and the contact pathway can initiate coagulation in vitro, the results from TEG assays lacking a contact pathway inhibitor such as CTI, are difficult to interpret [28;29]. In assays lacking such an inhibitor, the relative contribution of each pathway to procoagulant processes are impossible to dissect. For example, a previously devised TEG assay lacking CTI [30] had a shorter clot time (5.9 ± 1.0 minutes, men only) than we observed (7.4 ± 1.3 minutes, n = 5) despite using a smaller “theoretical” concentration of Tf. In a second study lacking CTI, a higher concentration of Tf was used and the R time was shortened to 4.3 ± 0.4 minutes [7]. In these studies, the extent that the clot time is shortened may reflect the degree of contact pathway activation or differential Tf reagent activities. Therefore, assays which monitor global hemostasis including TEG assays, are most informative if a well characterized Tf factor reagent is used in conjunction with CTI.
Previously, Mann et al. showed that the dynamics of coagulant processes in whole blood are altered when calcium must be added to overcome the citrate [31]. Our assay makes use of minimally altered whole blood (i.e. no citrate) meaning that our assay is not confounded by the recalcification process. The method, however, is easily adapted to make use of clinical samples that may contain citrate. Finally, the reagents used in this assay are identical to those used in a para vivo whole blood assay that has been used extensively to monitor thrombin generation, platelet activation, fibrinopeptide cleavage, fXIII activation and fV degradation by activated protein C [15;16;32] as well as determine the effect of oral anticoagulants [33]. This means that the assay described here can be easily coupled with the para vivo whole blood assay to identify how procoagulant events contribute to mechanical clot strength.
Overall, our data suggest that when a reliable and highly reproducible Tf reagent is used, TEG may be used to monitor global hemostasis in minimally altered whole blood. In addition, in conjunction with TEG, baseline PAI-1 levels should be determined in tPA-based assays to account for fibrinolytic variation.
Acknowledgments
We would like to thank Ruhin Yuridullah and Matt Whelihan for their technical assistance and Matt Gissel for statistical assistance. This work was supported by a grant from The National Institutes of Health, USA [HL46703, Project 5 (KBZ)].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nemerson Y. Tissue Factor And Hemostasis. Blood. 1988;71:1–8. [PubMed] [Google Scholar]
- 2.Gailani D, Renne T. The intrinsic pathway of coagulation: a target for treating thromboembolic disease? J Thromb Haemost. 2007;5:1106–1112. doi: 10.1111/j.1538-7836.2007.02446.x. [DOI] [PubMed] [Google Scholar]
- 3.Lapecorella M, Mariani G. Factor VII deficiency: defining the clinical picture and optimizing therapeutic options. Haemophilia. 2008;14:1170–1175. doi: 10.1111/j.1365-2516.2008.01844.x. [DOI] [PubMed] [Google Scholar]
- 4.Ratnoff OD, COLOPY JE. A familial hemorrhagic trait associated with a deficiency of a clot-promoting fraction of plasma. J Clin Invest. 1955;34:602–613. doi: 10.1172/JCI103109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Komiyama Y, Pedersen AH, Kisiel W. Proteolytic Activation of Human Factors IX and X by Recombinant Human Factor VIIa: Effects of Calcium, Phospholipids, and Tissue Factor. Biochemistry. 1990;29:9418–9425. doi: 10.1021/bi00492a016. [DOI] [PubMed] [Google Scholar]
- 6.Silverberg SA, Nemerson Y, Zur M. Kinetics of the activation of bovine coagulation factor X by components of the extrinsic pathway. Kinetic behavior of two-chain factor VII in the presence and absence of tissue factor. J Biol Chem. 1977;252:8481–8488. [PubMed] [Google Scholar]
- 7.Kupesiz A, Rajpurkar M, Warrier I, Hollon W, Tosun O, Lusher J, Chitlur M. Tissue plasminogen activator induced fibrinolysis: standardization of method using thromboelastography. Blood Coagul Fibrinolysis. 2010;21:320–324. doi: 10.1097/MBC.0b013e32833464e9. [DOI] [PubMed] [Google Scholar]
- 8.Rugeri L, Levrat A, David JS, Delecroix E, Floccard B, Gros A, Allaouchiche B, Negrier C. Diagnosis of early coagulation abnormalities in trauma patients by rotation thrombelastography. J Thromb Haemost. 2007;5:289–295. doi: 10.1111/j.1538-7836.2007.02319.x. [DOI] [PubMed] [Google Scholar]
- 9.van der Kamp KW, Hauch KD, Feijen J, Horbett TA. Contact activation during incubation of five different polyurethanes or glass in plasma. J Biomed Mater Res. 1995;29:1303–1306. doi: 10.1002/jbm.820291018. [DOI] [PubMed] [Google Scholar]
- 10.Barenholz Y, Gibbes D, Litman BJ, Goll J, Thompson TE, Carlson FD. A Simple Method for the Preparation of Homogeneous Phospholipid Vesicle. Biochemistry. 1977;16(12):2806–2810. doi: 10.1021/bi00631a035. [DOI] [PubMed] [Google Scholar]
- 11.Lawson JH, Krishnaswamy S, Butenas S, Mann KG. Extrinsic pathway proteolytic activity. Methods Enzymol. 1993;222:177–195. doi: 10.1016/0076-6879(93)22013-6. [DOI] [PubMed] [Google Scholar]
- 12.Hojima Y, Pierce JV, Pisano JJ. Hageman Factor Fragment Inhibitor in Corn Seeds: Purification and Characterization. Thromb Res. 1980;20:149–162. doi: 10.1016/0049-3848(80)90381-3. [DOI] [PubMed] [Google Scholar]
- 13.Butenas S, Kalafatis M, Mann KG. Analysis of tissue plasminogen activator specificity using peptidyl fluorogenic substrates. Biochemistry. 1997;36:2123–2131. doi: 10.1021/bi9617670. [DOI] [PubMed] [Google Scholar]
- 14.Krudysz-Amblo J, Jennings ME, Mann KG, Butenas S. Carbohydrates and activity of natural and recombinant tissue factor. J Biol Chem. 2010;285:3371–3382. doi: 10.1074/jbc.M109.055178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Willemse JL, Hendriks DF. A role for procarboxypepidase U (TAFI) in thrombosis. Front Biosci. 2007;12:1973–1987. doi: 10.2741/2203. [DOI] [PubMed] [Google Scholar]
- 16.Brummel KE, Paradis SG, Butenas S, Mann KG. Thrombin functions during tissue factor-induced blood coagulation. Blood. 2002;100:148–152. doi: 10.1182/blood.v100.1.148. [DOI] [PubMed] [Google Scholar]
- 17.Fraser CG, Harris EK. Generation and application of data on biological variation in clinical chemistry. Crit Rev Clin Lab Sci. 1989;27:409–437. doi: 10.3109/10408368909106595. [DOI] [PubMed] [Google Scholar]
- 18.Butenas S, Cawthern KM, van't Veer C, DiLorenzo ME, Lock JB, Mann KG. Antiplatelet agents in tissue factor-induced blood coagulation. Blood. 2001;97:2314–2322. doi: 10.1182/blood.v97.8.2314. [DOI] [PubMed] [Google Scholar]
- 19.Brummel KE, Butenas S, Mann KG. An Integrated Study of Fibrinogen During Blood Coagulation. J Biol Chem. 1999;274:22862–22870. doi: 10.1074/jbc.274.32.22862. [DOI] [PubMed] [Google Scholar]
- 20.Orfeo T, Butenas S, Brummel-Ziedins KE, Mann KG. The tissue factor requirement in blood coagulation. J Biol Chem. 2005;280:42887–42896. doi: 10.1074/jbc.M505506200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rand MD, Lock JB, van't Veer C, Gaffney DP, Mann KG. Blood clotting in minimally altered whole blood. Blood. 1996;88:3432–3445. [PubMed] [Google Scholar]
- 22.Walker JB, Nesheim ME. A kinetic analysis of the tissue plasminogen activator and DSPA 1 cofactor activities of untreated and TAFIa-treated soluble fibrin degradation products of varying size. J Biol Chem. 2001;276:3138–3148. doi: 10.1074/jbc.M005876200. [DOI] [PubMed] [Google Scholar]
- 23.Vaughan DE. PAI-1 and atherothrombosis. J Thromb Haemost. 2005;3:1879–1883. doi: 10.1111/j.1538-7836.2005.01420.x. [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto K, Takeshita K, Kojima T, Takamatsu J, Saito H. Aging and plasminogen activator inhibitor-1 (PAI-1) regulation: implication in the pathogenesis of thrombotic disorders in the elderly. Cardiovasc Res. 2005;66:276–285. doi: 10.1016/j.cardiores.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 25.Levin EG, Marzec U, Anderson J, Harker LA. Thrombin stimulates tissue plasminogen activator release from cultured human endothelial cells. J Clin Invest. 1984;74:1988–1995. doi: 10.1172/JCI111620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levin EG, Stern D, Nawroth PP, Marlar RA, Fair DS, Fenton JW, II, Harker LA. Specificity of the Thrombin-Induced Release of Tissue Plasminogen Activator From Cultured Human Endothelial Cells. Thromb Haemost. 1986;56:115–119. [PubMed] [Google Scholar]
- 27.Shatos MA, Orfeo T, Doherty JM, Penar PL, Collen D, Mann KG. Alpha Thrombin Stimulates Urokinase Production and DNA Synthesis in Cultured Human Cerebral Microvascular Endothelial Cells. Arterioscler Thromb Vasc Biol. 1995;15:903–911. doi: 10.1161/01.atv.15.7.903. [DOI] [PubMed] [Google Scholar]
- 28.Luddington R, Baglin T. Clinical measurement of thrombin generation by calibrated automated thrombography requires contact factor inhibition. J Thromb Haemost. 2004;2:1954–1959. doi: 10.1111/j.1538-7836.2004.00964.x. [DOI] [PubMed] [Google Scholar]
- 29.Tappenden KA, Gallimore MJ, Evans G, Mackie IJ, Jones DW. Thrombin generation: a comparison of assays using platelet-poor and -rich plasma and whole blood samples from healthy controls and patients with a history of venous thromboembolism. Br J Haematol. 2007;139:106–112. doi: 10.1111/j.1365-2141.2007.06732.x. [DOI] [PubMed] [Google Scholar]
- 30.Sorensen B, Johansen P, Christiansen K, Woelke M, Ingerslev J. Whole blood coagulation thrombelastographic profiles employing minimal tissue factor activation. J Thromb Haemost. 2003;1:551–558. doi: 10.1046/j.1538-7836.2003.00075.x. [DOI] [PubMed] [Google Scholar]
- 31.Mann KG, Whelihan MF, Butenas S, Orfeo T. Citrate anticoagulation and the dynamics of thrombin generation. J Thromb Haemost. 2007;5:2055–2061. doi: 10.1111/j.1538-7836.2007.02710.x. [DOI] [PubMed] [Google Scholar]
- 32.Campbell JE, Brummel-Ziedins KE, Butenas S, Mann KG. Cellular regulation of blood coagulation: a model for venous stasis. Blood. 2010;116:6082–6091. doi: 10.1182/blood-2010-01-266395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brummel KE, Paradis SG, Branda RF, Mann KG. Oral anticoagulation thresholds. Circulation. 2001;104:2311–2317. doi: 10.1161/hc4401.098492. [DOI] [PubMed] [Google Scholar]