

Abstract
Aims
Caveolae are membrane microdomains where important signalling pathways are assembled and molecular effects transduced. In this study, we hypothesized that shear stress-mediated vasodilation (SSD) of mouse small coronary arteries (MCA) is caveolae-dependent.
Methods and results
MCA (80–150 μm) isolated from wild-type (WT) and caveolin-1 null (Cav-1−/−) mice were subjected to physiological levels of shear stress (1–25 dynes/cm2) with and without pre-incubation of inhibitors of nitric oxide synthase (L-NAME), cyclooxygenase (indomethacin, INDO), or cytochrome P450 epoxygenase (SKF 525A). SSD was endothelium-dependent in WT and Cav-1−/− coronaries but that in Cav-1−/− was significantly diminished compared with WT. Pre-incubation with L-NAME, INDO, or SKF 525A significantly reduced SSD in WT but not in Cav-1−/− mice. Vessels from the soluble epoxide hydrolase null (Ephx2−/−) mice showed enhanced SSD, which was further augmented by the presence of arachidonic acid. In donor–detector-coupled vessel experiments, Cav-1−/− donor vessels produced diminished dilation in WT endothelium-denuded detector vessels compared with WT donor vessels. Shear stress elicited a robust intracellular Ca2+ increase in vascular endothelial cells isolated from WT but not those from Cav-1−/− mice.
Conclusion
Integrity of caveolae is critical for endothelium-dependent SSD in MCA. Cav-1−/− endothelium is deficient in shear stress-mediated generation of vasodilators including NO, prostaglandins, and epoxyeicosatrienoic acids. Caveolae plays a critical role in endothelial signal transduction from shear stress to vasodilator production and release.
Keywords: Caveolae, Shear stress, Coronary artery, Soluble epoxide hydrolase, Calcium
1. Introduction
Caveolae are 50- to 100 nm cholesterol- and sphingolipid-rich flask-shaped invaginations of the plasma membrane found in most cell types of the body that form in the presence of the structural scaffolding protein, caveolin.1 Caveolae form plasma membrane microdomains which act as platforms for assembling signal transduction molecules that include receptors, ion channels, and enzymes such as kinases and phosphatases.2,3 Three isoforms of caveolin are known: caveolin-1 (Cav-1), caveolin-2, and caveolin-3.1 In the vasculature, caveolae are abundant in endothelial cells and Cav-1 is the major coat protein that is required for caveolae formation in the endothelium.1–3 Genetic ablation of Cav-1 (Cav-1−/−) attenuates both pressure- and agonist-induced vasoconstriction, and decreases agonist-induced vasodilation, suggesting that caveolae play an important role in the regulation of vascular reactivity.4,5 Caveolae are also postulated to be mechanosensors and function as mechanotransducers in the vascular endothelium.6
Uniquely situated at the interface between luminal haemodynamic forces and the underlying smooth muscle, the vascular endothelium transduces mechanical and chemical signals into vasoreactivity by the release of vasoactive molecules, which govern vascular tone and tissue perfusion. Shear stress is a potent endothelium-dependent physiological vasodilatory signal that regulates most tissue beds.7,8 Shear stress-mediated vasodilation (SSD) is known to involve major endothelium-dependent vasodilators including nitric oxide (NO),9 prostacyclin (PGI2),10 and endothelium-derived hyperpolarizing factors (EDHFs).10 Epoxyeicosatrienoic acids (EET), the cytochrome P450 (CYP) epoxygenase metabolites of arachidonic acid (AA), are a major form of EDHF in coronary circulation.11 In this study, we examined the role of caveolae in mediating SSD through NO, prostaglandins (PG), and EET signalling in mouse coronary arteries (MCA).
2. Methods
2.1. Animals
Male wild-type (WT) and Cav-1−/− mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA) at 10 weeks of age. The epoxide hydrolase null (Ephx2−/−) mice were generated as previously described.12 Handling and care of animals, as well as all animal procedures, were conducted in conformity with the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH Publication no. 85–23, revised 1996) and approved by the Institutional Animal Care and Use Committee, Mayo Foundation.
2.2. Preparation of mouse coronary arteries and SSD
Mouse coronary arteries were prepared as previously described.13 Briefly, mice were anaesthetized with sodium pentobarbital (100 mg/kg, intraperitoneal). Their hearts were rapidly excised and placed in ice-cold Krebs' solution (in mmol/L): NaCl 118.3, KCl 4.7, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, and dextrose 11.1, pH 7.4. Isolated small MCA (80–150 μm in diameter) were mounted in a vessel chamber filled with Krebs' solution, secured between two borosilicate glass micropipettes, and placed on the stage of an inverted microscope (CK40, Olympus) that was coupled to a CCD camera (OLY-105, Olympus) and a video micrometer (VIA-100, Boeckeler Instruments, Tucson, AZ, USA). The vessel lumen was filled with Krebs' solution and maintained at a constant intraluminal pressure of 80 mmHg using a syringe microinjection pump and a pressure-servo controller (Living Systems, Burlington, VT, USA) as shown in Supplementary material online, Figure S1 and is similar to that used by other investigators.14 Incremental levels of shear stress (1, 5, 10, 15, 20, and 25 dynes/cm2) were applied to each vessel through the microinjection pump with flow rates calculated according to the following equation:
where Q is the flow rate, D is the vessel diameter, τ is the shear stress, and η is the viscosity of fluid. Examples of vessel diameter measurements using videomicroscopy are shown in the Supplementary material online, Figure S2. Vessel diameters were monitored and measured continuously. Flow rates were adjusted to achieve the next level of shear stress based on the diameter reached.
2.3. Pharmacological interventions
Vessels were equilibrated for 60 min in oxygenated (21% O2, 5% CO2, balanced with N2, 37 °C) Krebs' solution. Endothelin-1 (ET-1, up to 10 nmol/L) was applied to pre-contract the vessels to 50–70% of the passive diameter. ET-1 produced similar diameter changes in vessels from WT (76.0 ± 4.0 µm, n = 22) and Cav-1−/− (77.9 ± 7.2 µm, n = 18). The effects of ET-1 were sustained for at least 30 min which was the usual duration of the shear stress experiments (vessel diameters were 66.0 ± 4.3 μm 5 min after ET-1 exposure and were 67.7 ± 4.7 μm after 30 min, n = 6, P = NS). Results on the % vessel pre-contraction by ET-1 before shear stress or other vasodilation interventions are tabulated in Supplementary material online, Table S1. Dilation in response to different physiological levels of shear stress was compared between WT and Cav-1−/− vessels in the presence and absence of 100 μmol/L N(ω)-nitro-l-arginine methyl ester (L-NAME, eNOS inhibitor), 10 μmol/L indomethacin (INDO, cyclooxygenase (COX) inhibitor), and 10 μmol/L SKF 525A (SKF, CYP epoxygenase inhibitor). Incubation with these inhibitors did not produce any significant effects on vessel diameters (Supplementary material online, Table S2). At the end of each experiment, vessels were constricted with 100 mmol/L KCl and then maximally dilated with a Ca2+-free solution. Vessels were not acceptable for experiments if they showed leaks, failed to constrict by 50% to 10 nmol/L ET-1 or to 100 mmol/L KCl, or failed to dilate to zero Ca2+ or to nitroprusside. The vasodilatory response was calculated as a percentage of the maximum diameter as defined by the following equation:
where DSS is the vessel diameter at a specific level of shear stress, DET is the vessel diameter after application of ET-1 in the absence of shear stress, and DMAX is the vessel diameter in zero Ca2+ Krebs' solution in the absence of shear stress but in the presence of ET-1. Continuous recordings of vessel diameters were performed using a real-time edge-detection system (V94 Living Systems Instrumentation) that detected and tracked the luminal vessel diameter. Signal output was acquired by a 16-bit data acquisition system (DIGIDATA 1321A, Axon Instruments, Foster City, CA, USA) with a sampling frequency of 2 kHz and no filter, and was continuously recorded using Axoscope software. In this study, each vessel was used for only one intervention/condition, so different sets of vessels were used for determination of the effects of endothelium denudation or pre-incubation of pharmacological agents.
In some vessels, the endothelium was denuded by slowly perfusing 3–5 mL of air through the lumen of unpressurized vessels. The effectiveness of endothelium denudation was verified by demonstrating that the vessel: (i) failed to dilate to 1 μmol/L acetylcholine; (ii) constricted normally to ET-1; and (iii) dilated normally to 100 μmol/L sodium nitroprusside. In the donor–detector-coupled vessel experiments, the donor vessels had intact endothelium, whereas the detector vessels were denuded of endothelium. Donor and detector vessels were mounted in separate chambers connected by a glass micropipette (Supplementary material online, Figure S1B).
2.4. Measurements of intracellular calcium in endothelial cells
Vascular endothelial cells were obtained by explantation from control and Cav-1−/− mouse aortas using Matrigel enriched with endothelial growth factors.15 The outgrowing cells were dissociated by dispase (10 U/mL), collected, and passaged in endothelial growth medium-2 (EGM-2, Lonza) supplemented with 5% foetal bovine serum, fibroblast growth factor, vascular growth factor, insulin-like growth factor, ascorbic acid, and hydrocortisone. The isolated cells were used within four passages and were characterized by immunostaining with antibodies against von Willebrand factor (1:200 dilution), an endothelial marker, and with antibodies against α-actin (1:200 dilution), a smooth muscle marker. Results showed that >85% of the cells in the preparation were endothelial cells.
For calcium imaging, cells were seeded on a slide and loaded with fura-2 AM (3 μmol/L, Invitrogen) for 30 min at 37°C. The slide was then assembled in a shear stress chamber similar to that previously described16 and placed on the stage of an inverted Olympus IX71 microscope. The cells were exposed to 11 dynes/cm2 of shear stress and the intracellular calcium fluorescence signals were measured as a ratio of fluorescence intensities at 510 nm from excitations of 340/380 nm using a Hamamatsu ORCA-R2 CCD camera with a Sutter LB-LS/17 light source. Background fluorescence was subtracted and the calcium signal (F) was normalized to baseline fluorescence (F0) and expressed as a ratio (F/F0) using MetaFluor software.
2.5. Materials
Chemicals were purchased from Sigma-Aldrich Co. (St Louis, MO, USA).
2.6. Statistical analysis
All data are expressed as mean ± SEM. Vasodilation is expressed as the percentage of change in vessel diameter relative to the maximum vessel diameter measured in the presence of a Ca2+-free solution. Vasodilation data on key findings expressed as the change in vessel diameter, which is considered to be the most accurate representation of SSD, are provided in Supplementary material online, Figures S3, S5, and S6. Drug concentration at half maximum effect (EC50) was determined by curve fitting using Origin software (OriginLab Corp., Northamptonm, MA, USA). Efficacies of endothelium-dependent vasodilation were compared by area-under-the curve calculations. One-way ANOVA was used to compare multiple groups and statistical significance was defined as P < 0.05.
3. Results
3.1. Shear stress-induced endothelium-dependent vasodilation is impaired in Cav-1−/− coronary arteries
SSD in MCA was significantly diminished in vessels from Cav-1−/− mice. Shear stress of 25 dynes/cm2 resulted in a 92.2 ± 0.7% dilation in WT vessels (n = 22) but only a 41.8 ± 1.5% dilation in Cav-1−/− coronaries (n = 18, P < 0.001) (Figure 1A and C). Removal of the endothelium reduced SSD significantly to 22.3 ± 3.3% at 25 dynes/cm2 in WT (n = 8, vs. endothelium intact, P < 0.001) and to 14.9 ± 1.3% in Cav-1−/− (n = 8, vs. vessels with intact endothelium, P < 0.001) (Figure 1B and C). Vasodilation expressed as vessel diameter change in micrometres is shown in Supplementary material online, Figure S3A and B. Similar results were obtained when caveolae formation was disrupted by treatment with methyl-β-cyclodextrin (Supplementary material online, Figure S3C and D). These results suggest that SSD in MCA is endothelium- and caveolae-dependent.
Figure 1.

Role of caveolae and effects of endothelium denudation on SSD. (A) Representative continuous recordings of vessel diameters of isolated coronary arteries from WT (left panel) and Cav-1−/− (right panel) mouse vessels were pre-contracted with ET-1 followed by shear stress-induced dilation. At the end of the experiments, vessels were exposed to 100 mmol/L KCl and then to zero Ca2+ Kreb’ solution. (B) Endothelium-denuded WT (left panel) and Cav-1−/− (right panel) mouse coronary arteries failed to dilate in response to acetylcholine (ACh 1 μmol/L) while the effects of sodium nitroprusside (SNP 100 μmol/L) remained intact. After endothelium removal, shear stress-induced dilation was significantly attenuated in both WT and Cav-1−/− vessels. (C) Group data showing SSD in WT (n = 22) and Cav-1−/− mice (n = 18) and the effects of endothelium denudation. WT-endo and Cav-1−/−-endo are vessels without endothelium, n = 8 for both, #P < 0.05, +P < 0.01, and *P < 0.001 vs. vessels with intact endothelium.
3.2. Impaired endothelium-dependent vasodilator pathways in Cav-1−/− coronary arteries
Animal studies have shown that SSD is mediated by the release of endothelium-dependent vasodilators including NO, PG, and EETs.8,17,18 SSD by 25 dynes/cm2 was significantly reduced after individual incubation with L-NAME (100 μmol/L), INDO (10 μmol/L), and SKF 525A (10 μmol/L), from 92.2 ± 0.7% (n = 22) to 61.0 ± 2.8% (n = 6), 69.1 ± 1.7% (n = 8), and 77.6 ± 1.7% (n = 12), respectively in WT (P < 0.001 for all three inhibitors vs. without drug pre-treatment), but had no effects in Cav-1−/− vessels (Figure 2). Representative continuous tracings of SSD after pre-incubation with L-NAME, INDO, and SKF 525A in WT and Cav-1−/− MCA are shown in Supplementary material online, Figure S4. These results suggest that all three vasodilator pathways are important in mediating SSD in normal MCA with a quantitative contribution in the order of NO > PG > EETs, but their effects are lost in Cav-1−/− vessels. After incubation with L-NAME, INDO, and SKF 525A together, vasodilation produced by shear stress was at a level comparable to that of Cav-1−/− vessels (Figure 2D). These results suggest that the endothelium of Cav-1−/− mice is deficient in the generation and release of shear stress-induced vasodilators.
Figure 2.

Effects of eNOS, COX, and CYP inhibition on SSD. Vessels were pre-treated with (A) L-NAME (100 μmol/L) for 30 min, with (B) Indomethacin (10 μmol/L) for 30 min, with (C) SKF 525A (10 μmol/L) for 60 min, and with (D) L-NAME + INDO + SKF 525A. #P < 0.05, +P < 0.01, and *P < 0.001 vs. no treatment.
3.3. Shear stress-induced dilation of coronary arteries from Ephx2−/− mice
To further delineate the role of EETs in SSD, we used coronary arteries from soluble epoxide hydrolase (sEH) null (Ephx2−/−) mice.12 sEH converts EETs to dihydroxyeicosatrienoic acids and is responsible for removal of EETs thereby diminishing their beneficial cardiovascular properties.19 In Ephx2−/− mice, the effects of EETs should be potentiated. In the presence of eNOS and COX inhibition with L-NAME and INDO, shear stress of 25 dynes/cm2 produced dilations of 46.2 ± 3.3% (n = 8) dilation in WT vessels but 70.7 ± 3.4% (n = 6) in Ephx2−/− coronary arteries (P < 0.001) (Figure 3A). Endothelium denudation reduced SSD in both WT and Ephx2−/− to the same level (Figure 3A). Results with vasodilation expressed in vessel diameter change are shown in Supplementary material online, Figure S6A. These results indicate that the levels of EETs are important in mediating SSD.
Figure 3.

Role of soluble epoxide hydrolase on SSD. (A) In the presence of L-NAME (100 μmol/L) and INDO (10 μmol/L), the same level of shear stress produced greater vasodilation in Ephx2−/− compared with WT vessels (WT n = 8 and Ephx2−/− n = 6, *P < 0.001 vs. WT). After endothelium denudation, SSD was significantly reduced in both Ephx2−/− and WT mice (n = 6 for both Ephx2−/−-Endo and WT–Endo; +P < 0.01 and *P < 0.001 vs. vessels with intact endothelium). In the presence of arachidonic acid (AA 10 μmol/L) in vessels pre-incubated with with L-NAME and INDO, SSD is increased both in WT (n = 10) (B) and in Ephx2−/− vessels (n = 4) (C) #P < 0.05 and *P < 0.001 vs. no AA).
After a 15 min incubation with 10 μmol/L AA, in the presence of L-NAME and INDO, SSD by 25 dynes/cm2 was augmented significantly by AA in both WT and Ephx2−/− MCA to 80.2 ± 1.3% (n = 10, P < 0.001 vs. without AA) and 84.7 ± 3.8% (n = 4, P < 0.05 vs. without AA), respectively (Figure 3B and C). These results confirmed that EETs are important mediators of SSD and their contribution can be enhanced by AA supplementation.
3.4. Impaired acetylcholine- and arachidonic acid-induced dilation in Cav-1−/− coronary arteries
We further assessed the ability of AA to produce dilation in mouse vessels in the absence of sheer stress. We found that AA dilated WT MCA in a concentration-dependent manner with an EC50 of 3.46 μM. At 10 μmol/L, AA produced 55.3 ± 6.1% (n = 4) dilation in WT MCA (Figure 4A). However, the ability of AA to produce dilation in Cav-1−/− MCA is significantly diminished. At 10 μmol/L, AA produced only 21.0 ± 4.6% (n = 4, P < 0.001 vs. WT) dilation in Cav-1−/− MCA (Figure 4A). Vasodilation expressed in vessel diameter change is shown in Supplementary material online, Figure S6B.
Figure 4.

Impaired endothelial function in Cav-1−/− coronary arteries. (A) AA-induced (10−10 to 10−4 mol/L) vasodilation in WT and Cav-1−/− coronary arteries with and without intact endothelium. #P < 0.05, +P < 0.01, and *P < 0.001 for Cav-1−/− vs. WT vessels. ##P < 0.05, ++P < 0.01, and **P < 0.001 for WT vessels denuded of endothelium (WT-endo) vs. WT vessels. (B) ACh-induced (10−10 to 10−4 mol/L) vasodilation in WT and Cav-1−/− coronary arteries. +P < 0.01 and *P < 0.001 for Cav-1−/− vs. WT vessels. (C) Sodium nitroprusside-induced (10−10 to 10−4 mol/L) vasodilation in WT and Cav-1−/− coronary arteries.
To further examine the non-shear stress-dependent endothelial function, we determine the effects of acetylcholine (ACh) on the dilation of WT and Cav-1−/− MCA. In WT vessels, ACh produced a concentration-dependent vasodilation with an EC50 of 0.27 μM. At 1 μmol/L, ACh produced 52.7 ± 3.3% dilation in WT vessels (n = 7). The effects of ACh were significantly blunted in Cav-1−/− vessels producing only 21.0 ± 1.6% dilation (n = 6, P < 0.001 vs. WT) at 1 μmol/L (Figure 4B). Vasodilation expressed in vessel diameter change is shown in Supplementary material online, Figure S6C. These results indicate that both shear stress-dependent and -independent endothelial functions are abnormal in Cav-1−/− coronary arteries.
To determine whether the vasodilation abnormalities were endothelial or smooth muscle in origin, we determine the effects of sodium nitroprusside (SNP) on WT and Cav-1−/− vessels. SNP produced similar dilations in WT and Cav-1−/− MCA (Figure 4C), suggesting that the ability of the contractile elements to produce vasodilation is intact, supporting that the vascular abnormalities associated with the absence of caveolae are endothelium-related.
3.5. Donor-detector-coupled vessels studies confirmed deficiency in shear stress-induced vasodilation by Cav-1−/− vessels
To confirm the deficiency in shear stress-induced generation and release of vasodilators in Cav-1−/− vessels, we performed experiments with donor–detector coupled vessels (Supplementary material online, Figure S1B). Donor vessels had intact endothelium, whereas detector vessels had endothelium removed. With WT donor vessels, 25 dynes/cm2 induced 45.9 ± 3.5% vasodilation in WT detector vessels that were denuded of endothelium (n = 6). In contrast, Cav-1−/− donor vessels only induced 26.8 ± 2.3% vasodilation in WT detector vessels (n = 7, P < 0.001 vs. WT donor vessels) (Figure 5). These results confirmed that Cav-1−/− MCA are deficient in generating and/or releasing vasodilators in response to shear stress.
Figure 5.

Effect of shear stress-induced coronary vasodilation in donor-detector-coupled vessels with WT and Cav-1−/− as donor vessels and endothelium-denuded WT vessels as detectors. SSD in detector vessels with WT donor vessels (closed circle) and Cav-1−/− donor vessels (open circle). #P < 0.05, +P < 0.01, and *P < 0.001 vs. WT donor vessels.
3.6. Deficient shear stress-induced calcium response in Cav-1−/− endothelial cells
To determine the mechanisms that underlie the endothelial dysfunction in Cav-1−/− MCA, we measured the intracellular Ca2+ response to shear stress in endothelial cells explanted from WT and Cav-1−/− aortas. Endothelial cells explanted from mouse aortas were immunohistochemically positive for von Willebrand factor but negative for α-actin which is positive for vascular smooth muscle cells (Figure 6A). Endothelial cells from WT vessels showed a robust increase in intracellular Ca2+ upon exposure to 11 dynes/cm2 of shear stress. In contrast, endothelial cells from Cav-1−/− vessels showed a significantly blunted response, indicating that the absence of caveolae has profound effects on Ca2+ homeostasis in endothelial cells in response to shear stress (Figure 6B).
Figure 6.
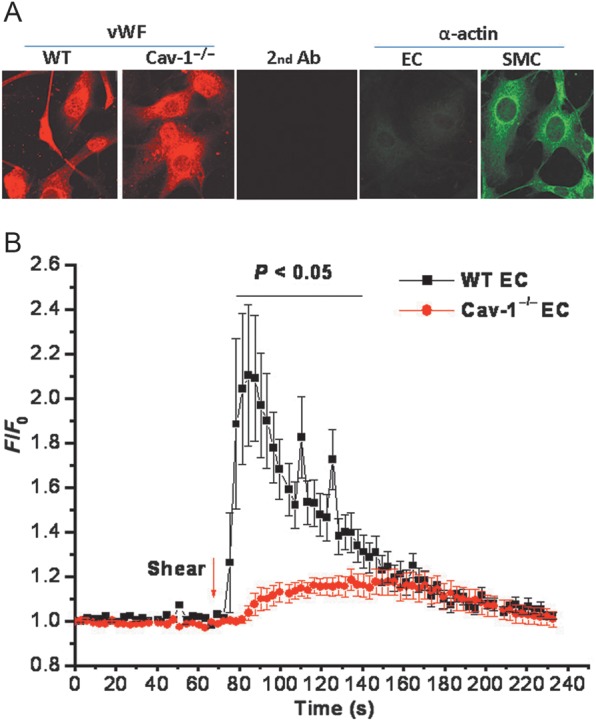
Impaired shear stress-induced Ca2+ response in vascular endothelial cells from Cav-1 −/− mice. (A) Characterization of endothelial cells explanted from WT and Cav-1−/− aortas. Endothelial cells were stained with antibodies against the endothelial cell marker, von Willebrand factor (vWF, red), and the smooth muscle cell marker, α-actin (green). (B) Increase in intracellular Ca2+ in response to shear stress (11 dynes/cm2) in fura-2 loaded endothelial cells from WT (n = 7) and Cav-1−/− (n = 8) vessels. Statistical difference between WT and Cav-1−/− cells was indicated by the bar above the tracings with P < 0.05.
4. Discussion
We have made several important observations in the present study. First, endothelium-dependent SSD of MCA is dependent on the integrity of caveolae. Secondly, shear stress-mediated activation of NO, PG, and EETs is caveolae-dependent. Thirdly, the contribution of EETs to SSD can be augmented by retardation of degradation or by supplementation with AA. Fourthly, in the absence of caveolae, endothelial function is abnormal with blunted Ca2+ response to shear stress. These novel important findings have significant physiological and clinical relevance.
Shear stress is an important physiological mechanical stimulus that regulates endothelial and vascular function. Endothelial cells are capable of transducing haemodynamic forces into intracellular signalling events.20 Our study shows that physiological levels of shear stress produce potent vasodilation in MCA, while vessels from Cav-1−/− mice only generate 38% of the response as that in WT (Figure 1). This finding reinforces the contention that caveolae may serve as the mechanical sensors that transduce the effects of shear stress.6,21
While NO has been found to be the dominant vasodilator in SSD in some previous reports,22,23 our study showed that in normal mouse coronary arteries, SSD involves all three major vasodilator pathways, which generate NO, PG, and EETs. eNOS is known to be a target of mechanotransduction mechanisms involving endothelial surface molecules such as integrin.24 Integrin activation phosphorylates eNOS at serine-1179, which results in eNOS activation and enhancement of NO production.25 Shear stress-induced activation of COX-mediated generation of PG is less well-understood. Studies have shown that flow-mediated vasodilation in eNOS-deficient mice is compensated by enhanced release of PG, and SSD is completely abolished by inhibitors of NO and PG.26 Other studies have reported that COX-2 is essential for both the shear stress response and maintenance of flow-mediated vasodilation in the absence of eNOS.27 The cross-talk between NO and PG is essential to maintain normal vasotone under physiological conditions. We found that the effects of eNOS and COX inhibition do not completely abolish SSD, which requires the additional inhibition of CYP epoxygenase. EETs are CYP epoxygenase metabolites of AA and have been shown to be EDHFs in a variety of vascular beds, including coronary arteries.10,28 Recent studies have shown that shear stress stimulates the release of EETs from endothelium, which directly hyperpolarizes smooth muscle in rat mesentery arteries.29 EETs dilate human coronary arteries through activation of BK channels.11
The quantitative contributions of these vasodilators are in the order of NO > PG > EETs in approximate ratios of 5:2:1 based on area-under-the-curve calculations for the endothelium-dependent vasodilations. These findings are different from previous reports that showed NO as the sole shear stress-induced vasodilator. The discrepancy in results may be due to differences in species,23 the different methods of baseline vasoconstriction,22 and the absence of caveolae in non-endothelial cells in the current study. However, non-NO dilators are involved in SSD is confirmed in Ephx2−/− vessels that the contribution of EETs to shear stress-induced endothelium-dependent vasodilation is augmented two-fold. Similarly, in the presence of L-NAME and INDO, supplementation with AA resulted in a 2.4-fold enhancement of SSD. These results suggest increased levels of AA may augment SSDs possibly through increased production of vasodilators that are non-prostaglandin metabolites of AA, possibly EETs.
It is rather striking that in the absence of caveolae, all three major vasodilator generating pathways lose their effectiveness in responding to shear stress. eNOS is targeted to caveolae and binding with Cav-1 inhibits eNOS and SSD through NO signalling is abnormal in Cav-1−/− vessels.2 In addition, basal eNOS phosphorylation of serine 1176, a key regulatory site of phosphorylation by many kinases including Akt, AMP kinase, and PKA, was reduced in Cav-1−/− vascular extracts, suggesting that flow activation of upstream kinases may be impaired.30
PG including PGI2 are also important endothelium-derived vasodilator released as a result of mechanotransduction by shear stress.27 In WT coronary arteries, COX-mediated generation of vasodilators accounts for about 25% of SSD. In human vascular endothelial cells, prostacyclin synthase (PGIS) is targeted to caveolae and colocalize with Cav-1 and the production of PGI2 in caveolae is important for the mediation of PGI2 cellular function.31,32 Our results suggest that the integrity of caveolae is critical in PG signalling. This is consistent with a previous observation that production of COX metabolites in caveolae is important for mediation of PG cellular function.31,32
EETs are EDHFs that produce vasodilation via activation of K+ channels, which in turn leads to membrane hyperpolarization and vasorelaxation through closure of Ca2+ influx pathways.33,34 Under basal conditions, shear stress-induced CYP epoxygenase-mediated vasodilation accounts for about 13% of endothelium-dependent vasodilation in WT coronary arteries (Figure 2), which is absent in Cav-1−/− vessels. CYP enzymes are not known to be targeted to caveolae microdomains. These findings raise the possibility that caveolae-dependent regulation of the vasodilator pathways may be mediated through a more fundamental mechanism than simple physical localization within caveolae microdomains. We speculate that in Cav-1−/− vessels, SSD is impaired due to loss of the spatial organization that is critical for the efficient generation of shear stress-induced vasodilators and for the coupling between vasodilators and effectors such as BK channels. In addition, our finding that shear stress-induced Ca2+ response in endothelial cells from Cav-1−/− vessels is profoundly abnormal elucidates a fundamental mechanism that underlies the shear stress-induced vascular dysfunction in Cav-1−/− coronary arteries.
Experiments involving donor–detector-coupled vessels confirmed that Cav-1−/− MCA are deficient in the generation and release of shear stress-induced endothelium-dependent vasodilators (Figure 5). With WT donor vessels, detector vessels showed a dose-dependent response to increasing levels of physiological shear stress with 45.9 ± 3.5% dilation by 25 dynes/cm2. In comparison, the ability of Cav-1−/− donor vessels to produce shear stress-induced dilation in detector vessels was significantly diminished (Figure 5).
Intracellular Ca2+ is a major determinant of vascular endothelial function.35 The dynamic spatiotemporal control of intracellular Ca2+ levels in vascular endothelial cells facilitates the modulation of multiple signalling pathways. Shear stress is the most potent stimulus in the activation of endothelial NOS and this process is Ca2+ dependent.36 In addition, Ca2+ activates kinases that phosphorylate specific serine residues and augment eNOS activities. Production of AA-derived vasodilators is also Ca2+ dependent. AA from intracellular stores in phospholipids is released by the rate-determining hydrolytic action of phospholipase A2 which is activated by Ca2+.37 AA serves as a precursor for eicosanoids including PGI2 through the cyclooxygenase pathway and EETs through the CYP pathway.38 Hence, disturbance in intracellular Ca2+ homeostasis as a result of loss of caveolae would affect the generation of NO, PG, and EET from all three vasodilator pathways. In support of our findings, a previous study showed that flow-induced Ca2+ response in endothelial cells starts at the caveolae and propagates as Ca2+ waves through the entire cell.39 The shear stress-induced endothelial Ca2+ increase is dependent on extracellular Ca2+ entry.40 The major Ca2+ entry pathways including the voltage-gated Ca2+ channels, the transient receptor potentialchannels, and the store-operated Ca2+ entry Orai channels are known to be localized to lipid rafts/caveolae.41 Loss of the caveolae microdomains may result in the abnormal activation of the Ca2+ entry mechanisms. Our findings in this study provide further support that caveolae are mechanosensors and are important in mediating shear stress-induced endothelial function, but other mechanisms of mechanosensing are also known including the role of endothelial cell glycocalyx.42
In this study, we have presented compelling evidence that shear stress-induced coronary vasodilation is caveolae-dependent. Although we are unable to measure directly the generation of vasodilators in the mouse coronary arteries, our results suggest that with the loss of caveolae, NO, PG, and EET-mediated SSD is abolished. In addition, in the absence of caveolae, shear stress fails to elicit a normal endothelial intracellular Ca2+ increase which is critical for the generation of vasodilators. Our results support the notion that caveolae are critical determinants that transduce haemodynamic signals into vascular responses.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This study was supported by funding from the National Institute of Health, National Institute of Heart Lung and Blood Institute (HL080118 and HL74180), and the Intramural Research program of the National Institute of Environmental Health Sciences (Z01 ES025034).
Supplementary Material
References
- 1.Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev. 2002;54:431–467. doi: 10.1124/pr.54.3.431. [DOI] [PubMed] [Google Scholar]
- 2.Gratton JP, Bernatchez P, Sessa WC. Caveolae and caveolins in the cardiovascular system. Circulation Research. 2004;94:1408–1417. doi: 10.1161/01.RES.0000129178.56294.17. [DOI] [PubMed] [Google Scholar]
- 3.Patel HH, Murray F, Insel PA. Caveolae as organizers of pharmacologically relevant signal transduction molecules. Annu Rev Pharmacol Toxicol. 2008;48:359–391. doi: 10.1146/annurev.pharmtox.48.121506.124841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adebiyi A, Zhao G, Cheranov SY, Ahmed A, Jaggar JH. Caveolin-1 abolishment attenuates the myogenic response in murine cerebral arteries. Am J Physiol. 2007;292:H1584–H1592. doi: 10.1152/ajpheart.00584.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaw L, Sweeney MA, O'Neill SC, Jones CJ, Austin C, Taggart MJ. Caveolae and sarcoplasmic reticular coupling in smooth muscle cells of pressurised arteries: the relevance for Ca2+ oscillations and tone. Cardiovasc Res. 2006;69:825–835. doi: 10.1016/j.cardiores.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 6.Yu J, Bergaya S, Murata T, Alp IF, Bauer MP, Lin MI, et al. Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. J Clin Invest. 2006;116:1284–1291. doi: 10.1172/JCI27100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jazuli F, Pyke KE. The impact of baseline artery diameter on flow-mediated vasodilation: a comparison of brachial and radial artery responses to matched levels of shear stress. Am J Physiol. 2011;301:H1667–H1677. doi: 10.1152/ajpheart.00487.2011. [DOI] [PubMed] [Google Scholar]
- 8.Mendoza SA, Fang J, Gutterman DD, Wilcox DA, Bubolz AH, Li R, et al. TRPV4-mediated endothelial Ca2+ influx and vasodilation in response to shear stress. Am J Physiol. 2010;298:H466–H476. doi: 10.1152/ajpheart.00854.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Resnick N, Yahav H, Shay-Salit A, Shushy M, Schubert S, Zilberman LC, et al. Fluid shear stress and the vascular endothelium: for better and for worse. Prog Biophys Mol Biol. 2003;81:177–199. doi: 10.1016/s0079-6107(02)00052-4. [DOI] [PubMed] [Google Scholar]
- 10.Edwards G, Feletou M, Weston AH. Endothelium-derived hyperpolarising factors and associated pathways: a synopsis. Pflugers Arch. 2010;459:863–879. doi: 10.1007/s00424-010-0817-1. [DOI] [PubMed] [Google Scholar]
- 11.Huang A, Sun D, Jacobson A, Carroll MA, Falck JR, Kaley G. Epoxyeicosatrienoic acids are released to mediate shear stress-dependent hyperpolarization of arteriolar smooth muscle. Circ Res. 2005;96:376–383. doi: 10.1161/01.RES.0000155332.17783.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng Y, Edin ML, Theken KN, Schuck RN, Flake GP, Kannon MA, et al. Endothelial CYP epoxygenase overexpression and soluble epoxide hydrolase disruption attenuate acute vascular inflammatory responses in mice. FASEB J. 2011;25:703–713. doi: 10.1096/fj.10-171488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu T, Zhang DM, Wang XL, He T, Wang RX, Chai Q, et al. Regulation of coronary arterial BK channels by caveolae-mediated angiotensin II signaling in diabetes mellitus. Circ Res. 2010;106:1164–1173. doi: 10.1161/CIRCRESAHA.109.209767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koller A, Sun D, Kaley G. Role of shear stress and endothelial prostaglandins in flow- and viscosity-induced dilation of arterioles in vitro. Circ Res. 1993;72:1276–1284. doi: 10.1161/01.res.72.6.1276. [DOI] [PubMed] [Google Scholar]
- 15.Suh SH, Vennekens R, Manolopoulos VG, Freichel M, Schweig U, Prenen J, et al. Characterisation of explanted endothelial cells from mouse aorta: electrophysiology and Ca2+ signalling. Pflugers Arch. 1999;438:612–620. doi: 10.1007/s004249900085. [DOI] [PubMed] [Google Scholar]
- 16.Wang XL, Fu A, Raghavakaimal S, Lee HC. Proteomic analysis of vascular endothelial cells in response to laminar shear stress. Proteomics. 2007;7:588–596. doi: 10.1002/pmic.200600568. [DOI] [PubMed] [Google Scholar]
- 17.Merkus D, Sorop O, Houweling B, Boomsma F, van den Meiracker AH, Duncker DJ. NO and prostanoids blunt endothelin-mediated coronary vasoconstrictor influence in exercising swine. Am J Physiol. 2006;291:H2075–H2081. doi: 10.1152/ajpheart.01109.2005. [DOI] [PubMed] [Google Scholar]
- 18.Loot AE, Popp R, Fisslthaler B, Vriens J, Nilius B, Fleming I. Role of cytochrome P450-dependent transient receptor potential V4 activation in flow-induced vasodilatation. Cardiovasc Res. 2008;80:445–452. doi: 10.1093/cvr/cvn207. [DOI] [PubMed] [Google Scholar]
- 19.Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov. 2009;8:794–805. doi: 10.1038/nrd2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davies PF, Polacek DC, Shi C, Helmke BP. The convergence of haemodynamics, genomics, and endothelial structure in studies of the focal origin of atherosclerosis. Biorheology. 2002;39:299–306. [PMC free article] [PubMed] [Google Scholar]
- 21.Park H, Go YM, Darji R, Choi JW, Lisanti MP, Maland MC, et al. Caveolin-1 regulates shear stress-dependent activation of extracellular signal-regulated kinase. Am J Physiol. 2000;278:H1285–H1293. doi: 10.1152/ajpheart.2000.278.4.H1285. [DOI] [PubMed] [Google Scholar]
- 22.Bagi Z, Koller A, Kaley G. PPARgamma activation, by reducing oxidative stress, increases NO bioavailability in coronary arterioles of mice with Type 2 diabetes. Am J Physiol. 2004;286:H742–H748. doi: 10.1152/ajpheart.00718.2003. [DOI] [PubMed] [Google Scholar]
- 23.Csiszar A, Ungvari Z, Edwards JG, Kaminski P, Wolin MS, Koller A, et al. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res. 2002;90:1159–1166. doi: 10.1161/01.res.0000020401.61826.ea. [DOI] [PubMed] [Google Scholar]
- 24.Shyy JY, Chien S. Role of integrins in endothelial mechanosensing of shear stress. Circ Res. 2002;91:769–775. doi: 10.1161/01.res.0000038487.19924.18. [DOI] [PubMed] [Google Scholar]
- 25.Koshida R, Rocic P, Saito S, Kiyooka T, Zhang C, Chilian WM. Role of focal adhesion kinase in flow-induced dilation of coronary arterioles. Arterioscler Thromb Vasc Biol. 2005;25:2548–2553. doi: 10.1161/01.ATV.0000188511.84138.9b. [DOI] [PubMed] [Google Scholar]
- 26.Sun D, Liu H, Yan C, Jacobson A, Ojaimi C, Huang A, et al. COX-2 contributes to the maintenance of flow-induced dilation in arterioles of eNOS-knockout mice. Am J Physiol. 2006;291:H1429–H1435. doi: 10.1152/ajpheart.01130.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Di Francesco L, Totani L, Dovizio M, Piccoli A, Di Francesco A, Salvatore T, et al. Induction of prostacyclin by steady laminar shear stress suppresses tumor necrosis factor-alpha biosynthesis via heme oxygenase-1 in human endothelial cells. Circ Res. 2009;104:506–513. doi: 10.1161/CIRCRESAHA.108.191114. [DOI] [PubMed] [Google Scholar]
- 28.Zhou W, Wang XL, Lamping KG, Lee HC. Inhibition of protein kinase Cbeta protects against diabetes-induced impairment in arachidonic acid dilation of small coronary arteries. J Pharmacol Exp Ther. 2006;319:199–207. doi: 10.1124/jpet.106.106666. [DOI] [PubMed] [Google Scholar]
- 29.Larsen BT, Miura H, Hatoum OA, Campbell WB, Hammock BD, Zeldin DC, et al. Epoxyeicosatrienoic and dihydroxyeicosatrienoic acids dilate human coronary arterioles via BK(Ca) channels: implications for soluble epoxide hydrolase inhibition. Am J Physiol. 2006;290:H491–H499. doi: 10.1152/ajpheart.00927.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fisslthaler B, Dimmeler S, Hermann C, Busse R, Fleming I. Phosphorylation and activation of the endothelial nitric oxide synthase by fluid shear stress. Acta Physiol Scand. 2000;168:81–88. doi: 10.1046/j.1365-201x.2000.00627.x. [DOI] [PubMed] [Google Scholar]
- 31.Spisni E, Bianco MC, Griffoni C, Toni M, D'Angelo R, Santi S, et al. Mechanosensing role of caveolae and caveolar constituents in human endothelial cells. J Cell Physiol. 2003;197:198–204. doi: 10.1002/jcp.10344. [DOI] [PubMed] [Google Scholar]
- 32.Spisni E, Griffoni C, Santi S, Riccio M, Marulli R, Bartolini G, et al. Colocalization prostacyclin (PGI2) synthase–caveolin-1 in endothelial cells and new roles for PGI2 in angiogenesis. Exp Cell Res. 2001;266:31–43. doi: 10.1006/excr.2001.5198. [DOI] [PubMed] [Google Scholar]
- 33.Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- 34.Isshiki M, Anderson RG. Function of caveolae in Ca2+ entry and Ca2+-dependent signal transduction. Traffic. 2003;4:717–723. doi: 10.1034/j.1600-0854.2003.00130.x. [DOI] [PubMed] [Google Scholar]
- 35.Sandow SL, Senadheera S, Grayson TH, Welsh DG, Murphy TV. Calcium and endothelium-mediated vasodilator signaling. Adv Exp Med Biol. 2012;740:811–831. doi: 10.1007/978-94-007-2888-2_36. [DOI] [PubMed] [Google Scholar]
- 36.Fleming I, Busse R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1–R12. doi: 10.1152/ajpregu.00323.2002. [DOI] [PubMed] [Google Scholar]
- 37.Rosa AO, Rapoport SI. Intracellular- and extracellular-derived Ca(2+) influence phospholipase A(2)-mediated fatty acid release from brain phospholipids. Biochim Biophys Acta. 2009;1791:697–705. doi: 10.1016/j.bbalip.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O'Donnell VB, Maskrey B, Taylor GW. Eicosanoids: generation and detection in mammalian cells. Methods Mol Biol. 2009;462:5–23. [PubMed] [Google Scholar]
- 39.Isshiki M, Ando J, Korenaga R, Kogo H, Fujimoto T, Fujita T, et al. Endothelial Ca2+ waves preferentially originate at specific loci in caveolin-rich cell edges. Proc Natl Acad Sci USA. 1998;95:5009–5014. doi: 10.1073/pnas.95.9.5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ungvari Z, Sun D, Huang A, Kaley G, Koller A. Role of endothelial [Ca2+]i in activation of eNOS in pressurized arterioles by agonists and wall shear stress. Am J Physiol. 2001;281:H606–H612. doi: 10.1152/ajpheart.2001.281.2.H606. [DOI] [PubMed] [Google Scholar]
- 41.Pani B, Singh BB. Lipid rafts/caveolae as microdomains of calcium signaling. Cell Calcium. 2009;45:625–633. doi: 10.1016/j.ceca.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mochizuki S, Vink H, Hiramatsu O, Kajita T, Shigeto F, Spaan JA, et al. Role of hyaluronic acid glycosaminoglycans in shear-induced endothelium-derived nitric oxide release. Am J Physiol. 2003;285:H722–H726. doi: 10.1152/ajpheart.00691.2002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.