

Abstract
Cryptosporidium parasites infect intestinal cells, causing cryptosporidiosis. Despite its high morbidity and association with stunting in the developing world, current therapies for cryptosporidiosis have limited efficacy. Calcium-dependent protein kinases (CDPKs) are essential enzymes in the biology of protozoan parasites. CDPK1 was cloned from the genome of Cryptosporidium parvum, and potent and specific inhibitors have been developed based on structural studies. In this study, we evaluated the anti-Cryptosporidium activity of a novel CDPK1 inhibitor, 1294, and demonstrated that 1294 significantly reduces parasite infection in vitro, with a half maximal effective concentration of 100 nM. Pharmacokinetic studies revealed that 1294 is well absorbed, with a half-life supporting daily administration. Oral therapy with 1294 eliminated Cryptosporidium parasites from 6 of 7 infected severe combined immunodeficiency–beige mice, and the parasites did not recur in these immunosuppressed mice. Mice treated with 1294 had less epithelial damage, corresponding to less apoptosis. Thus, 1294 is an important lead for the development of drugs for treatment of cryptosporidiosis.
Keywords: Cryptosporidium parvum, cryptosporidiosis, Calcium-dependant Protein kinase, CpCDPK-1, bumped kinase inhibitor, antiparasitic
The apicomplexan parasites of the genus Cryptosporidium infect human intestinal epithelial cells. Infections by Cryptosporidium species are important causes of diarrheal diseases worldwide [1]. While cryptosporidiosis causes a self-limiting, acute diarrheal disease in immunocompetent individuals, it can cause persistent diarrhea with severe stunting when malnourished young children are infected [1–3]. Furthermore, cryptosporidiosis is a leading cause of life-threatening, chronic diarrheal disease in immunocompromised patients. Cryptosporidium is found in about 16% of patients who have AIDS and persistent diarrhea [4, 5].
Current treatment of cryptosporidiosis is unsatisfactory. While some agents may shorten the course of infection in immunocompetent hosts, there is no antimicrobial chemotherapeutic agent that will reliably eradicate the organism from immunocompromised hosts. Paromomycin is an aminoglycoside antibiotic poorly absorbed from the gut epithelium [6]. In cell culture, it is weakly effective against Cryptosporidium and has only limited efficacy in Cryptosporidium-infected gnotobiotic piglets and mice [7, 8]. Controlled trials of paromomycin in Cryptosporidium-infected AIDS patients have demonstrated that Cryptosporidium is seldom eliminated from stools, and relapse of diarrhea occurs in over half of the patients [5, 9]. Nitazoxanide is licensed for the treatment of cryptosporidiosis in nonimmunodeficient children and adults after being shown in controlled trials to reduce the time of cryptosporidial diarrhea. However, nitazoxanide is expensive and thus not widely available to the highest risk groups in the developing world. Nitazoxanide does not work well in AIDS patients, the group at highest risk in the United States [5]. Thus, new drugs are needed to treat patients, especially those with the highest risk for infection.
Apicomplexan parasites rely on calcium-mediated signaling for a variety of vital functions [10]. These functions are controlled in part by a number of specialized systems for uptake and release of calcium, which acts as a secondary messenger, and on the functions of calcium-dependent proteins [11]. For the apicomplexan Toxoplasma gondii, calcium-dependent protein kinase 1 (TgCDPK1) is involved in secretion, invasion, and gliding motility [11]. Structural studies for drug development have shown that TgCDPK1 contains an enlarged adenosine triphosphate (ATP)–binding pocket relative to that observed in human kinases, with a glycine gatekeeper residue, which makes it uniquely sensitive to bumped kinase inhibitors (BKIs) that target this structural feature [12]. We and others have successfully developed a number of potent inhibitors of TgCDPK1 activity [13, 14]. Interestingly, Cryptosporidium CDPK1(CpCDPK1) also contains an enlarged ATP-binding pocket with a glycine gatekeeper residue that is very similar to that of TgCDPK1. A gene that is 99% identical to CpDCPK1, with a glycine gatekeeper residue, is present in the Cryptosporidium hominis genome, and we expect will have the exact same inhibitor-binding mode as CpCDPK1. In recent studies, we showed that BKIs based on a pyrazolopyrimidine scaffold are also able to potently block the activity of CpCDPK1. In addition, we demonstrated that pyrazolopyrimidine-based inhibitors are able to block the infection of Cryptosporidium parvum in cell culture [12]. On the basis of these structural, biochemical, and pharmacokinetics studies, we hypothesized that a recently synthesized pyrazolopyrimidine-based compound, 1294, would be useful to treat cryptosporidiosis. Here, we show the anti-cryptosporidial efficacy of 1294 in human cells, as well as in a severe combined immunodeficiency (SCID)–beige mouse cryptosporidiosis model.
METHODS
Cell Culture In Vitro Infection and Drug Activity Assays
Human cells were prepared as follows. HCT-8 cells (ATCC) were seeded on 24-well plates with 500 µL of complete medium (Roswell Park Memorial Institute [RPMI], 10% fetal bovine serum [FBS], and 1% antibiotic antimycotic solution [penicillin/streptomycin/amphotericin B; Life Technologies, Grand Island, NY]) and incubated at 37°C in 5% CO2 overnight. After reaching 90% of confluence, the medium was removed, and the cells were infected as described below. Parasites were prepared as follows. For the infection, 1 × 108 Cryptosporidium oocysts (Iowa strain obtained from Sterling parasitology laboratory at the University of Arizona) were diluted and washed 3 times at 500 g for 10 minutes with sterile phosphate-buffered saline (PBS). The pellet was resuspended in 500 µL acidic water (pH 2–3) and incubated for 10 minutes on ice. After centrifugation at 500 ×g for 10 minutes, the supernatant was discarded, and the pellet was resuspended in infection medium (complete medium supplemented with 0.8% taurocholate) and incubated for 1 hour at 37°C. Microscopy was used to confirm that at least 95% of the sporozoites were excysted, before continuing with the experiment. The sporozoites were diluted at a concentration of approximately 2 × 105 sporozoites/mL of complete medium. The 1294 compound was prepared as follows. Compound was diluted in 500 µL of dimethyl sulfoxide (DMSO; 0.5%) at final concentrations of 0, 0.01, 0.1, 1, and 5 µM. The solution containing the drug was mixed with the sporozoites and then added to the HCT-8 cells and cultured for 2 hours at 37°C in 5% CO2. Controls included infected and uninfected HCT-8 cells in medium with DMSO. After incubation, the medium was removed, cells were washed 3 times with PBS, and fresh medium without drug was added. After overnight incubation at 37°C in 5% CO2, medium was removed, and the cells were incubated with trypsin (0.025% for 15 minutes at 37°C) and then diluted with RPMI and 10% FBS. Cells were washed 3 times with complete medium by centrifugation at 500 ×g for 10 minutes. Cell pellets were stored at −20°C until quantitative real-time polymerase chain reaction (qRT-PCR) analysis. Kinase assays were performed as previously described [12, 14].
Quantitation of Cryptosporidium by qRT-PCR
Quantitation of Cryptosporidium was performed by qRT-PCR as previously described [12]. Briefly, RNA was isolated from the pellet, using a commercial kit (RNeasy Plus Kit, Qiagen, Valencia, CA). The final RNA concentration and quality were determined with a spectrophotometer (Nanodrop 1000, Thermo Scientific, Wilmington, DE). The parasite numbers were monitored by qRT-PCR, using the Applied Biosystems 7 obtained 500 Real-Time PCR System (Life Technologies, Grand Island, NY). For all reactions, the 1-step RT-PCR Super Script III was used, with approximately 250 ng of each sample and specific primers for C. parvum Cp-F (CAA TCA GCA ACC AAG CTC AA) and Cp-R (TTG TTG AGC AGC AGG TTC AG). Quantitation was normalized to host 18 s RNA (CCG ATA ACG AAC GAG ACT CTG G-3′, forward) and 5′-(TAG GGT AGG CAC ACG CTG AGC C, reverse). The qRT-PCR conditions were 1 cycle at 95°C for 5 minutes, followed by 60 cycles at 95°C for 15 seconds and then 66°C for 1 minute. The specificity of the primers was confirmed by melting curve analysis. For parasite quantitation, a standard curve with serial dilutions of RNA from a known number of parasites was included in each reaction plate. Results with this method were similar to those obtained by lectin staining or immunofluorescence.
Infection and Treatment of and Stool Collection From Mice
To test the anti-Cryptosporidium activity in an animal model, 7-week-old SCID-beige mice were infected with 1 × 106 C. parvum oocysts (Iowa strain) by oral gavage [15]. Weight, food consumption, and stool characteristics were monitored daily. Beginning on day 4 after infection, 100 mg/kg of 1294 (diluted in vehicle [7% Tween 80, 3% ethanol, and 90% normal saline]) or vehicle control (placebo) was administered by oral gavage daily for 10 days. To determine the efficacy of the treatment, we evaluated the presence of parasites in stool specimens by qRT-PCR. Stool (approximately 25 mg) was collected every day for the first 13 days and then every 3–6 days after the second week, up to day 33; all samples were stored at −20°C until subsequent analysis. Negative controls for qRT-PCR and for histological analysis included specimens from uninfected animals.
DNA Extraction and qRT-PCR Assays
For DNA extractions, we used 250 mg (approximately 10 pellets) of stool from each mouse. DNA was extracted and purified using the QIAamp DNA Stool Mini Kit (Qiagen). The purity and concentration were determined by spectrophotometry (Nanodrop 1000). The samples were stored at −20°C until subsequent analysis. The parasite burden was determined by qRT-PCR (Applied Biosystems 7500 Real-Time PCR System), using the Platinum SYBR Green qPCR SuperMix-UDG Kit (Life Technologies) with primers for C. parvum Cp-F and Cp-R (described above). The qRT-PCR conditions were 1 cycle at 95°C for 5 minutes, and subsequently 60 cycles at 95°C for 15 seconds followed by 66°C for 1 minute. To test the specificity of the primers, an additional dissociation stage was added at the end of the reaction for dissociation curve analysis. A standard curve was generated from serial dilutions of DNA from a known number of parasites spiked in mice stool specimens and was included in each reaction plate.
Histological Analysis
To determine the intestinal damage, we analyzed sections of treated, untreated, and uninfected mice. The mice were euthanized on day 35, and the terminal ileum of 3 mice from each group was collected and fixed in formalin. The tissue was transferred to 70% methyl and isopropyl alcohol solution (Flex 100, Thermo Fisher Scientific, Waltham, MA) to minimize overdehydration, and then the samples were submitted to the University of Texas Medical Branch histopathology core laboratory for sectioning and staining. All tissue sections were 5-µm thick. Apoptosis was assayed by immunofluorescence with the TUNEL apoptosis detection kit (Millipore, Billerica, MA). For quantitative analysis of the histological damages, we evaluated the number of apoptotic cells by fluorescent microscopy as described before with some modifications [16]. Briefly, to evaluate the apoptosis level in the intestine, the number of apoptotic cells was assessed semiquantitatively (score, 0–3) by an observer masked to the treatment group, as follows: no apoptosis, 0; low apoptosis (1–5 apoptotic cells/field), 1; moderate apoptosis (6–10 apoptotic cells/field), 2; and heavy apoptosis (>10 apoptotic cells/field), 3. The average of the number of apoptotic cells from a total of 250 cells counted in 3 representative fields of each sample was taken as the total score of apoptotic damage.
RESULTS
Development, Pharmacokinetics, and Mammalian Cell Toxicity of 1294
We cloned, expressed, and determined the structure of C. parvum CDPK1 and showed that the enzyme is uniquely susceptible to BKIs because this kinase possesses an extremely rare glycine gatekeeper residue [12]. BKIs also inhibit the growth of C. parvum in vitro in mammalian cells and work better if added at the time of infection than if added after infection [12]. Initially, our most promising CpCDPK1 inhibitor was BKI-1 [17] (referred to as compound 3b in table 1 of the article by Larson et al [18]). However, we found that BKI-1 was cleared from the bloodstream of rodents too quickly to provide continuous exposure when administered daily. On the basis of the hypothesis that BKI-1 is likely metabolized by CyP450 enzymes, we determined BKI-1 metabolism by liver microsomes in the presence of NADPH. We found that the piperidine group of BKI-1 was oxidized and reasoned that alkylation of the secondary nitrogen of BKI-1 would slow this metabolism without affecting binding to CpCPDK1. Thus, we synthesized compound 1294 (Figure 1A), an N-methyl-piperidine analog of BKI-1. Compound 1294 retained efficacy against CpCDPK1, with a half maximal effective concentration (IC50) of 1 nM against the enzyme. We found that the mouse plasma exposure of 1294 after oral dosing with 10 mg/kg was 8-fold greater than that of another BKI-1 administered by the same route (Ojo et al, unpublished data). With 100 mg/kg oral daily dosing of 1294, a peak plasma concentration of 7.2 µM was obtained, with a half-life of >13 hours. In addition, we administered 100 mg/kg of 1294 to mice twice daily for 5 days to determine whether toxicity would emerge with high exposure. Mice appeared healthy during the 5-day period and lost no weight, and results of histological and blood chemistry analyses remained normal. Thus, there was no apparent toxicity of 1294 administration, even with the high exposures provided by 100 mg/kg administered orally twice daily. Furthermore, 1294 was >1000 times more active in inhibiting CpCDPK1 than human kinases with the smallest gatekeeper residue. For example, the IC50 values for p38, EPHA3, EGFR, Csk, and SRC were >10 µM, and the IC50 for ABL (referred to as compound 15o by Johnson et al [14]) was 4.6 µM. Thus, selectivity for kinase inhibition is unlikely to be a problem. Thus, administration of 1294 daily provides outstanding exposure with low toxicity.
Figure 1.
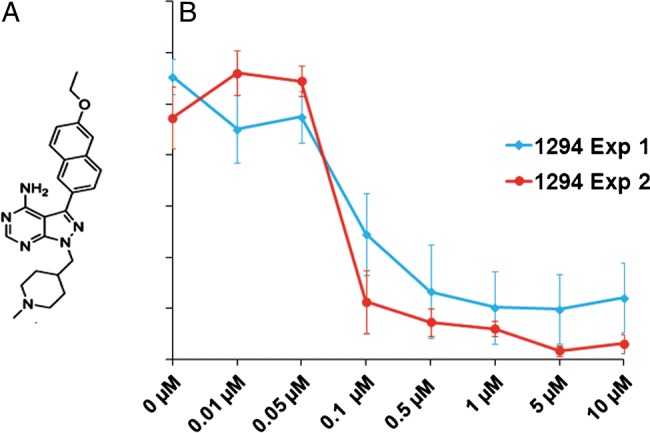
In vitro anti-Cryptosporidium activity of 1294. A, Chemical structure of 1294. B, Five initial studies demonstrated consistent submicromolar efficacy of 1294. The 2 independent experiments shown in the figure used a wider range of concentrations to quantify the half maximal effective concentration of 1294 against Cryptosporidium parvum in HCT-8 cells. Mean values±SD (indicated with vertical bars) are shown.
In Vitro Activity of 1294 Against Cryptosporidium
We evaluated the potency of 1294 against Cryptosporidium by preincubating sporozoites with several concentrations of the compound before infecting the HCT-8 cells. The number of viable parasites after infection was determined by qRT-PCR. In repeated experiments, 1294 demonstrated potent inhibition of Cryptosporidium (IC50, approximately 100 nM; Figure 1B). Doses of ≥500 nM led to almost complete eradication of the parasite. There was no evidence of damage or morphological alterations in the HCT-8 cells from exposure to 1294.
In Vivo Activity of 1294 Against Cryptosporidium in Mice
SCID-beige mice were infected by oral gavage with C. parvum oocysts. In this model, the infection is biphasic, with an initial acute increase in parasites resulting in a peak at day 8, followed by a second wave of parasite shedding from day 20 up to day 33 (Figure 2). Parasites were detected in stool as early as 4 days after the infection, and no significant difference was observed when we compared the 2 groups of mice before treatment on day 4. By day 8, mice treated with 1294 shed significantly fewer parasites, compared with controls treated with vehicle alone (Figure 2), demonstrating a significant effect of 1294 on Cryptosporidium during the acute phase of infection. During the second wave of Cryptosporidium shedding, which began after day 12, all but 1 of the placebo-treated mice produced significant numbers of parasites. In sharp contrast, only two 1294-treated mice had detectable oocysts on day 20, and only 1 remained infected on day 33. Neither of these mice had oocysts detected before day 20.
Figure 2.
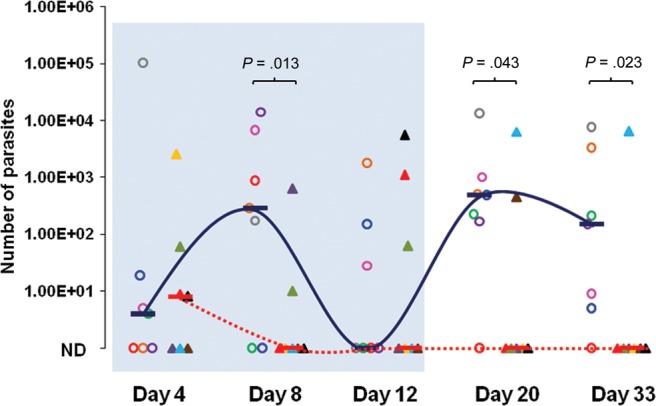
The effects of 1294 treatment in a severe combined immunodeficiency–beige mouse model of Cryptosporidium infection. Median oocyst shedding by mice treated with 1294 (dotted line) or placebo (solid line) is shown for each time point. Findings of stool specimen assays for each mouse are represented with the same color and shape through the experiment. Mice treated with 1294 (triangles [n = 7]; treatment regimen, 1 dose of 100 mg/kg by oral gavage once daily on days 4–14 after infection) or placebo (circles [n = 7]; treatment regimen, vehicle only by oral gavage once daily on days 4–14 after infection) are indicated, and horizontal bars represent median values. The sky-blue box indicates the treatment period, stool specimens from day 4 collected before initiation of dosing. The number of parasites shed in per 25 mg of stool among treated and untreated groups was quantified by real-time polymerase chain reaction and normalized to the number of oocysts, using a standard curve. Differences between groups were compared at each time point, and P values were calculated by Kruskal-Wallis analysis. Only 1 of seven 1294-treated mice had detectable parasites on day 33 after treatment, yet 6 of 7 placebo-treated mice had parasites after treatment. Abbreviation: ND, none detected.
Therapeutic Effects of 1294 in Treated Mice
We evaluated clinical signs such as weight loss and diarrhea. We did not note significant differences between the groups during the experiment, as typical of this model. Despite persistent Cryptosporidium shedding, SCID-beige mice do not become detectably ill with cryptosporidial infection [19]. We analyzed the damage present in the intestinal tissue by comparing sections of mice treated with 1294 and then infected (Figure 3A), mice treated with placebo and then infected (Figure 3B), and uninfected mice (Figure 3C). As demonstrated in placebo-treated mice, infection by Cryptosporidium caused villous atrophy, crypt hyperplasia, and epithelial damage. Findings for 1294-treated mice appeared similar to those for uninfected controls (Figure 3A and 3C, respectively) and did not exhibit the signs of intestinal damage found in placebo-treated mice (Figure 3B).
Figure 3.
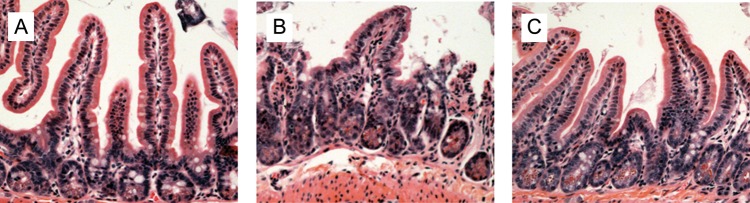
Intestinal damage in the cryptosporidiosis severe combined immunodeficiency (SCID)–beige mouse model. A–C, Microscopic analysis of representative hematoxylin-eosin–stained sections of intestine from 1294-treated and infected (A), vehicle-treated and infected (B), and uninfected (C) SCID-beige mice. Note that findings for 1294-treated and infected mice (A) and uninfected controls (C) are essentially identical, but intestines of vehicle-treated and infected mice had profoundly blunted villi, epithelial loss, damaged villi, and villous inflammation (B).
We also evaluated the number of apoptotic cells in the small intestines of each group and found that the 1294-treated and infected group had minimal apoptosis (Figure 4B). In contrast, the placebo-treated and infected mice had significantly more apoptosis (Figure 4C–E), correlating villous damage with persistent cryptosporidial infection (Figure 4E). Control, uninfected SCID-beige animals showed minimal apoptosis (Figure 4A), similar to that observed in 1294-treated and infected animals, and were the normalizing group (Figure 4E).
Figure 4.
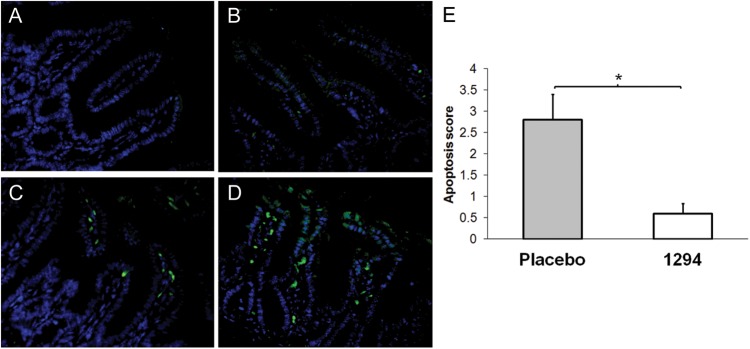
Determination of apoptotic damage in intestinal tissue. Intestinal sections were prepared and double-stained for apoptotic cells by the TUNEL technique (green) and counterstained with DAPI (blue). A–D, Representative terminal small intestinal areas of uninfected controls (A), 1294-treated and infected mice (B), and placebo (ie, vehicle)–treated and infected controls (C and D). E, An apoptotic score was determined for each section, and the score was normalized to the rate of TUNEL-positive cells found in untreated matched mouse intestines (apoptosis score, 1.0). Data are mean ± SD. *P < .05, by the Student t test.
DISCUSSION
In this study, we explored the efficacy of a novel CpCDPK1 inhibitor, 1294, during cryptosporidiosis. We have previously demonstrated the efficacy of pyrazolopyrimidine-based CDPK inhibitors as Cryptosporidium growth inhibitors in vitro [12]. Of a number of compounds characterized in vitro, we selected 1294 for further characterization because of its ability to potently inhibit the catalytic activity of CpCDPK1, its favorable pharmacokinetics, its lack of toxicity to mice in vivo, and its lack of activity against human protein kinases. In addition, similar pyrazolopyrimidine inhibitors show potent activity against TgCDPK1 in the protozoan parasite T. gondii [14]. The ability of 1294 to potently block Cryptosporidium infection of HCT-8 cells confirms our previous observations that selective pyrazolopyrimidine-based inhibitors possess anticryptosporidial activity (Figure 1B). To evaluate the anticryptosporidial activity of 1294 in an animal model, we first conducted experiments to determine possible routes of administration in mice. These studies revealed that 1294 was well absorbed after oral administration and persisted in the animal for >24 hours after an oral dose of 100 mg/kg. Since the acute phase of infection by Cryptosporidium occurs during the first week, we treated mice for 10 days and continued collecting stool specimens for up to 1 month, with the aim of evaluating relapses after the treatment. These experiments showed that daily oral 1294 doses of 100 mg/kg over 10 days reduced parasite infection significantly during the acute phase, as well as during the chronic phase (Figure 2). Interestingly, the two 1294-treated mice that were not shedding oocysts at the time of therapy (day 4) subsequently shed oocysts after therapy was halted. By contrast, none of the mice treated after oocyst shedding had begun relapsed. The numbers are low, but this observation suggests that the drug might be more effective for patent infections than when used in the prepatent period. We hypothesize that the rebound may be due to differential effects of the drug on parasite stages; for instance, the drug may work predominantly on merozoite invasion.
Our animal model is only one of a number of models used to test compounds for activity in cryptosporidiosis. Models have included neonatal mice, mice with inactivation of immune molecules (eg, SCID mice and interferon γ–knockout mice), animals with experimentally induced immunosuppression (eg, mice treated with steroids), and a recently described model of malnourished mice [19, 20]. Unfortunately, at this time, there is no standardized way to assess efficacy. We used a model with inactivation of T cells and natural killer cells as a rigorous test of drug efficacy.
Although this study demonstrates the efficacy of 1294 at reducing the parasite burden in the Cryptosporidium-immunosuppressed mouse model, further experiments will be needed to determine optimal routes of administration, drug concentrations, and treatment duration. To evaluate the efficacy of 1294 to reduce the proportion of infected mice, we used a placebo (ie, vehicle only)–treated and infected group. We observed a marked reduction of parasites in 1294-treated mice, compared with placebo-treated mice. Previous observations in mouse models have showed that anticryptosporidial drugs such as paromomycin confer partial protection that is limited to the acute phase of the infection [7]. In addition, studies of paromomycin in AIDS patients with cryptosporidiosis have been associated with frequent relapses [9]. In contrast, we did not observe relapses in 5 of 7 animals treated with 1294, with the relapse clearing spontaneously in one of the relapsing animals. Thus, we hypothesize that 1294 is more potent at suppressing relapse during the chronic phase, because of its fast and potent anticryptosporidial activity during the acute phase. Its potency may be related to blocking cell entry, motility, or other unrecognized processes.
Since intestinal apoptosis is related to active cryptosporidiosis [21], the histological analysis presented here demonstrated a significant reduction of apoptosis in 1294-treated and infected mice, compared with placebo-treated and infected mice. The increased apoptosis in placebo-treated mice correlated with the persistence of parasites in the stool and villous damage to the small intestine, effects not seen in 1294-treated and infected animals. Thus, although Cryptosporidium infection did not lead to clinical illness in the SCID-beige mice, infection did induce intestinal damage that was prevented by 1294.
In summary, these data demonstrate that 1294 is active against Cryptosporidium infection in vitro and in vivo. Pharmacologic studies suggest that it displays favorable pharmacokinetics, suggesting that this may be an effective lead compound for further development.
Notes
Financial support. This work was supported by the National Institutes of Health (grants R01AI089441, R01GM086858, and KL2TR000072). K. R. K. was funded by a scholarship through the University of Washington Plein Endowment for Geriatric Pharmacy Research.
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.White AC., Jr . Cryptosporidium species. In: Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases. 7. Philadelphia: Churchill-Livingstone/Elsevier; 2010. pp. 3547–3560. [Google Scholar]
- 2.Kotloff KL, Nataro JP, Blackwelder WC, et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet. 2013;382:209–22. doi: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 3.Shirley DA, Moonah SN, Kotloff KL. Burden of disease from cryptosporidiosis. Curr Opin Infect Dis. 2012;25:555–63. doi: 10.1097/QCO.0b013e328357e569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lima AAM, Samie A, Guerrant RL. Cryptosporidiosis. In: Guerrant RL, Walker DH, Weller PF, editors. Tropical infectious diseases. Principles, pathogens, and practice. Philadelphia: Elsevier-Churchill Livingston; 2011. pp. 640–63. [Google Scholar]
- 5.Cabada MM, White AC., Jr Treatment of cryptosporidiosis: do we know what we think we know? Curr Opin Infect Dis. 2010;23:494–9. doi: 10.1097/QCO.0b013e32833de052. [DOI] [PubMed] [Google Scholar]
- 6.Rossignol JF. Cryptosporidium and Giardia: treatment options and prospects for new drugs. Exp Parasitol. 2010;124:45–53. doi: 10.1016/j.exppara.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 7.Theodos CM, Griffiths JK, D'Onfro J, Fairfield A, Tzipori S. Efficacy of nitazoxanide against Cryptosporidium parvum in cell culture and in animal models. Antimicrob Agents Chemother. 1998;42:1959–65. doi: 10.1128/aac.42.8.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Healey MC, Yang S, Rasmussen KR, Jackson MK, Du C. Therapeutic efficacy of paromomycin in immunosuppressed adult mice infected with Cryptosporidium parvum. J Parasitol. 1995;81:114–6. [PubMed] [Google Scholar]
- 9.Hashmey R, Smith NH, Cron S, Graviss EA, Chappell CL, White AC., Jr Cryptosporidiosis in Houston, Texas. A report of 95 cases. Medicine (Baltimore) 1997;76:118–39. doi: 10.1097/00005792-199703000-00004. [DOI] [PubMed] [Google Scholar]
- 10.Nagamune K, Sibley LD. Comparative genomic and phylogenetic analyses of calcium ATPases and calcium-regulated proteins in the apicomplexa. Mol Biol Evol. 2006;23:1613–27. doi: 10.1093/molbev/msl026. [DOI] [PubMed] [Google Scholar]
- 11.Kisaburo Nagamune SNM, Chini EN, Sibley LD. Calcium regulation and signaling in apicomplexan parasites. New York: Springer; 2008. pp. 70–81. [DOI] [PubMed] [Google Scholar]
- 12.Murphy RC, Ojo KK, Larson ET, et al. Discovery of potent and selective inhibitors of calcium-dependent protein kinase 1 (CDPK1) from C. parvum and T. gondii. ACS Med Chem Lett. 2010;1:331–5. doi: 10.1021/ml100096t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ojo KK, Larson ET, Keyloun KR, et al. Toxoplasma gondii calcium-dependent protein kinase 1 is a target for selective kinase inhibitors. Nat Struct Mol Biol. 2010;17:602–7. doi: 10.1038/nsmb.1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson SM, Murphy RC, Geiger JA, et al. Development of Toxoplasma gondii calcium-dependent protein kinase 1 (TgCDPK1) inhibitors with potent anti-toxoplasma activity. J Med Chem. 2012;55:2416–26. doi: 10.1021/jm201713h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takeuchi D, Jones VC, Kobayashi M, Suzuki F. Cooperative role of macrophages and neutrophils in host antiprotozoan resistance in mice acutely infected with Cryptosporidium parvum. Infect Immun. 2008;76:3657–63. doi: 10.1128/IAI.00112-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pagel JM, Laugen C, Bonham L, et al. Induction of apoptosis using inhibitors of lysophosphatidic acid acyltransferase-beta and anti-CD20 monoclonal antibodies for treatment of human non-Hodgkin's lymphomas. Clin Cancer Res. 2005;11:4857–66. doi: 10.1158/1078-0432.CCR-04-2352. [DOI] [PubMed] [Google Scholar]
- 17.Ojo KK, Pfander C, Mueller NR, et al. Transmission of malaria to mosquitoes blocked by bumped kinase inhibitors. J Clin Invest. 2012;122:2301–5. doi: 10.1172/JCI61822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larson ET, Ojo KK, Murphy RC, et al. Multiple determinants for selective inhibition of apicomplexan calcium-dependent protein kinase CDPK1. J Med Chem. 2012;55:2803–10. doi: 10.1021/jm201725v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tzipori S, Widmer G. Animal models. Cryptosporidium and cryptosporidiosis. Boca Raton: CRC Press; 2008. [Google Scholar]
- 20.Costa LB, Noronha FJ, Roche JK, et al. Novel in vitro and in vivo models and potential new therapeutics to break the vicious cycle of Cryptosporidium infection and malnutrition. J Infect Dis. 2012;205:1464–71. doi: 10.1093/infdis/jis216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sasahara T, Maruyama H, Aoki M, et al. Apoptosis of intestinal crypt epithelium after Cryptosporidium parvum infection. J Infect Chemother. 2003;9:278–81. doi: 10.1007/s10156-003-0259-1. [DOI] [PubMed] [Google Scholar]