

Summary
Background
The vascular and gastrointestinal effects of non-steroidal anti-inflammatory drugs (NSAIDs), including selective COX-2 inhibitors (coxibs) and traditional non-steroidal anti-inflammatory drugs (tNSAIDs), are not well characterised, particularly in patients at increased risk of vascular disease. We aimed to provide such information through meta-analyses of randomised trials.
Methods
We undertook meta-analyses of 280 trials of NSAIDs versus placebo (124 513 participants, 68 342 person-years) and 474 trials of one NSAID versus another NSAID (229 296 participants, 165 456 person-years). The main outcomes were major vascular events (non-fatal myocardial infarction, non-fatal stroke, or vascular death); major coronary events (non-fatal myocardial infarction or coronary death); stroke; mortality; heart failure; and upper gastrointestinal complications (perforation, obstruction, or bleed).
Findings
Major vascular events were increased by about a third by a coxib (rate ratio [RR] 1·37, 95% CI 1·14–1·66; p=0·0009) or diclofenac (1·41, 1·12–1·78; p=0·0036), chiefly due to an increase in major coronary events (coxibs 1·76, 1·31–2·37; p=0·0001; diclofenac 1·70, 1·19–2·41; p=0·0032). Ibuprofen also significantly increased major coronary events (2·22, 1·10–4·48; p=0·0253), but not major vascular events (1·44, 0·89–2·33). Compared with placebo, of 1000 patients allocated to a coxib or diclofenac for a year, three more had major vascular events, one of which was fatal. Naproxen did not significantly increase major vascular events (0·93, 0·69–1·27). Vascular death was increased significantly by coxibs (1·58, 99% CI 1·00–2·49; p=0·0103) and diclofenac (1·65, 0·95–2·85, p=0·0187), non-significantly by ibuprofen (1·90, 0·56–6·41; p=0·17), but not by naproxen (1·08, 0·48–2·47, p=0·80). The proportional effects on major vascular events were independent of baseline characteristics, including vascular risk. Heart failure risk was roughly doubled by all NSAIDs. All NSAID regimens increased upper gastrointestinal complications (coxibs 1·81, 1·17–2·81, p=0·0070; diclofenac 1·89, 1·16–3·09, p=0·0106; ibuprofen 3·97, 2·22–7·10, p<0·0001; and naproxen 4·22, 2·71–6·56, p<0·0001).
Interpretation
The vascular risks of high-dose diclofenac, and possibly ibuprofen, are comparable to coxibs, whereas high-dose naproxen is associated with less vascular risk than other NSAIDs. Although NSAIDs increase vascular and gastrointestinal risks, the size of these risks can be predicted, which could help guide clinical decision making.
Funding
UK Medical Research Council and British Heart Foundation.
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are among the most widely used drugs in the world. They are chiefly used to treat pain, but their long-term use is limited by serious gastrointestinal side-effects. NSAIDs inhibit the two recognised forms of prostaglandin G/H synthase (also referred to as cyclo-oxygenase [COX]), namely COX-1 and COX-2.1 Since the analgesic and anti-inflammatory effects of NSAIDs are mediated by inhibition of COX-2, and their gastrointestinal side effects mostly by inhibition of COX-1, NSAIDs which selectively inhibit COX-2 might reduce the risk of gastrointestinal toxicity compared with other NSAIDs. Several such COX-2 selective drugs (collectively known as coxibs) were developed in the 1990s, and early trials comparing coxibs versus traditional NSAIDs (tNSAIDS) seemed to confirm that coxibs at doses with similar analgesic efficacy had less gastrointestinal toxicity.2,3 Unfortunately, however, subsequent placebo-controlled trials also showed unequivocally that coxibs were associated with an increased risk of atherothrombotic vascular events.4,5
Soon after these placebo-controlled trials were reported, a meta-analysis of randomised trials comparing a coxib versus placebo or a coxib versus tNSAID indicated that some tNSAIDs might also have adverse effects on atherothrombotic events, but that these hazards might depend on the degree and duration of suppression of platelet COX-1.6 In these analyses, high-dose naproxen (generally 500 mg twice a day), which is alone among NSAID regimens in being able to induce near-complete suppression of platelet thromboxane biosynthesis throughout the 12-h dosing interval in some individuals,7 did not seem to increase the risk of atherothrombosis, but other high-dose tNSAID regimens with only transient effects on platelet COX-1 were associated with a small, but definite, vascular hazard.6 Similar findings have emerged in non-randomised observational studies of NSAIDs.8,9 The US Food and Drug Administration requires that the summaries of product characteristics of all NSAIDs carry a boxed warning about the risks of cardiovascular disease,10 whereas the European Medicines Agency's Committee for Medicinal Products for Human Use (CHMP) decided that coxibs (but not tNSAIDs11) should be contraindicated in patients with coronary heart disease or stroke, and used with caution in patients with risk factors for coronary heart disease.12 Because randomised trials avoid selection bias, they could provide more reliable estimates of the size, timing, and severity of any moderate cardiovascular hazards of NSAID regimens than observational studies (which are better suited to detecting large effects). Accordingly, we initiated a collaborative meta-analysis of individual participant data (or, if not available, tabular data) from randomised trials of NSAIDs (the Coxib and traditional NSAID Trialists' [CNT] Collaboration). The main objective was to characterise and quantify the cardiovascular and gastrointestinal risks of particular NSAID regimens among different types of patients, particularly those at increased risk of vascular disease.
Methods
Identification of trials and eligibility assessment
Searches of Medline and EMBASE were done using the Cochrane strategy13 (see appendix p 27 for details of search terms), with searches up to January, 2009, supplemented by subsequent periodic scrutiny of clinical trial registers (including www.clinicaltrials.gov and www.clinicaltrialresults.org), review of reference lists of relevant papers, and enquiry among collaborators and pharmaceutical companies. For the present analyses, trials with results available prior to January, 2011, were eligible if they were properly randomised (ie, they used a randomisation method with robust allocation concealment), of at least 4 weeks duration, and: involved a comparison of an NSAID versus placebo (or open control) or one NSAID regimen versus another NSAID regimen; and no other systematic differences in drug treatment between treatment arms were planned. All trials were reviewed for eligibility by two authors and information on key trial characteristics, including information pertaining to the risk of bias (method of randomisation, treatment masking, and publication status) were extracted and recorded. The secretariat sought individual participant data (or, where not available, aggregate data) from all eligible trials. Aggregate data in a standard format were either provided by trialists or, more commonly, data fields were extracted from publications and checked by at least two authors. Four companies agreed to provide individual participant data from published and unpublished trials, including those involving celecoxib (Pfizer), rofecoxib or etoricoxib (Merck), lumiracoxib (Novartis), and GW403681 (GlaxoSmithKline). Individual participant data from trials of valdecoxib (Pfizer) were requested but not provided, although aggregate data from these trials were included in our analyses. The US National Cancer Institute and the European Organisation for Research and Treatment of Cancer also provided individual participant data from any trials of NSAIDs they had sponsored.
Prespecified analyses
Intention-to-treat analyses of first events during the scheduled treatment periods were planned. Wherever available, adjudicated outcomes were used, but in a few trials only un-adjudicated outcomes based on standard Medical Dictionary of Regulatory Authorities (MedDRA) codes were available. The primary vascular outcome was major vascular events, defined as non-fatal myocardial infarction, non-fatal stroke, or death from a vascular cause; subsidiary vascular outcomes included major coronary events (non-fatal myocardial infarction or death from coronary disease); stroke (subdivided into haemorrhagic, ischaemic, or unknown types), and hospitalisation for heart failure. Deaths were subdivided into vascular, non-vascular, and unknown causes. The primary gastrointestinal outcome was upper gastrointestinal complications, defined as an upper gastrointestinal perforation, obstruction, or bleed. For subgroup analyses of the effects of NSAIDs or for defining ulcer risk categories, we used symptomatic upper gastrointestinal events, defined as a symptomatic ulcer or upper gastrointestinal complication, to supplement statistical power.
Statistical analysis
Meta-analyses of each comparison were done using standard logrank methods where individual patient data were available, or standard methods for 2×2 contingency tables otherwise.14,15 For each trial, the observed minus expected statistic (o–e) and its variance (v) were calculated. These (o–e) values, one from each trial, were summed to produce a grand total (G), with variance (V) equal to the sum of their separate variances. The one-step estimate of the log of the event rate ratio is G/V. The χ2n–1 statistic for heterogeneity between the effects in n different trials is S–(G2/V), where S is the sum over all the trials of [o–e]2/v. To help allow for multiple subdivisions of the data, only summary rate ratios (indicated by open diamonds in figures) have 95% CI; all other rate ratios have 99% CIs. Rate ratios in different subgroups were compared by standard χ2 tests for heterogeneity or, where the subgroups could be arranged in some meaningful order (eg, by dose), χ2 tests for trend.
Rate ratios for the comparison tNSAID versus placebo were obtained by combining estimates obtained directly (from the small number of trials including such a comparison) with estimates obtained indirectly (from a comparison of trials of coxib vs tNSAID with trials of coxib vs placebo). For the calculation of indirect estimates of rate ratios for a tNSAID versus placebo, we used the following method.16 Let A be the set of trials involving a direct randomised comparison of a coxib versus placebo (but not also including the tNSAID of interest as a third group) and B the set of trials involving a direct randomised comparison of a coxib versus the tNSAID of interest (but not also including placebo as a third group). From A, we calculated the average log event rate ratio GA/VA for coxib versus placebo and, from B, the average log event rate ratio GB/VB for coxib versus tNSAID. These two results are independent of one another because A and B are non-overlapping sets of trials, so (subject to certain regularity assumptions) the log event rate ratio for tNSAID vs placebo can then be estimated indirectly by GA/VA–GB/VB (with variance 1/VA+1/VB). The overall (combined) estimate of the effect of tNSAID versus placebo was calculated as the inverse variance weighted average of the direct and indirect estimates.
For each comparison, we assessed heterogeneity of treatment effect in subgroups defined by: demographic features (eg, age, sex); past medical history; physical measurements (eg, blood pressure); concomitant treatments at baseline (eg, aspirin); and 5-year predicted risks of major vascular events (low [<5%], intermediate [5–10%], or high [>10%]) or of symptomatic upper gastrointestinal events (low [<5%], intermediate [5–10%], or high [>10%]). The predicted risks of each of the primary outcomes were modelled using Poisson regression, following a method described previously (appendix p28).17 Bonferroni corrections were applied for tests of heterogeneity to allow for multiple comparisons.
Role of the funding source
The sponsor of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
We found 24 278 titles and abstracts, from which we identified 639 randomised trials for analysis (appendix p 4). The main NSAID regimens contributing information on major vascular events, and their key pharmacological properties, are shown in the appendix (p 1). Data from comparisons of coxib versus placebo were available in 184 trials (88 367 participants, 52 466 person-years), and coxib versus tNSAID in 113 trials (diclofenac in 33 trials, 61 572 participants, 90 644 person-years; ibuprofen in 22 trials, 22 225 participants, 11 668 person-years; naproxen in 48 trials, 48 706 participants, 31 631 person-years; and another tNSAID in 14 trials, 6192 participants, 928 person-years; table). Almost all (roughly 99%) of primary outcomes occurred in trials involving a coxib or high-dose tNSAID (diclofenac 150 mg daily, ibuprofen 2400 mg daily, or naproxen 1000 mg daily), and most such trials provided individual participant data (table).
Table.
Availability of data for analyses
|
Data available |
No data available* | Total | ||||
|---|---|---|---|---|---|---|
| IPD Provided | Tabular data only | Total data available | ||||
| Coxib vs placebo | ||||||
| Number of trials | 113 | 71 | 184 | 6 | 190 | |
| Number of participants | 73 635 (83%) | 14 732 (17%) | 88 367 (>99%) | 238 (<1%) | 88 605 | |
| Person-years† | 46 407 (88%) | 6 059 (12%) | 52 466 (>99%) | 164 (<1%) | 52 630 | |
| Number of major vascular events (number of upper gastrointestinal complications) | 436 (91) | 46 (6) | 482 (97) | .. | .. | |
| tNSAID vs placebo | ||||||
| Number of trials | 47 | 111 | 158 | 30 | 188 | |
| Number of participants | 18 018 (43%) | 20 063 (48%) | 38 081 (91%) | 3 756 (9%) | 41 837 | |
| Person-years† | 8 253 (49%) | 7 964 (47%) | 16 217 (96%) | 700 (4%) | 16 917 | |
| Number of major vascular events (number of upper gastrointestinal complications) | 45 (34) | 25 (26) | 70 (60) | .. | .. | |
| Coxib vs tNSAID | ||||||
| Diclofenac | ||||||
| Number of trials | 27 | 6 | 33 | 2 | 35 | |
| Number of participants | 58 891 (95%) | 2681 (4%) | 61 572 (>99%) | 240 (<1%) | 61 812 | |
| Person-years† | 89 311 (99%) | 1333 (1%) | 90 644 (>99%) | 21 (<1%) | 90 665 | |
| Number of major vascular events (number of upper gastrointestinal complications) | 762 (211) | 11 (11) | 773 (222) | .. | .. | |
| Ibuprofen | ||||||
| Number of trials | 20 | 2 | 22 | 0 | 22 | |
| Number of participants | 21 398 (96%) | 827 (4%) | 22 225 (100%) | 0 | 22 225 | |
| Person-years† | 11 508 (99%) | 160 (1%) | 11 668 (100%) | 0 | 11 668 | |
| Number of major vascular events (number of upper gastrointestinal complications) | 81 (82) | 2 (0) | 83 (82) | .. | .. | |
| Naproxen | ||||||
| Number of trials | 34 | 14 | 48 | 1 | 49 | |
| Number of participants | 42 222 (87%) | 64 84 (13%) | 48 706 (>99%) | 66 (<1%) | 48 772 | |
| Person-years† | 30 040 (95%) | 1591 (5%) | 31 631 (>99%) | 20 (<1%) | 31 651 | |
| Number of major vascular events (number of upper gastrointestinal complications) | 254 (213) | 14 (12) | 268 (225) | .. | .. | |
| Any tNSAID vs any other tNSAID | ||||||
| Number of trials | 1 | 334 | 335 | 49 | 384 | |
| Number of participants | 733 (1%) | 67 774 (89%) | 68 507 (90%) | 7247 (10%) | 75 754 | |
| Person-years† | 134 (1%) | 22 284 (94%) | 22 418 (94%) | 1323 (6%) | 23 741 | |
| Number of major vascular events (number of upper gastrointestinal complications) | 3 (0) | 21 (105) | 24 (105) | .. | .. | |
| Coxib vs other coxib | ||||||
| Number of trials | 32 | 3 | 35 | 0 | 35 | |
| Number of participants | 25 442 (98%) | 489 (2%) | 25 931 (100%) | 0 | 25 931 | |
| Person-years† | 9033 (99%) | 60 (1%) | 9093 (100%) | 0 | 9093 | |
| Number of major vascular events (number of upper gastrointestinal complications) | 59 (19) | 1 (0) | 60 (19) | .. | .. | |
IPD=individual participant data. tNSAIDS=traditional non-steroidal anti-inflammatory drugs.
There were also seven trials involving a comparison of a coxib versus placebo, seven trials involving a comparison of a tNSAID versus placebo, one trial involving a comparison of a coxib versus ibuprofen, four trials involving a comparison of two different tNSAIDs, and one trial involving a comparison of two different coxibs for which the number of randomised patients was unknown.
Person-years for mortality.
In trials providing individual participant data, the mean age at randomisation was 61 years, about two-thirds were female, and 79% were white (appendix p 2). Few patients had a history of atherosclerosis (9%), of diabetes (9%), or of upper gastrointestinal peptic ulcer (7%). Mean body-mass index was 29 kg/m2, blood pressure was 132/79 mm Hg, haemoglobin 137 g/L, creatinine 79 μmol/L, and total cholesterol 5·3 mmol/L. About a fifth of participants reported using aspirin at randomisation, 17% a proton-pump inhibitor, and 13% were current smokers. Overall, the indication for treatment with an NSAID was rheumatoid arthritis or osteoarthritis in around four-fifths of participants, but in trials of a coxib versus placebo the indication was the prevention of colorectal adenomata or of Alzheimer's disease in around a quarter of participants.
Compared with placebo (or, in a few cases, allocation to no NSAID treatment), the risk of major vascular events was increased by about a third in those allocated to a coxib (307 [1·15% per annum] coxib vs 175 [0·82% per annum] placebo; rate ratio [RR] 1·37, 95% CI 1·14–1·66, p=0·0009) or diclofenac (1·41, 1·12–1·78, p=0·0036), chiefly due to an increase of about three-quarters in the risk of major coronary events (coxibs 1·76, 1·31–2·37, p=0·0001; diclofenac 1·70, 1·19–2·41, p=0·0032; figures 1, 2). Ibuprofen also significantly increased major coronary events (2·22, 1·10–4·48, p=0·0253), but not major vascular events (1·44, 0·89–2·33, p=0·14; figure 3). By contrast with other tNSAIDs (heterogeneity p=0·04), high-dose naproxen was not associated with any significant excess risk of major vascular events (0·93, 0·69–1·27; figure 4), and nor was there an increase in major coronary events (0·84, 0·52–1·35). There was no evidence that any NSAID significantly increased the risk of stroke (figures 1–4).
Figure 1.
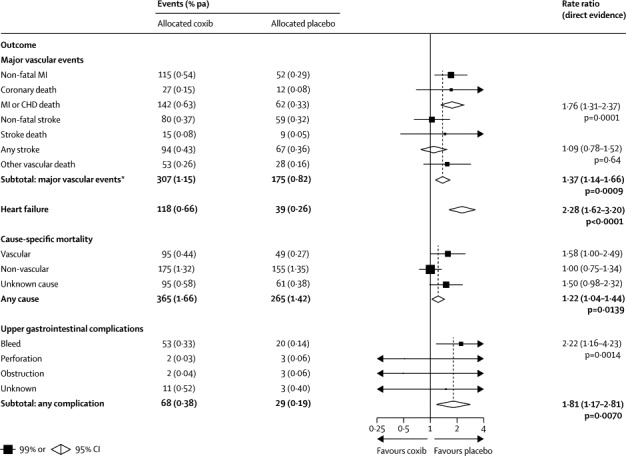
Effects of coxib therapy on major vascular events, heart failure, cause-specific mortality, and upper gastrointestinal complications
Actual numbers for participants are presented, together with the corresponding mean yearly event rate (in parentheses). Participants can contribute only once to the total of major vascular events. Rate ratios (RRs) for all outcomes are indicated by squares and their 99% CIs by horizontal lines. Subtotals and their 95% CIs are represented by diamonds. Squares or diamonds to the left of the solid line indicate benefit. MI=myocardial infarction. CHD=coronary heart disease. Major vascular event=myocardial infarction, stroke, or vascular death. *Includes a further 25 vs 21 major vascular events in patients randomised into trials for which only tabular information was available.
Figure 2.
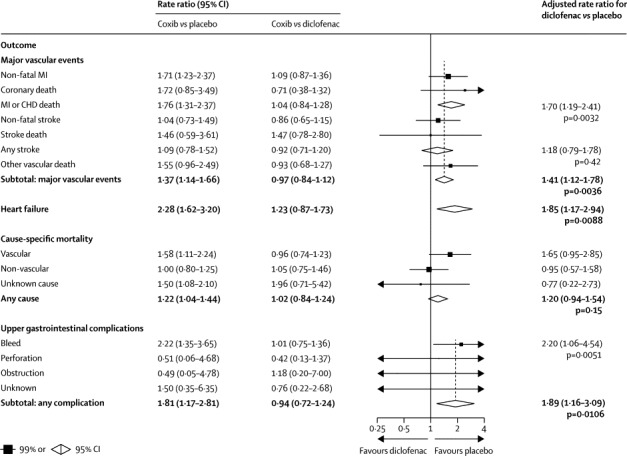
Effects of diclofenac on major vascular events, heart failure, cause-specific mortality, and upper gastrointestinal complications (indirect comparisons)
Rate ratios (RRs) are for comparisons of a tNSAID versus placebo, calculated indirectly from ratio of RRs for a coxib versus placebo and RRs for a coxib versus tNSAID, each of which is shown in the vertical columns (see statistical methods). MI=myocardial infarction. CHD=coronary heart disease.
Figure 3.
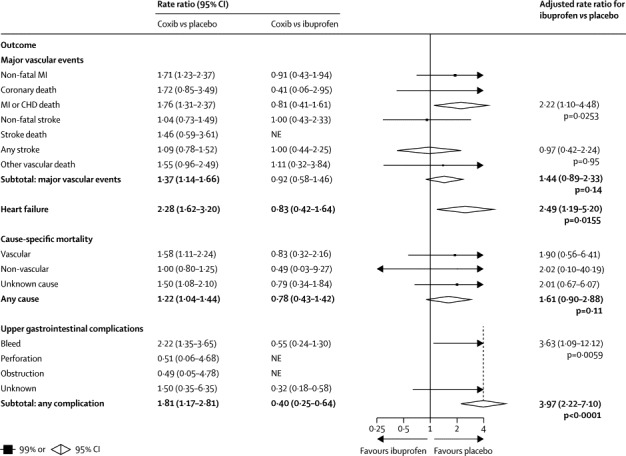
Effects of ibuprofen on major vascular events, heart failure, cause-specific mortality, and upper gastrointestinal complications (indirect comparisons)
MI=myocardial infarction. CHD=coronary heart disease. NE=not estimated.
Figure 4.
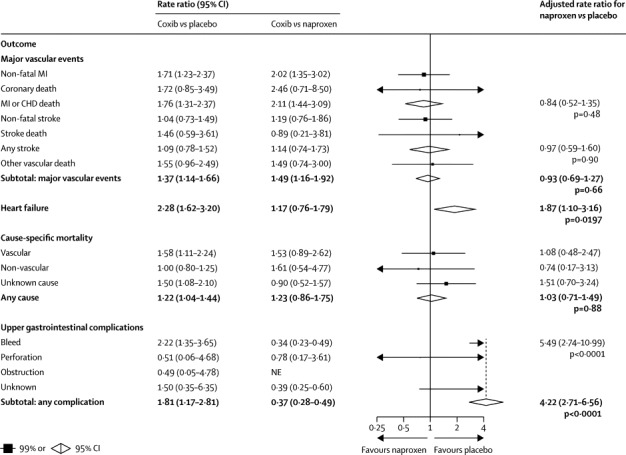
Effects of naproxen on major vascular events, heart failure, cause-specific mortality, and upper gastrointestinal complications (indirect comparisons)
MI=myocardial infarction. CHD=coronary heart disease.
The risk of hospitalisation due to heart failure was roughly doubled by all NSAID regimens studied (coxib 2·28, 95% CI 1·62–3·20, p<0·0001; diclofenac 1·85, 1·17–2·94, p=0·0088; ibuprofen 2·49, 1·19–5·20, p=0·0155; naproxen 1·87, 1·10–3·16, p=0·0197; figures 1–4).
The risk of vascular death was significantly increased by coxibs (1·58, 99% CI 1·00–2·49, p=0·0103) and diclofenac (1·65, 0·95–2·85, p=0·0187), non-significantly increased by ibuprofen (1·90, 0·56–6·41, p=0·17), but not increased by naproxen (1·08, 0·48–2·47, p=0·80; figures 1–4). The risk of death from any cause was significantly increased by around a quarter by allocation to a coxib (1·22, 1·04–1·44, p=0·0139), but despite a clear excess of vascular deaths the corresponding excess was not significant for diclofenac (1·20, 0·94–1·54, p=0·15), and nor were there significant excesses of death from any cause for ibuprofen (1·61, 0·90–2·88, p=0·11) or naproxen (1·03, 0·71–1·49, p=0·88).
Compared with placebo, there was an increased risk of upper gastrointestinal complications (most of which were bleeds) in association with allocation to a coxib (68 [0·38% per annum] coxib vs 29 [0·19% per annum] placebo; 1·81, 1·17–2·81, p=0·0070), diclofenac (1·89, 1·16–3·09, p=0·0106), ibuprofen (3·97, 2·22–7·10, p<0·0001), and naproxen (4·22, 2·71–6·56, p<0·0001; appendix p 3 and figures 1–4). Only 2% of upper gastrointestinal complications were recorded as being fatal.
There was very little power to assess variation in treatment effects on major vascular events or on symptomatic upper gastrointestinal events in patient subgroups; however, for each of the main categories of NSAIDs studied, after allowance for multiple comparisons, the proportional effects on each specific outcome seemed similar in different types of patients, including those at low, intermediate, and high risk of major vascular events and those at differing risk of symptomatic upper gastrointestinal events (Bonferroni-adjusted heterogeneity p values all >0·1; appendix pp 5–14).
There was only limited evidence for an increased risk of major vascular events during the first 6 months for coxibs (p=0·06) and diclofenac (p=0·0329), and no evidence that any proportional excess increased with greater exposure to treatment (p values all non-significant; appendix p 15). For symptomatic upper gastrointestinal ulcers, however, a more definite pattern of excess within the first 6 months was seen for coxibs (2·55, 99% CI 1·49–4·35), diclofenac (3·93, 2·16–7·13), ibuprofen (5·73, 3·24–10·14), and naproxen (6·31, 3·81–10·44; appendix p 16).
Overall, celecoxib and rofecoxib significantly increased the risks of major vascular events (celecoxib 1·36, 95% CI 1·00–1·84; rofecoxib 1·38, 1·07–1·80; appendix pp 17, 18). There was a smaller proportional excess risk of major vascular events with lower celecoxib doses in placebo-controlled trials (p for trend=0·0117; appendix p 18). Etoricoxib had not been extensively studied in placebo-controlled trials (appendix p 17), but the effects of etoricoxib, rofecoxib, and celecoxib seemed similar (heterogeneity p=0·21; appendix p 19) in trials of a coxib versus diclofenac (where the same diclofenac regimen was used in each trial). Similarly, trials of lumiracoxib versus placebo provided little useful information, whereas trials of lumiracoxib versus ibuprofen or lumiracoxib versus naproxen (1000 mg in seven trials, 440 mg in one trial) were consistent with the vascular risks of lumiracoxib being similar to other coxibs (Bonferroni-adjusted heterogeneity p values all >0·1; appendix pp 20, 21).
In comparable analyses of symptomatic upper gastrointestinal events, there was also a lack of evidence of heterogeneity between coxibs in comparisons with placebo, diclofenac, ibuprofen, and naproxen (Bonferroni-adjusted heterogeneity p values all >0·1; appendix pp 22–26), suggesting that each of the coxibs yielded similar ulcer risks. For several of them, however, there was evidence that higher doses yielded larger proportional excesses in ulcer risk (celecoxib: p for trend=0·0043; rofecoxib: p for trend=0·0350; appendix p 25; etoricoxib: heterogeneity p=0·0135; appendix p 24).
Discussion
Meta-analyses of randomised trials and of observational studies have shown that coxibs and tNSAIDs are associated with an increased risk of cardiovascular disease and upper gastrointestinal complications,5,6,8,18–20 but there has been uncertainty about the nature and magnitude of these risks, and the relative safety of different NSAID regimens, especially in those at increased risk of coronary heart disease.10–12
Our meta-analysis, which is unaffected by selection and other biases inherent in observational studies, showed clearly that the vascular risks of diclofenac, and possibly ibuprofen, are similar to coxibs, but that naproxen is not associated with an increased risk of major vascular events. However, it also showed that the excess risk of both vascular and gastrointestinal events can be predicted once the baseline risks of such hazards are known, which could help clinical decision-making.
Most of the information available for the estimation of vascular risks was derived from trials involving four coxibs (celecoxib, rofecoxib, etoricoxib, and lumiracoxib) and three high-dose tNSAID regimens (daily doses: diclofenac 150 mg, ibuprofen 2400 mg, and naproxen 1000 mg [table and appendix]). Overall, coxibs increased the risk of major vascular events by around a third, as previously reported in meta-analyses of summary trial data,6 but these analyses show that the excess risk was mainly attributable to an increase of about three quarters in the risk of major coronary events. These results are similar to those previously reported for coxibs, diclofenac, ibuprofen, and naproxen in observational studies8 but, by contrast with the present meta-analysis of randomised trials, the observational studies used a wide range of vascular outcomes and tNSAID doses, so precise comparisons between these different types of studies are not possible.
This meta-analysis showed clearly that high-dose diclofenac has similar vascular risks to the average coxib regimen studied. The absolute excess risks were small but serious: compared with placebo, allocation to a coxib or diclofenac caused around three additional major vascular events per 1000 participants per year, with one such event causing death. High-dose ibuprofen also significantly increased the risk of major coronary events, but there were many fewer relevant events in trials of coxib versus ibuprofen, so its safety (including the possible relevance of its interaction with aspirin21) requires further study. Naproxen 500 mg twice a day did not seem to increase the risk of major vascular events, consistent with experimental studies showing that this naproxen regimen is capable of producing COX-1 inhibition that is sufficiently prolonged and intense to result in platelet inhibition in some individuals, which could attenuate any adverse vascular effects of COX-2 inhibition.7
There was no evidence of an increased risk of stroke for any of the NSAIDs studied, but few strokes were recorded and the absence of any stroke risk for drug regimens known to increase blood pressure is implausible. All NSAIDs doubled the risk of heart failure causing hospital admission (ie, not just ankle oedema), consistent with this being a COX-2 dependent hazard unrelated to variable platelet inhibition. As expected, NSAIDs increased the risk of upper gastrointestinal complications by around 2–4 times and, as previously shown by individual trials,2,3 coxibs yielded the lowest risk of such complications.
Our analyses do not allow definite conclusions about whether particular NSAIDs increase vascular risk immediately after starting treatment, but evidence for an early hazard of coxibs would have been enhanced if data had been included from two trials that indicated vascular hazard from intravenous parecoxib followed by oral valdecoxib during a 2-week period after coronary artery bypass surgery.22,23 There was, however, clear evidence that NSAIDs increase the early risk of upper gastrointestinal complications. Since the average trial duration was less than 1 year, our analyses do not provide reliable information about whether the risks of NSAIDs persist with prolonged treatment (and since events occurring more than a few weeks after patients discontinued treatment were not generally recorded, our analyses might underestimate those risks).
Overall, at the daily doses studied most frequently, the vascular risks of different coxib regimens seemed similar. Little information was available on whether the vascular hazards of coxibs were dose-dependent. Although there was a trend towards less risk with lower celecoxib doses, the vascular effects of celecoxib 200 mg daily (the most widely used coxib regimen) were statistically uncertain. The tNSAID regimens studied were all high-dose, with little variation between trials, so comparable analyses of tNSAIDs were not possible. However, since vascular hazard is probably related to the degree of COX-2 inhibition, which increases with dose,9 such dose-dependency seems likely.24
The potential for bias has been minimised in this meta-analysis by obtaining access to detailed individual data from most trials recording vascular and gastrointestinal outcomes (including some that were unpublished). Since most events occurred in a small number of recent trials that used secure randomisation methods and treatment blinding, sensitivity analyses (available on request) indicated that our results were not materially influenced by uncertainties about the quality of older trials. There was also no evidence that our results depended on whether participating trials had been published, although some unpublished trials of which we were unaware might have affected particular findings. A novel element of our analyses was that treatment effects were estimated by comparing the results of trials of a coxib versus placebo and trials of a coxib versus tNSAID. The conditions under which such indirect comparisons might be expected to yield valid results25 are satisfied, since the two sets of trials involved similar doses of coxibs and similar populations, and different studies used the same (high-dose) tNSAID regimens as comparators.
A key objective was to quantify the hazards of NSAIDs in patients with an increased risk of vascular disease. The results of a previous meta-analysis suggested that the proportional increase in vascular risk might be highest for celecoxib in those at greatest risk of coronary heart disease.26 In our meta-analysis, however, the proportional effects of coxibs and tNSAIDs seemed similar irrespective of baseline characteristics, and in particular were similar at all levels of risk of major vascular events (<5%, 5–10%, >10% over 5 years), although there were limited data among patients with a history of atherosclerosis. Assuming that proportional effects are indeed similar in different patients, we undertook hypothetical calculations (appendix) of the annual excess risks of each of the main NSAIDs as compared with placebo (figure 5). Excess risks were calculated for major vascular events in patients at high (2% per annum) or low (0·5%) risk of major vascular events (left panel), and for upper gastrointestinal complications in patients at moderate (0·5% a year) or low (0·2% a year) risk of such complications (right panel). For each outcome, the fraction of fatal events is shown in darker shading. Among those at low risk of vascular disease (the majority of participants in these trials), the predicted absolute risks of major vascular events were small irrespective of the particular regimen chosen. For high-risk individuals (about 40% of whom were taking aspirin), for every 1000 patients allocated to a year of treatment with a coxib regimen or high-dose diclofenac regimen, about seven or eight more would have a major vascular event, of which two would be fatal. High-dose ibuprofen may be associated with a similar risk, but is also likely to yield a higher risk of upper gastrointestinal complications than either a coxib or diclofenac.
Figure 5.
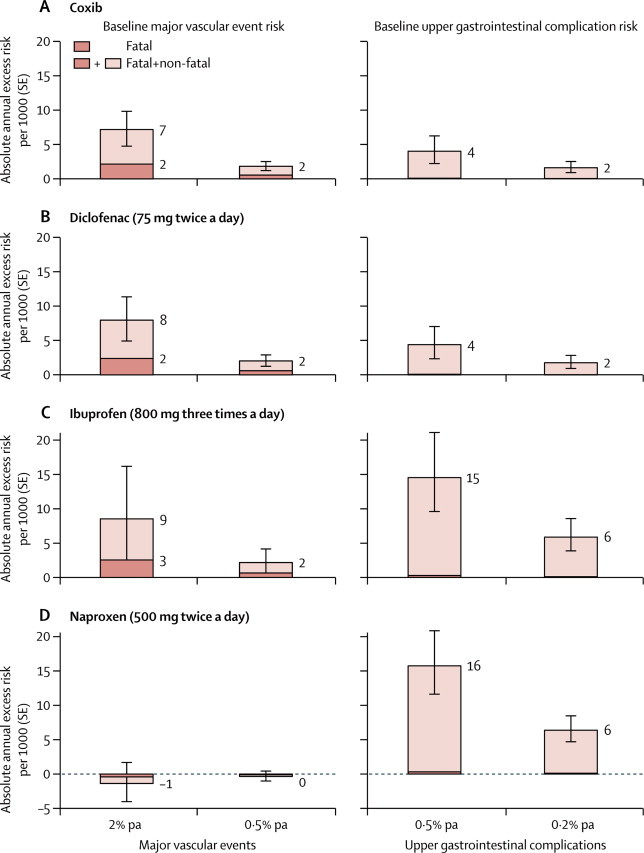
Annual absolute effects per 1000 of coxibs and tNSAIDs at different baseline risks of major vascular events and upper gastrointestinal complications
For each category of drug (coxib, diclofenac, ibuprofen, and naproxen), the predicted annual absolute risks of major vascular events (±1 SE) are shown (left) for patients with predicted risk of 2·0% or 0·5% per annum of a major vascular event. For comparison, predicted annual absolute risks of upper gastrointestinal complications (±1 SE) are shown for patients with predicted risks of 0·5% or 0·2% per annum (right). Absolute annual risks for placebo-allocated patients are assumed to be those of a hypothetical patient after all appropriate forms of prophylactic treatment (eg, antihypertensive therapy, statin therapy, proton-pump inhibitors) have been instituted.
Our analyses suggest that naproxen might not be associated with an increased risk of major vascular events, but this result should be interpreted with caution. First, we do not know whether this would be true in patients treated with aspirin, in whom naproxen will not result in any additional inhibition of COX-1 and might actually interfere with the antiplatelet effect of low-dose aspirin.27,28 Secondly, the effects of lower naproxen doses, such as those typically used in over-the-counter preparations (eg, 220 mg twice a day), are uncertain since they would be less likely to mimic the aspirin-like effect of 500 mg twice a day.29 Thirdly, the apparent advantage of naproxen regimens might not be preserved after longer term use. Finally, naproxen substantially increases the risk of upper gastrointestinal complications (although such bleeds are less likely than vascular events to result in disability30 and such hazards could be mitigated with proton-pump inhibitors31).
This meta-analysis of individual participant data helps to characterise and quantify the vascular and gastrointestinal hazards of coxibs and tNSAIDs. It shows that high-dose diclofenac has vascular risks similar to coxibs, but also raises the possibility that high-dose ibuprofen has similar vascular effects. High-dose naproxen seems to be associated with less vascular hazard, although whether this is true of the lower doses most commonly used in clinical practice is unclear. Although NSAIDs increase vascular and gastrointestinal risks to a varying extent, our analyses indicate that the effects of different regimens in particular patients can be predicted, which could help in guiding decisions about the clinical management of inflammatory disorders.
Correspondence to: Prof Colin Baigent, CNT Secretariat, Clinical Trial Service Unit and Epidemiological Studies Unit (CTSU), Nuffield Department of Clinical Medicine, Richard Doll Building, Old Road Campus, Roosevelt Drive, Oxford OX3 7LF, UK cnt@ctsu.ox.ac.uk
Acknowledgments
Acknowledgments
The study was funded by core grants from the UK Medical Research Council and the British Heart Foundation. This collaboration is coordinated by the Clinical Trial Service Unit & Epidemiological Studies Unit (CTSU) at the University of Oxford, UK. This work is supported at the CTSU by the UK Medical Research Council and the British Heart Foundation. N Bhala was funded by a MRC Health of the Public Research Fellowship. J Emberson acknowledges support from the BHF Centre of Research Excellence, Oxford, UK (RE/08/04). These organisations had no role in study design, data collection, data analysis, data interpretation or writing of the report. The authors thank the European Organization for Research and Treatment of Cancer for permission to use the data from EORTC trial 40015 for this research.
Contributors
The writing committee accepts full responsibility for the content of this paper. All of the members contributed to the collection and analysis of the data, and to the preparation of the manuscript. All collaborators had an opportunity to contribute to the interpretation of the results and to drafting the manuscript. The pharmaceutical companies providing data were invited to comment on the study results and draft manuscripts, but the Writing Committee had full control of all editorial decisions.
The CNT Collaborative Group
Writing committee: N Bhala*, J Emberson*, A Merhi, Prof S Abramson, Prof N Arber, Prof J A Baron, Prof C Bombardier, Prof C Cannon, M E Farkouh, Prof GA FitzGerald, Prof P Goss, H Halls, Prof E Hawk, Prof C Hawkey, Prof C Hennekens, Prof M Hochberg, L E Holland, P M Kearney, Prof L Laine, Prof A Lanas, Prof P Lance, Prof A Laupacis, Prof J Oates, Prof C Patrono, Prof T J Schnitzer, S Solomon, Prof P Tugwell, K Wilson, J Wittes, Prof C Baigent.
*Contributed equally.
Other investigators who provided data: O Adelowo, P Aisen, A Al-Quorain, R Altman, G Bakris, H Baumgartner, C Bresee, M Carducci, D-M Chang, C-T Chou, D Clegg, M Cudkowicz, L Doody, Y El Miedany, C Falandry, J Farley, L Ford, M GarcíLosa, M González-Ortiz, M Haghighi, M Hála, T Iwama, Z Jajić, D Kerr, H-S Kim, C Köhne, B-K Koo, B Martin, C Meinert, N Müller, G Myklebust, D Neustadt, R Omdal, S Ozgocmen, A Papas, P Patrignani, F Pelliccia, V Roy, I Schlegelmilch, A Umar, O Wahlström, F Wollheim, S Yocum, X Y Zhang.
Others (not already listed) present at the initial presentation of results in January, 2011: E Hall, P McGettigan, R Midgley, R A Moore, GSK: R Philipson; Merck: S Curtis, A Reicin; Novartis: J Bond, A Moore; Pfizer: M Essex, J Fabule, B Morrison, L Tive.
Secretariat: C Baigent, N Bhala, K Davies, J Emberson, H Halls, L E Holland, P M Kearney, A Merhi, C Patrono, K Wilson, and F Yau.
Conflicts of interest
The Clinical Trial Service Unit and Epidemiological Studies Unit (CTSU), where the CNT Secretariat (C Baigent, N Bhala, J Emberson, H Halls, L Holland, A Merhi, K Wilson) is located, has a policy of staff not accepting fees, honoraria, or paid consultancies. CTSU staff are, however, involved in clinical trials of lipid-modifying therapy funded by research grants from Merck to the University of Oxford, with the University the trial sponsor in all cases. In particular, C Baigent was chief investigator of the Study of Heart and Renal Protection of ezetimibe/simvastatin prior to 2011. N Arber, M Farkouh, P M Kearney, P Lance and S Solomon declare that they have no conflicts of interest. S Abramson serves as a consultant to Abbott and Pfizer. J A Baron was formerly a consultant to Merck and is currently a consultant to Bayer and to Pfizer (as a member of a safety and data monitoring committee). C Bombardier is a consultant to Abbott, AstraZeneca, Bristol-Myers Squibb (Canada and USA), and UCB, and receives research support from Abbott, Amgen, Bristol-Myers Squibb, Janssen, Hoffman La-Roche, Pfizer, and UCB Canada. C Cannon reports receiving research grants or support from Accumetrics, AstraZeneca, CSL Behring, Essentialis, GlaxoSmithKline, Merck, Regeneron, Sanofi, and Takeda. He is clinical advisor to, and holds equity in, Automedics Medical Systems. He also sits on advisory boards (but donates funds to charity) for Alnylam, Bristol-Myers Squibb, Lipimedix, and Pfizer. G FitzGerald has consulted for Boehringer Ingelheim, Lilly, Merck, BMS, Johnson and Johnson, Genentech, and Takeda. P Goss has received speaker honoraria from GlaxoSmithKline and Novartis. E Hawk reports a paid consultancy from Pozen Pharmaceuticals and is an unpaid consultant to Cancer Prevention Pharmaceuticals and PLx Pharmaceuticals. C Hawkey reports previously having received grants or honoraria from Merck, Pfizer, and AstraZeneca and at present advises Bayer, GlaxoSmithKline, and Novartis. C Hennekens reports serving on data and safety monitoring boards for Actelion, Amgen, AstraZeneca, Bayer, Bristol-Myers Squibb, British Heart Foundation, Cadila, Canadian Institutes of Health Research, Lilly, Sunovion, and the Wellcome Foundation, and also serves as an advisor to legal counsel for Stryker. M Hochberg serves as a consultant or member of an advisory board for Abbott, Bioiberica SA, Boehringer Ingelheim, Bristol-Myers Squibb, Covidien, Eli Lilly, Genentech/Roche, Iroko Pharmaceuticals, Merck, Pfizer, and Regeneron. He chaired a data and safety monitoring board for Novartis and has received speaker honoraria from Bioiberica SA and IBSA. L Laine reports serving on data and safety monitoring boards for Bayer, Merck, and Eisai and has previously consulted for Pfizer, AstraZeneca, and Horizon. A Lanas is an advisor to Bayer and AstraZeneca Spain. A Laupacis reports serving on data and safety monitoring boards for Novartis and advisory boards for Pfizer and Eli Lilly, but none of these activities involve discussions about non-steroidal anti-inflammatory drugs. J Oates was a member of the scientific advisory board of Merck, concluding in 2006. C Patrono has received, during the past 2 years, consultant and speaker fees from Astra Zeneca, Bayer, Eli Lilly, Merck, NicOx, Novartis, and Servier, as well as an institutional grant from Bayer for investigator-initiated research. T Schnitzer reports paid consultancies with Merck, Janssen, Regeneron, Abbott, and McNeil, as well as research support (to Northwestern University) from Novartis, Merck, Eli Lilly, and Nuvo. P Tugwell reports paid consultancies with Bristol-Myers Squibb, Chelsea, and UCB, reports that OMERACT (Outcomes measures in Rheumatology), whose Executive he serves on in an unpaid capacity, receives support from Actellion, Alderbio, Amgen, Ardea Biosciences, Astra Zeneca, Bristol-Myers Squibb, Celgene, Centocor, Cypress/Forest, Eli Lilly, Boehringer Ingelheim, Genentech, Genzyme, Jass Pharmaceuticals, Merck, Novartis, Novo Nordisk, Pfizer, Regeneron, Savient, Takeda, UCB, and reports that the Ontario Biologics Research Initiative (OBRI) Industry Council receives support from Abbott, Roche, Schering Plough/Merck, UCB, and Bristol-Myers Squibb. J Wittes reports that Statistics Collaborative, a company in which she holds majority ownership, has consulting agreements with Merck, Bristol-Myers Squibb, AbbVie, Amgen, and Pfizer.
Supplementary Material
References
- 1.FitzGerald GA, Patrono C. The coxibs, selective inhibitors of cyclooxygenase-2. New Engl J Med. 2001;345:433–442. doi: 10.1056/NEJM200108093450607. [DOI] [PubMed] [Google Scholar]
- 2.Bombardier C, Laine L, Reicin A. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. New Engl J Med. 2000;343:1520–1528. doi: 10.1056/NEJM200011233432103. [DOI] [PubMed] [Google Scholar]
- 3.Schnitzer TJ, Burmester GR, Mysler E. Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), reduction in ulcer complications: randomised controlled trial. Lancet. 2004;364:665–674. doi: 10.1016/S0140-6736(04)16893-1. [DOI] [PubMed] [Google Scholar]
- 4.Bresalier RS, Sandler RS, Quan H. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. New Engl J Med. 2005;352:1092–1102. doi: 10.1056/NEJMoa050493. [DOI] [PubMed] [Google Scholar]
- 5.Solomon SD, McMurray JJ, Pfeffer MA. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. New Engl J Med. 2005;352:1071–1080. doi: 10.1056/NEJMoa050405. [DOI] [PubMed] [Google Scholar]
- 6.Kearney PM, Baigent C, Godwin J, Halls H, Emberson JR, Patrono C. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ. 2006;332:1302–1308. doi: 10.1136/bmj.332.7553.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Capone ML, Tacconelli S, Sciulli MG. Clinical pharmacology of platelet, monocyte, and vascular cyclooxygenase inhibition by naproxen and low-dose aspirin in healthy subjects. Circulation. 2004;109:1468–1471. doi: 10.1161/01.CIR.0000124715.27937.78. [DOI] [PubMed] [Google Scholar]
- 8.McGettigan P, Henry D. Cardiovascular risk with non-steroidal anti-inflammatory drugs: systematic review of population-based controlled observational studies. PLoS Med. 2011;8:e1001098. doi: 10.1371/journal.pmed.1001098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia Rodriguez LA, Tacconelli S, Patrignani P. Role of dose potency in the prediction of risk of myocardial infarction associated with nonsteroidal anti-inflammatory drugs in the general population. J Am Coll Cardiol. 2008;52:1628–1636. doi: 10.1016/j.jacc.2008.08.041. [DOI] [PubMed] [Google Scholar]
- 10.US Food and Drug Administration Analysis and recommendations for agency action regarding non-steroidal anti-inflammatory drugs and cardiovascular risk. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ (accessed May 20, 2013).
- 11.European Medicines Agency Press release: European Medicines Agency review concludes positive benefit-risk balance for non-selective NSAIDs. 24/10/2006. http://www.ema.europa.eu/docs/en_GB/document_library/Press_release/2009/12/WC500017362.pdf (accessed May 20, 2013).
- 12.European Medicines Agency Press release: European Medicines Agency concludes action on COX-2 inhibitors. 27/06/2005. http://www.ema.europa.eu/docs/en_GB/document_library/Press_release/2010/01/WC500059088.pdf (accessed May 20, 2013).
- 13.Robinson KA, Dickersin K. Development of a highly sensitive search strategy for the retrieval of reports of controlled trials using PubMed. Int J Epidemiol. 2002;31:150–153. doi: 10.1093/ije/31.1.150. [DOI] [PubMed] [Google Scholar]
- 14.Early Breast Cancer Trialists' Collaborative Group . Treatment of early breast cancer. Volume 1, worldwide evidence 1985–90. Oxford University Press; Oxford: 1990. [Google Scholar]
- 15.Antiplatelet Trialists' Collaboration Collaborative overview of randomised trials of antiplatelet therapy—I: prevention of death, myocardial infarction, and stroke by prolonged antiplatelet therapy in various categories of patients. BMJ. 1994;308:81–106. [PMC free article] [PubMed] [Google Scholar]
- 16.Glenny AM, Altman DG, Song F. Indirect comparisons of competing interventions. Health Technol Assess. 2005;9:1–134. doi: 10.3310/hta9260. iii–iv. [DOI] [PubMed] [Google Scholar]
- 17.Baigent C, Blackwell L, Emberson J. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–1681. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trelle S, Reichenbach S, Wandel S. Cardiovascular safety of non-steroidal anti-inflammatory drugs: network meta-analysis. BMJ. 2011;342:c7086. doi: 10.1136/bmj.c7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ofman JJ, MacLean CH, Straus WL. A metaanalysis of severe upper gastrointestinal complications of nonsteroidal antiinflammatory drugs. J Rheumatol. 2002;29:804–812. [PubMed] [Google Scholar]
- 20.Rostom A, Muir K, Dube C. Gastrointestinal safety of cyclooxygenase-2 inhibitors: a Cochrane Collaboration systematic review. Clin Gastroenterol Hepatol. 2007;5:818–828. doi: 10.1016/j.cgh.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 21.Catella-Lawson F, Reilly MP, Kapoor SC. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. New Engl J Med. 2001;345:1809–1817. doi: 10.1056/NEJMoa003199. [DOI] [PubMed] [Google Scholar]
- 22.Ott E, Nussmeier NA, Duke PC. Efficacy and safety of the cyclooxygenase 2 inhibitors parecoxib and valdecoxib in patients undergoing coronary artery bypass surgery. J Thorac Cardiovasc Surg. 2003;125:1481–1492. doi: 10.1016/s0022-5223(03)00125-9. [DOI] [PubMed] [Google Scholar]
- 23.Nussmeier NA, Whelton AA, Brown MT. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. New Engl J Med. 2005;352:1081–1091. doi: 10.1056/NEJMoa050330. [DOI] [PubMed] [Google Scholar]
- 24.Grosser T, Yu Y, Fitzgerald GA. Emotion recollected in tranquility: lessons learned from the COX-2 saga. Annu Rev Med. 2010;61:17–33. doi: 10.1146/annurev-med-011209-153129. [DOI] [PubMed] [Google Scholar]
- 25.Song F, Altman DG, Glenny AM, Deeks JJ. Validity of indirect comparison for estimating efficacy of competing interventions: empirical evidence from published meta-analyses. BMJ. 2003;326:472. doi: 10.1136/bmj.326.7387.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Solomon SD, Wittes J, Finn PV. Cardiovascular risk of celecoxib in 6 randomized placebo-controlled trials: the cross trial safety analysis. Circulation. 2008;117:2104–2113. doi: 10.1161/CIRCULATIONAHA.108.764530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Capone ML, Sciulli MG, Tacconelli S. Pharmacodynamic interaction of naproxen with low-dose aspirin in healthy subjects. J Am Coll Cardiol. 2005;45:1295–1301. doi: 10.1016/j.jacc.2005.01.045. [DOI] [PubMed] [Google Scholar]
- 28.Meek IL, Vonkeman HE, Kasemier J, Movig KL, van de Laar MA. Interference of NSAIDs with the thrombocyte inhibitory effect of aspirin: a placebo-controlled, ex vivo, serial placebo-controlled serial crossover study. Eur J Clin Pharmacol. 2013;69:365–371. doi: 10.1007/s00228-012-1370-y. [DOI] [PubMed] [Google Scholar]
- 29.Capone ML, Tacconelli S, Sciulli MG. Human pharmacology of naproxen sodium. J Pharmacol Exp Ther. 2007;322:453–460. doi: 10.1124/jpet.107.122283. [DOI] [PubMed] [Google Scholar]
- 30.Maetzel A, Krahn M, Naglie G. The cost-effectiveness of celecoxib and rofecoxib in patients with osteoarthritis or rheumatoid arthritis. Technology Report No. 23. Canadian Coordinating Office for Health Technology Assessment; Ottawa: 2001. [Google Scholar]
- 31.Bhatt DL, Scheiman J, Abraham NS. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. J Am Coll Cardiol. 2008;52:1502–1517. doi: 10.1016/j.jacc.2008.08.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.