

Abstract
Complex I (CI) deficiency is a frequent cause of mitochondrial disorders and, in most cases, is due to mutations in CI subunit genes encoded by mitochondrial DNA (mtDNA). In this study, we establish the pathogenic role of the heteroplasmic mtDNA m.3890G>A/MT-ND1 (p.R195Q) mutation, which affects an extremely conserved amino acid position in ND1 subunit of CI. This mutation was found in a young-adult male with optic atrophy resembling Leber's hereditary optic neuropathy (LHON) and bilateral brainstem lesions. The only previously reported case with this mutation was a girl with fatal infantile Leigh syndrome with bilateral brainstem lesions. Transfer of the mutant mtDNA in the cybrid cell system resulted in a marked reduction of CI activity and CI-dependent ATP synthesis in the presence of a normally assembled enzyme.
These findings establish the pathogenicity of the m.3890G>A/MT-ND1 mutation and remark the link between CI mutations affecting the mtDNA-encoded ND subunits and LHON-like optic atrophy, which may be complicated by bilateral and symmetric lesions affecting the central nervous system. Peculiar to this mutation is the distribution of the brainstem lesions, with sparing of the striatum in both patients.
Abbreviations: CI, Complex I; CII, Complex II; CIII, Complex III; CIV, Complex IV; LHON, Leber's hereditary optic neuropathy; mtDNA, mitochondrial DNA; nDNA, nuclear DNA; OS, Oculus Sinister (left eye); OD, Oculus Dexter (right eye)
Keywords: Mitochondrial disorder, Vision loss, MT-ND1, Complex I
Highlights
► Heteroplasmic m.3890G>A/MT-ND1 mutation causes optic atrophy and bilateral brainstem lesions ► This mutation affects a conserved amino acid in a functional domain of ND1 subunit ► This mutation significantly affects CI redox activity and function
1. Introduction
Mitochondrial complex I (NADH: ubiquinone oxidoreductase, CI, EC:1.6.5.3) is the first energy-transducing enzyme of the mitochondrial respiratory chain. By oxidizing NADH and transferring electrons to the final acceptor ubiquinone coupled with proton translocation across the mitochondrial inner membrane, CI contributes to generate the mitochondrial membrane potential utilized to produce ATP through the process of oxidative phosphorylation (OXPHOS) [1]. CI is a large multi-subunit respiratory enzyme built assembling seven ND subunits encoded by mitochondrial DNA (mtDNA) and about 38 subunits encoded by nuclear DNA (nDNA). Thus, this is the only respiratory enzyme with such a large component dependent on mtDNA and, considering the high mutation rate of this small circular genome, over the last 20 years an increasing number of pathogenic mutations has been described in association with human diseases [2].
Historically, the first nucleotide changes affecting ND subunits of complex I have been associated with Leber's hereditary optic neuropathy (LHON, OMIM 535000) [3–5]. Currently, three common pathogenic point mutations m.11778G>A/MT-ND4, m.3460G>A/MT-ND1 and m.14484T>C/MT-ND6 are found in over 90% of LHON cases worldwide [6]. Syndromic forms of LHON-like optic atrophy and basal ganglia lesions or stroke-like episodes have been also associated with severe pathogenic mutations in ND subunit genes, linking in a continuum LHON with Leigh syndrome (LS), bilateral striatal necrosis (BSN) and mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes (MELAS) [6]. These phenotypes may coexist in the same maternal lineage in relation with different tissue loads of heteroplasmic mutation, thus indicating different thresholds for clinical phenotypes. Examples of well-established mutations with variable phenotype are the m.14459G>A/MT-ND6 mutation associated with LHON and spastic dystonia, or the m.13513G>A/MT-ND5 mutation associated with LHON and MELAS, both also reported as causing infantile LS [6].
We here report the second patient carrying the heteroplasmic mutation m.3890G>A/MT-ND1 (p.R195Q), a young adult affected with LHON-like severe optic neuropathy and bilateral, partially reversible, lesions of the brainstem [7]. By transferring the mutant mtDNA in transmitochondrial cytoplasmic hybrid (cybrid) cells we established the pathogenic role of this mutation, which affects CI function in homoplasmic mutant cybrids.
2. Materials and methods
2.1. Case report
The proband is a 35 years old male who suffered, when he was 31, a sudden and painless loss of vision in OS and, after two weeks, in OD. The patient reported blurring of the central vision associated with photopsias, and a first ophthalmologic exam showed hyperemic optic disc with teleangectatic vessels in the absence of fluorescein leakage at fluorangiography. Pattern visual evoked potentials (PVEPs) revealed abnormal morphology with normal latency of cortical responses in OD and absent responses in OS. He was treated with corticosteroids and vitamin B12 without any benefit. Three months later a second ophthalmologic exam reported that initial temporal pallor of the optic disc and visual acuity was decreased to 1/20 both eyes with bilateral cecocentral scotoma.
The hypothesis of an autoimmune disorder was then pursued based on a preceding history of recurrent abdominal pain and diarrhea complained by the patient since the age of 29, and recurrent oral aphtae and joints pain since he was 14. Three months before visual loss the patient presented persistent vertigo and brain MRI showed bilateral and symmetrical focal hyperintensities on T2-weighted images affecting the vestibular nuclei (Fig. 1A). These abnormalities were completely resolved at an MRI scan performed 3 months later, at the time of visual loss, with only evidence of a mild left optic nerve hyperintensity on T2-weighted images. Thus, on the basis of clinical and neuroradiological presentation, although all blood and cerebrospinal fluid tests for autoimmune disorders were normal, a diagnosis of Bechet vasculitis with central nervous system involvement was postulated and a therapy with corticosteroids and cyclosporine was started without benefits.
Fig. 1.

Brain MRI: (A) 31-year-old, FLAIR T2-weighted axial image shows bilateral and symmetrical hyperintensities of the vestibular nuclei (arrows); (B) 33-year-old, FLAIR T2-weighted axial image shows increased volume and increased signal intensity of the right inferior colliculus (arrow); (C) 35-year-old, FSPGR-T1-weighted axial image shows hypointense signal changes of both inferior colliculi (arrows), consistent with cystic necrotic lesions. OCT analysis of RNFL thickness (Cirrus, Carl Zeiss Meditec) and fundus color pictures of the patient at 3 years follow-up. OCT shows a diffuse thinning of RNFL thickness corresponding to complete atrophy of the optic nerve as shown by pallor of the optic disc at fundus pictures (D).
We observed this patient 9 months after the onset of vision loss. At the ophthalmologic exam he showed bilateral severe reduction of visual acuity (counting finger in both eyes), cecocentral scotoma and bilateral temporal optic disc pallor. The infero-temporal retinal nerve fiber layer thickness was reduced in both eyes at the optical coherence tomography (OCT). The neurological exam showed mild bilateral postural tremor at upper limbs, brisk deep tendon reflexes with right rotuleus and achilles clonus. Autoimmune and celiac screenings were negative. PVEPs showed the absence of cortical responses bilaterally. Electromyography identified slight myopathic changes. Somatosensorial and motor evoked potentials were normal. The standard genetic screening for the three common mtDNA LHON mutations (m.11778G>A/MT-ND4, m.3460G>A/MT-ND1 and m.14484T>C/MT-ND6) was negative. Idebenone therapy was started after 10 months from the onset of visual loss (405 mg/day).
At 33 years of age the patient complained of recurrent tinnitus in the right ear and underwent a new brain MRI that showed, on T2-weighted images, increased signal intensity of the right inferior colliculus that was of increased volume without post-contrast enhancement (Fig. 1B). The brain MRI performed for follow-up at the age of 35 years showed signal changes (hyperintensities on T2-weighted and hypointensity on T1-weighted images) affecting both inferior colliculi consistent with bilateral necrotic cystic lesions, and the normalization of the volume of the right inferior colliculus (Fig. 1C), in the absence of brainstem, cerebellar or cerebral atrophy.
Over the last 3 years OCT analysis showed a progressive thinning of the retinal nerve fiber layer thickness in all quadrants (Fig. 1D) with pale optic discs bilaterally at fundus exam and further reduction of the patient visual function. He continued on therapy with idebenone (540 mg/day) without any subjective or measurable improvement of visual acuity.
2.2. Muscle, biochemical and magnetic resonance spectroscopy investigations
Venous serum lactate was measured by a standardized exercise protocol, as previously detailed [8], in the proband and, for comparison, in a cohort of twenty LHON patients with one of the three canonical mutations.
Proton Magnetic Resonance Spectroscopy (1H-MRS) and skeletal muscle phosphorus Magnetic Resonance Spectroscopy (31P-MRS) studies were carried out on a 1.5-T whole body magnet (Signa Horizon General Electrics Medical Systems Milwaukee, Wisconsin). MRS investigations were performed in order to assess the bioenergetics of brain and skeletal muscle. Brain single voxel 1H-MRS was performed using the point resolved spectroscopy sequence. In order to maximize the detection of lactate, a volume of interest was selected in the lateral ventricles to include mostly the CSF (TE = 288, TR = 1500, number of acquisitions = 384) [9]. Suppressed water spectra were pre-processed with Gaussian filtering of 2 Hz followed by exponential filtering of − 1 Hz, and lactate fitted by the time domain semi-parametric algorithm QUEST. Skeletal muscle 31P -MRS was performed under resting conditions, as well as during an aerobic exercise and after recovery, as previously described [10]. A sample of 15 healthy volunteers age-matched was also studied with the same protocol.
Tibialis anterior muscle biopsy was carried out after informed consent of the patient. Histological and histoenzymatic analysis including hematoxylin & eosin (H&E) staining, cytochrome oxidase (COX) activity, succinic dehydrogenase (SDH) activity and double COX/SDH staining were performed [11].
Respiratory chain complexes activities on skeletal muscle homogenates were assessed with a dual-wavelength spectrophotometer (V550 Jasco Europe, Italy) in a stirred cuvette at 37 °C containing 50mMKH2PO4 (pH 7.6) 1 mM EDTA. CI activity (NADH:DB oxidoreductase) was assessed in the presence of 0.3 mM KCN, 50 μM DB and 100 μM NADH (λ 340–380 nm; εNADH: 6.2 mM− 1 cm− 1), after subtraction of 1 μM rotenone-insensitive activity. CII + CIII (succinate:cytochrome c oxidoredutase) and CIII (DBH2:cytochrome c oxidoreductase) activities were measured following the antimycin A sensitive reduction of cytochrome c (λ: 550–540 nm; εcyt c = 19.1 mM− 1 cm− 1) in presence of 0.3 mM KCN, 40 μM cytochrome c, 10 mM succinate or 50 μM DBH2. The CIV activity was determined adding 60 μM of reduced bovine heart cytochrome c, after subtraction of 0.3 mM KCN-insensitive activity. Data were corrected for citrate synthase activity and protein content [12,13].
2.3. Mitochondrial DNA analysis
Total DNA was extracted by standard methods from blood cells, urinary sediment epithelium and skeletal muscle after informed consent and approval of the internal review board. Direct sequence analysis of the entire mtDNA molecule was performed on total DNA extracted from skeletal muscle, as previously reported [14]. The m.3890G>A/MT-ND1 mutation was confirmed by restriction fragment length polymorphism (RFLP) analysis (primers and conditions are available upon request).
2.4. Quantification of the m.3890G>A/MT-ND1 mutation load by real time amplification refractory mutation system (ARMS) quantitative PCR
The ARMS qPCR assay was performed with a LightCycler® 480 Real-Time PCR System (Roche) using SYBR Green chemistry, according to published procedures [15]. Primers were designed introducing two mismatches, according to Bai and Wong [16]. The primer sequences used were as follows: ARMS forward primer for wild-type m.3890G: 5′-CACACTAGCAGAGACCAAggG-3′; ARMS forward primer for mutant m.3890A: 5′-CACACTAGCAGAGACCAAggA-3′; ARMS reverse primer: 5′-TGTGTATTCGGCTATGAAGAATAGG-3′, which is common for both wild-type and mutant alleles; forward primer for total mtDNA: 5′-CCCTAAAACCCGCCACATCT-3′; reverse primer for total mtDNA: 5′-GAGCGATGGTGAGAGCTAAGGT-3′. The lowercase letter in the primer indicates a mismatch nucleotide being introduced to increase the primer specificity [16]. The percentage of mutation heteroplasmy was calculated by absolute quantification through calibration curves generated by serial dilutions of the target sequence PCR products. Two different mtDNA amplicons were produced and inserted in a pGEM-T Easy Vector (Promega). The two plasmids contained respectively 3890G (wild-type) and 3890A (mutant), both encompassing the entire MT-ND1gene sequence.
2.5. Cybrids studies
Cybrid cell lines were generated from patient's skin fibroblasts (heteroplasmic with about 50% mutant load) as previously described [17]. Cybrids were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (South America source from Gibco, Invitrogen, Italy), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, in an incubator with a humidified atmosphere of 5% CO2 at 37 °C. For cell viability experiments, cells (4 × 104 cells/cm2) were seeded in 24 well plates and incubated for different times in glucose-free DMEM supplemented with 5 mmol/L galactose, 5 mmol/L Na-pyruvate and 5% FBS (DMEM-galactose). Viability was determined using the colorimetric sulforhodamine B assay [18], by measuring the sulforhodamine B absorbance at 570 nm with a VICTOR3 Multilabel Plate Counter (PerkinElmer Life and Analytical Sciences, Zaventem Belgium).
The rate of mitochondrial ATP synthesis was measured in digitonin-permeabilized cybrids using the previously described luciferin/luciferase assay [19], with minor modifications [20]. The rates were normalized to protein content and citrate synthase activity [12,13].
Isolation of mitochondrial-enriched fraction was carried out essentially as previously described [21]. Respiratory chain complexes activities were performed in 50 mM KH2PO4 buffer (pH 7.6), 3,5 mg/ml BSA and 20 μg of mitochondrial protein. CI activity (NADH:DB:DCIP oxidoreductase) was assessed in the presence of 60 μM DCIP (λ 600 nm; εDCIP: 19.1 mM− 1 cm− 1), 70 μM DB and 200 μM NADH, as previously described [21], after subtraction of 1 μM rotenone-insensitive activity. CI + CIII activity (NADH:cytochrome c reductase) was performed following cytochrome c reduction in the presence of 200 μM NADH and 20 μM bovine heart cytochrome c, after subtraction of 1 μM rotenone- and 1 μM antimycin A-insensitive activity. The CIII activity was determined as the antimycin A-sensitive DBH2:cytochrome c reductase activity, in the presence of 50 μM DBH2 and 20 μM bovine heart cytochrome c, as previously described [22]. CIV activity was measured adding 20 μM of reduced bovine heart cytochrome c, essentially as described in ref. [22]. The rates were normalized to citrate synthase activity and protein content [12,13]. CI assembly and in-gel activity were determined after Blue-Native electrophoresis of isolated mitochondria as previously described [23].
2.6. Statistical analysis
Data are presented as means ± SD. Statistical significance, determined by the Student t test for unpaired data, was considered at p ≤ 0.05. When data were not normally distributed the Mann–Whitney test was applied.
3. Results
3.1. Evidence of mitochondrial dysfunction with CI defect
Brain 1H-MRS, performed 9 months after onset of vision loss, disclosed a mild pathological accumulation of lactate, in the lateral ventricles, confirmed 2 years later by a second brain 1H-MRS exam (Fig. 2A–B). Muscle 31P-MRS failed to show abnormalities of energy metabolism either at rest or after aerobic exercise. Notwithstanding this, two independent assessments of serum venous lactate after standardized exercise and after recovery showed a persistently elevated lactic acid in the proband (after exercise 60.0 ± 19.2 mg/dl; after recovery 46.7 ± 8.8 mg/dl; normal range 5.8–22 mg/dl), which was significantly higher compared to the levels measured by the same protocol in a cohort of LHON patients (n = 20) carrying one of the canonical mutations (Fig. 2C).
Fig. 2.

Brain proton MR spectroscopy: (A) mild pathological accumulation of lactate was detected by 1H-MRS (single voxel PRESS localization sequence, TR = 1500 ms, TE = 288 ms) in the cerebrospinal fluid of the lateral ventricles (B). Venous serum lactate in LHON patients (n = 20) and in the proband (normal range 5.8–22 mg/dl) (C). Muscle histopathology: H&E, COX/SDH and SDH stains show some variability of fiber caliber and slight subsarcolemmal increase of SDH (D). Respiratory chain complexes activity in skeletal muscle is normalized for citrate synthase (CS) activity (E). Asterisks indicate statistical significance (p < 0.05).
Muscle histology (H&E) and histoenzymatic stains (COX, SDH and COX/SDH) showed minimal changes with fibers size variability and increased subsarcolemmal SDH (Fig. 2D). Assessment of respiratory chain activities on skeletal muscle homogenate revealed that CI was 50% reduced compared to controls (proband 8.04 ± 0.70 nmol/min mg, controls 16.05 ± 1.80 nmol/min mg) (Fig. 2E). Non-significant differences were observed for the integrated CII + CIII activity, which was increased in the proband, and CIII activity, which was reduced in the proband; CIV activity was normal (Fig. 2E).
3.2. Mitochondrial DNA analysis and protein modeling
Based on the LHON-like phenotype, the brain pathological accumulation of lactate at 1H-MRS, the pathologically high serum lactic acid after exercise, the reduction of CI activity in muscle, and having excluded the three common mutations associated with LHON we completely sequenced the proband's mtDNA. This resulted in the detection of the previously reported non-synonymous transition at position m.3890G>A/MT-ND1 [7] on a haplogroup H1 background (GenBank ID JQ408982) (Table 1) [24]. The quantification of the m.3890G>A/MT-ND1 mutation load by ARMS qPCR in different tissues showed that it was almost homoplasmic in the skeletal muscle and heteroplasmic, with different mutational loads, in blood cells and urinary epithelium (Fig. 3A). The mutation was also found in the proband's unaffected mother, with a substantially lower mutant load, and was absent in the other maternal relatives (Fig. 3A–B).
Table 1.
All mtDNA changes in patient's complete mtDNA sequence (GenBank ID JQ408982) compared to the Cambridge Reference Sequence (rCRS, GenBank ID NC_012920). All changes, except m.3890G>A, are reported as neutral [39].
| Nucleotide differences relative to rCRS | Gene | Amino-acid change |
|---|---|---|
| m.263A>G | MT-DLOOP | – |
| m.315insC | MT-DLOOP | – |
| m.750A>G | MT-RNR1 | – |
| m.961T>C | MT-RNR1 | – |
| m.1438 A>G | MT-RNR1 | – |
| m.3010G>A | MT-RNR2 | – |
| m.3890G>A | MT-ND1 | p.R195Q |
| m.4769A>G | MT-ND2 | Synonymous |
| m.7765A>G | MT-CO2 | Synonymous |
| m.8860A>G | MT-ATP6 | p.Y112A |
| m.13836A>G | MT-ND5 | Synonymous |
| m.15326A>G | MT-CYB | p.Y194A |
| m.16519T>C | MT-DLOOP | – |
Fig. 3.
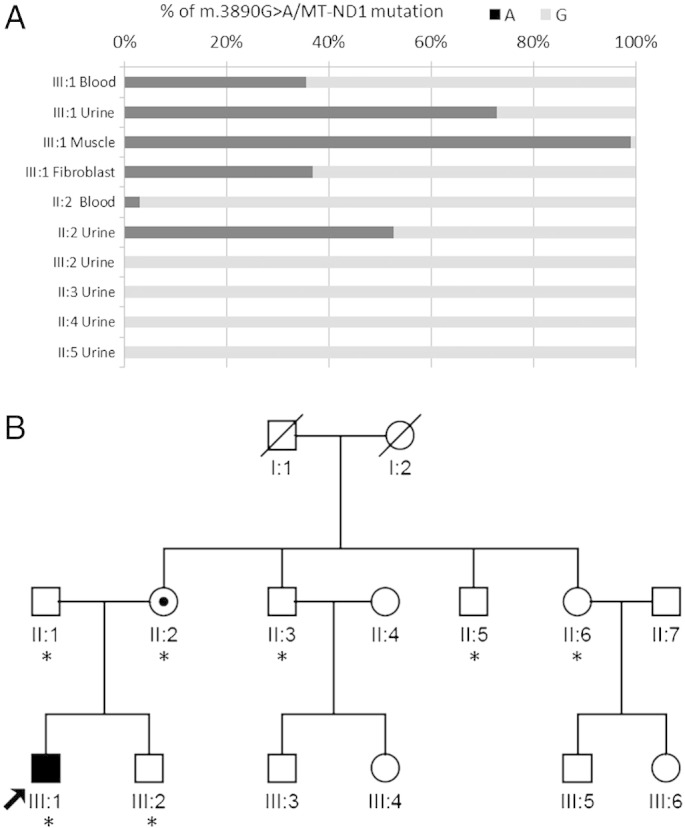
Quantification of the m.3890G>A/MT-ND1 mutation load by ARMS qPCR (A) in proband's skeletal muscle, blood cells and urinary epithelium, and in family (B) members' urinary epithelium, expressed as % of mutant 3890A compared to wild-type (wt) 3890G. The arrow indicates the proband, while asterisks indicate family members who were investigated.
The m.3890G>A/MT-ND1 mutation changes a basic arginine with a polar glutamine at the highly conserved amino acid position 195 (p.R195Q) in the ND1 subunit of CI (Fig. 4A). As shown in Fig. 4B this residue is located in the extra-membrane loop EF, which is also affected by the amino acid substitutions p.E214K and p.Y215H, respectively caused by the MELAS pathogenic mutations m.3946G>A and m.3949T>C [25]. In Fig. 4C and D all the mutations studied in the EF loop of the bacterial subunit corresponding to ND1 are reported. They are all associated with a decreased NADH-oxidation activity (40–98% of reduction), a partial CI disassembly and, in particular, the substitutions corresponding to p.E214K and p.Y215H abolished the membrane potential generated by the respiratory chain [26–28].
Fig. 4.

Global alignment of ND1 protein sequences from a wide range of eukaryotes and prokaryotes (A). Amino acid residues with a percentage of conservation ranging between 70.0% and 79.9% are highlighted in light gray, those between 80.0% and 99.9% are highlighted in dark gray and those invariant (100%) are highlighted in black. Localization of human pathogenic ND1 mutations in a bi-dimensional model (B). Note that most of the trans-membrane domains are free from mutations. Table of bacterial mutations versus human positions, in EF loop, which affect complex I activity (C). Bi-dimensional representation of EF loop with bacterial mutations (D). The bacteria residue R209 corresponds to R195 in human.
3.3. Cybrids studies
To assess the causal pathogenic role of the m.3890G>A/MT-ND1 mutation we generated cybrid cell lines, by transferring only the patient mtDNA into the rho mtDNA-devoid recipient cells, derived from the osteosarcoma 143B.TK-cell line. After cybridization we selected for further studies three syngeneic clones differing only at the np 3890, one homoplasmic wild type, one 50% heteroplasmic and one homoplasmic mutant. The CI redox activity was reduced in both heteroplasmic and homoplasmic mutant cybrids, but only the homoplasmic mutant clone showed a significant decrease (~ 50%) compared to wild-type (Fig. 5A). In addition, the CI + CIII integrated activity revealed a non-significant decrease in both heteroplasmic and homoplasmic mutant clones, whereas CIII activity was not affected, indicating a defective function limited to the respiratory CI (Fig. 5A). Complex IV activity was normal in all clones (Fig. 5A). Furthermore, we investigated the possible role played by the 3890G>A homoplasmic mutation on CI assembly. The in-gel-activity after BN-gel electrophoresis showed that this mutation did not affect the assembly of CI (Fig. 5B). We also assessed the overall mitochondrial energetic function by growing the cybrid clones in a glucose-free medium containing galactose. Under these conditions, the rate of glycolysis is markedly reduced and cells are forced to use oxidative phosphorylation for ATP production [29]. After 48 hours incubation in DMEM-galactose medium, the number of viable homoplasmic mutant cybrids was significantly reduced (~ 75%) compared to the wild-type or the heteroplasmic clones (Fig. 5C). Furthermore, the rate of mitochondrial ATP synthesis driven by CI substrates (pyruvate/malate) was significantly reduced in both the heteroplasmic (~ 20%) and homoplasmic (~ 50%) mutant cell lines, whereas the rate of ATP synthesis with the complex II substrate (succinate) was unaffected (Fig. 5D).
Fig. 5.

Activity of respiratory chain complexes normalized for citrate synthase (CS) activity (A), CI in-gel-activity (B), indicated by an arrow, time course of cell viability after incubation in galactose medium (C) and ATP synthesis rates normalized for CS activity (D) in three syngeneic cybrid clones: wild-type (0%), heteroplasmic (50%) and homoplasmic (100%) for m.3890G>A/MT-ND1 mutation. Asterisks indicate statistical significance (*p < 0.05; **p < 0.001).
4. Discussion
This study reports the second patient carrying the m.3890G>A/MT-ND1 mutation, characterized by a complex I defect in muscle and LHON-like severe optic atrophy associated with bilateral brainstem lesions. Our results, obtained by transferring the mutant mtDNA from heteroplasmic fibroblasts into cybrids, after selecting mtDNA isogenic clones except for the m.3890G>A/MT-ND1 mutant position, establish the pathogenic role of this rare mutation in affecting CI function.
The previously reported patient carrying the m.3890G>A/MT-ND1 mutation presented an early-onset severe encephalopathy leading to death at about 3 years of age. The clinical diagnosis was of Leigh syndrome, due to the occurrence of bilateral lesions in the subthalamic nuclei, lower pons and brainstem [7]. Respect to this case, our patient presented a milder phenotype consisting in a severe form of LHON-like visual loss with onset in adult age and without other major clinical manifestations in the 4 years of follow-up. Similarly to the other case, he developed bilateral and symmetrical focal alterations affecting the brainstem, hyperintense on T2-weighted images. These brain MRI abnormalities firstly involved the vestibular nuclei and were associated with transient episodes of vertigo, disappearing along with the symptoms after a few months. Subsequently, other lesions with similar characteristics involved the inferior colliculi, in association with recurrent tinnitus, evolving in necrotic cavities.
The presence of MR alterations and corresponding neuropathological changes in different structures of brainstem, including the inferior colliculi, are well recognized in LS, in association with lesions located in the basal ganglia and/or diencephalon [30]. On the other hand, a few patients, carrying the common LHON mutations or the m.11777C>A/MT-ND4 mutation affecting the same amino acid position of the LHON mutation m.11778/MT-ND4 (p.R340H) with a different amino acid change (p.R340S), presented bilateral brainstem lesions [31,32]. A recent study reviewing a large cohort of LS patients suggested that brainstem lesions are more typical for mtDNA-related complex I deficiency, in most cases with the concurrent striatal involvement [33]. Peculiar to both patients carrying the m.3890G>A/MT-ND1 mutation is the sparing of the striatum. Furthermore, in our patient the neuroradiological evolution also recalls the cerebral lesions characterizing the stroke-like episodes in MELAS syndrome, which can fully resolve or leave residual MRI abnormalities [34]. However, reversibility of the brain lesion has also been documented in LS [30].
Overall, our patient differed from the previous one for a milder phenotype. Optic atrophy was the main clinical feature of our patient, resembling a severe version of LHON. It is of note that the initial diagnostic hypothesis of Bechet vasculitis with central nervous system involvement delayed the identification of mitochondrial dysfunction as the pathogenic mechanism for optic neuropathy. However, the typical hallmarks of a mitochondrial optic neuropathy, with preferential involvement of the papillomacular bundle, with central scotoma and initial temporal pallor, re-directed the diagnosis towards LHON, leading, by complete mtDNA sequence analysis, to the identification of the rare m.3890G>A/MT-ND1 mutation. The OXPHOS impairment associated with this mutation was more severe than normally seen in LHON, as documented by higher values of serum lactic acid levels and pathological presence of lactic acid in the ventriculi at brain 1H− MRS. This impairment was associated with a virtually homoplasmic mutant mtDNA in the skeletal muscle and about 50% mutational load in blood cells. The previously reported patient had 95% mutant mtDNA in the skeletal muscle [7]. Thus, based on these results it is difficult to state that differences in clinical severity between the two patients are clearly ascribed to a different heteroplasmic load of the mutation in cerebral tissue. Nuclear modifying genes may also be involved in modulating the severity of mtDNA mutations affecting ND subunits of complex I.
The cybrid cell model has been exploited to characterize the pathologic features of the m.3890G>A/MT-ND1 mutation, which affects a highly conserved region of the ND1 subunit of CI. As recently demonstrated by X-ray crystallographic studies on CI from T. thermophilus, the ND1 subunit forms the interface between the two arms of CI, the hydrophobic membrane and the hydrophilic matrix-protruding regions [35]. Furthermore, the ND1 subunit most probably participates in forming the quinone-binding pocket together with the nuclear-encoded subunits NDUFS7 and NDUFS2 [36]. The 3D structure of the bacterial CI has been completely resolved, with the only remarkable exception of the ND1 subunit, for which only the overall number of transmembrane helices is available [37]. The extra-membrane domains of ND1, constituted by more than 100 amino acids, are essential for CI function and structure, as demonstrated by the presence of numerous pathogenic mutations in mitochondrial patients. Compatibly, the m.3890G>A/MT-ND1 mutation decreased CI redox activity and CI driven ATP synthesis, without affecting CI assembly, as studied in patient's mutant cybrids. Interestingly, other mutations in the same region of ND1 subunit have been reported in patients affected by MELAS and optic atrophy [2,6]. Noticeably, the CI activity, as well as the CI-driven ATP synthesis were also affected in the heteroplasmic cybrid clone (50% mutant) suggesting the absence of a clear cut threshold, as frequently seen for other mtDNA pathogenic mutations [38]. Thus, this mutation seems to be rate limiting on the CI-driven ATP synthesis. However, the parallel pathway through CII may function as compensatory, as suggested by the tendency to increased CII + CIII activity seen in both proband's muscle and mutant cybrids, and unaffected CII-substrate driven ATP synthesis in mutant cybrids.
5. Conclusions
In this study we identified the m.3890G>A/MT-ND1 (p.R195Q) mutation in a patient affected by LHON-like optic atrophy and bilateral brainstem lesions. This mutation, absent from population surveys and public datasets of more than ten thousand complete mtDNAs (GenBank database was searched on March 2nd 2012), was previously detected in a patient with fatal infantile LS with bilateral brainstem lesions [7]. The conservation analysis of the corresponding amino acid position and the functional characterization of cybrid cells carrying the mutant mtDNA compared to syngeneic wild-type cybrids provided evidence on the pathogenic role of this mutation clearly compromising the function of CI.
Acknowledgements
The authors are grateful to the proband and his family for participating in this study. This project received support from Telethon-Italy grants GUP09004 and GGP06233 (to VC), ERARE European consortium on optic atrophies (to AM), Fondazione Alma Mater Ticinensis (to AT), the Italian Ministry of the University: Progetti Ricerca Interesse Nazionale 2009 (to AA and AT), FIRB-Futuro in Ricerca 2008 (to AA). Calvaruso MA was a recipient of a fellowship supported by the Italian Ministry of the University project FIRB “Futuro in Ricerca” to Dr. Giuseppe Gasparre.
References
- 1.Efremov R.G., Sazanov L.A. Respiratory complex I: ‘steam engine’ of the cell? Curr. Opin. Struct. Biol. 2011;21:532–540. doi: 10.1016/j.sbi.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 2.Distelmaier F., Koopman W.J., van den Heuvel L.P., Rodenburg R.J., Mayatepek E., Willems P.H., Smeitink J.A. Mitochondrial complex I deficiency: from organelle dysfunction to clinical disease. Brain. 2009;132:833–842. doi: 10.1093/brain/awp058. [DOI] [PubMed] [Google Scholar]
- 3.Wallace D.C., Singh G., Lott M.T., Hodge J.A., Schurr T.G., Lezza A.M., Elsas L.J., II, Nikoskelainen E.K. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science. 1988;242:1427–1430. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- 4.Howell N., Bindoff L.A., McCullough D.A., Kubacka I., Poulton J., Mackey D., Taylor L., Turnbull D.M. Leber hereditary optic neuropathy: identification of the same mitochondrial ND1 mutation in six pedigrees. Am. J. Hum. Genet. 1991;49:939–950. [PMC free article] [PubMed] [Google Scholar]
- 5.Johns D.R., Neufeld M.J., Park R.D. An ND-6 mitochondrial DNA mutation associated with Leber hereditary optic neuropathy. Biochem. Biophys. Res. Commun. 1992;187:1551–1557. doi: 10.1016/0006-291x(92)90479-5. [DOI] [PubMed] [Google Scholar]
- 6.Fraser J.A., Biousse V., Newman N.J. The neuro-ophthalmology of mitochondrial disease. Surv. Ophthalmol. 2010;55:299–334. doi: 10.1016/j.survophthal.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moslemi A.R., Darin N., Tulinius M., Wiklund L.M., Holme E., Oldfors A. Progressive encephalopathy and complex I deficiency associated with mutations in MTND1. Neuropediatrics. 2008;39:24–28. doi: 10.1055/s-2008-1076739. [DOI] [PubMed] [Google Scholar]
- 8.Montagna P., Plazzi G., Cortelli P., Carelli V., Lugaresi E., Barboni P., Fiocchi M. Abnormal lactate after effort in healthy carriers of Leber's hereditary optic neuropathy. J. Neurol. Neurosurg. Psychiatry. 1995;58:640–641. doi: 10.1136/jnnp.58.5.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grimaldi D., Tonon C., Cevoli S., Pierangeli G., Malucelli E., Rizzo G., Soriani S., Montagna P., Barbiroli B., Lodi R., Cortelli P. Clinical and neuroimaging evidence of interictal cerebellar dysfunction in FHM2. Cephalalgia. 2010;30:552–559. doi: 10.1111/j.1468-2982.2009.01979.x. [DOI] [PubMed] [Google Scholar]
- 10.Lodi R., Tonon C., Valentino M.L., Manners D., Testa C., Malucelli E., La Morgia C., Barboni P., Carbonelli M., Schimpf S., Wissinger B., Zeviani M., Baruzzi A., Liguori R., Barbiroli B., Carelli V. Defective mitochondrial adenosine triphosphate production in skeletal muscle from patients with dominant optic atrophy due to OPA1 mutations. Arch. Neurol. 2011;68:67–73. doi: 10.1001/archneurol.2010.228. [DOI] [PubMed] [Google Scholar]
- 11.Dubovitz V., Sewry C.A. third ed. Saunders; Philadelphia: 2007. Muscle Biopsy. [Google Scholar]
- 12.Trounce I.A., Kim Y.L., Jun A.S., Wallace D.C. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- 13.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 14.Torroni A., Rengo C., Guida V., Cruciani F., Sellitto D., Coppa A., Calderon F.L., Simionati B., Valle G., Richards M., Macaulay V., Scozzari R. Do the four clades of the mtDNA haplogroup L2 evolve at different rates? Am. J. Hum. Genet. 2001;69:1348–1356. doi: 10.1086/324511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Genasetti A., Valentino M.L., Carelli V., Vigetti D., Viola M., Karousou E.G., Melzi d'Eril G.V., De Luca G., Passi A., Pallotti F. Assessing heteroplasmic load in Leber's hereditary optic neuropathy mutation 3460G->A/MT-ND1 with a real-time PCR quantitative approach. J. Mol. Diagn. 2007;9:538–545. doi: 10.2353/jmoldx.2007.060183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bai R.K., Wong L.J. Detection and quantification of heteroplasmic mutant mitochondrial DNA by real-time amplification refractory mutation system quantitative PCR analysis: a single-step approach. Clin. Chem. 2004;50:996–1001. doi: 10.1373/clinchem.2004.031153. [DOI] [PubMed] [Google Scholar]
- 17.King M.P., Attardi G. Isolation of human cell lines lacking mitochondrial DNA. Methods Enzymol. 1996;264:304–313. doi: 10.1016/s0076-6879(96)64029-4. [DOI] [PubMed] [Google Scholar]
- 18.Scarlatti F., Sala G., Somenzi G., Signorelli P., Sacchi N., Ghidoni R. Resveratrol induces growth inhibition and apoptosis in metastatic breast cancer cells via de novo ceramide signalling. FASEB J. 2003;17:2339–2341. doi: 10.1096/fj.03-0292fje. [DOI] [PubMed] [Google Scholar]
- 19.Manfredi G., Yang L., Gajewski C.D., Mattiazzi M. Measurements of ATP in mammalian cells. Methods. 2002;26:317–326. doi: 10.1016/S1046-2023(02)00037-3. [DOI] [PubMed] [Google Scholar]
- 20.Giorgio V., Petronilli V., Ghelli A., Carelli V., Rugolo M., Lenaz G., Bernardi P. The effects of idebenone on mitochondrial bioenergetics. Biochim. Biophys. Acta. 2012;1817:363–369. doi: 10.1016/j.bbabio.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janssen A.J., Trijbels F.J., Sengers R.C., Smeitink J.A., van den Heuvel L.P., Wintjes L.T., Stoltenborg-Hogenkamp B.J., Rodenburg R.J. Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin. Chem. 2007;53:729–734. doi: 10.1373/clinchem.2006.078873. [DOI] [PubMed] [Google Scholar]
- 22.Bénit P., Goncalves S., Philippe Dassa E., Brière J.J., Martin G., Rustin P. Three spectrophotometric assays for the measurement of the five respiratory chain complexes in minuscule biological samples. Clin. Chim. Acta. 2006;374:81–86. doi: 10.1016/j.cca.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 23.Porcelli A.M., Ghelli A., Ceccarelli C., Lang M., Cenacchi G., Capristo M., Pennisi L.F., Morra I., Ciccarelli E., Melcarne A., Bartoletti-Stella A., Salfi N., Tallini G., Martinuzzi A., Carelli V., Attimonelli M., Rugolo M., Romeo G., Gasparre G. The genetic and metabolic signature of oncocytic transformation implicates HIF1alpha destabilization. Hum. Mol. Genet. 2010;19:1019–1032. doi: 10.1093/hmg/ddp566. [DOI] [PubMed] [Google Scholar]
- 24.Achilli A., Rengo C., Magri C., Battaglia V., Olivieri A., Scozzari R., Cruciani F., Zeviani M., Briem E., Carelli V., Moral P., Dugoujon J.M., Roostalu U., Loogväli E.L., Kivisild T., Bandelt H.J., Richards M., Villems R., Santachiara-Benerecetti A.S., Semino O., Torroni A. The molecular dissection of mtDNA haplogroup H confirms that the Franco-Cantabrian glacial refuge was a major source for the European gene pool. Am. J. Hum. Genet. 2004;75:910–918. doi: 10.1086/425590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirby D.M., McFarland R., Ohtake A., Dunning C., Ryan M.T., Wilson C., Ketteridge D., Turnbull D.M., Thorburn D.R., Taylor R.W. Mutations of the mitochondrial ND1 gene as a cause of MELAS. J. Med. Genet. 2004;41:784–789. doi: 10.1136/jmg.2004.020537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinha P.K., Torres-Bacete J., Nakamaru-Ogiso E., Castro-Guerrero N., Matsuno-Yagi A., Yagi T. Critical roles of subunit NuoH (ND1) in the assembly of peripheral subunits with the membrane domain of Escherichia coli NDH-1. J. Biol. Chem. 2009;284:9814–9823. doi: 10.1074/jbc.M809468200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kervinen M., Hinttala R., Helander H.M., Kurki S., Uusimaa J., Finel M., Majamaa K., Hassinen I.E. The MELAS mutations 3946 and 3949 perturb the critical structure in a conserved loop of the ND1 subunit of mitochondrial complex I. Hum. Mol. Genet. 2006;15:2543–2552. doi: 10.1093/hmg/ddl176. [DOI] [PubMed] [Google Scholar]
- 28.Kurki S., Zickermann V., Kervinen M., Hassinen I., Finel M. Mutagenesis of three conserved Glu residues in a bacterial homologue of the ND1 subunit of complex I affects ubiquinone reduction kinetics but not inhibition by dicyclohexylcarbodiimide. Biochemistry. 2000;39:13496–13502. doi: 10.1021/bi001134s. [DOI] [PubMed] [Google Scholar]
- 29.Robinson B.H., Petrova-Benedict R., Buncic J.R., Wallace D.C. Nonviability of cells with oxidative defects in galactose medium: a screening test for affected patient fibroblasts. Biochem. Med. Metab. Biol. 1992;48:122–126. doi: 10.1016/0885-4505(92)90056-5. [DOI] [PubMed] [Google Scholar]
- 30.Arii J., Tanabe Y. Leigh syndrome: serial MR imaging and clinical follow-up. AJNR Am. J. Neuroradiol. 2000;21:1502–1509. [PMC free article] [PubMed] [Google Scholar]
- 31.Funalot B., Reynier P., Vighetto A., Ranoux D., Bonnefont J.P., Godinot C., Malthièry Y., Mas J.L. Leigh-like encephalopathy complicating Leber's hereditary opticNeuropathy. Ann. Neurol. 2002;52:374–377. doi: 10.1002/ana.10299. [DOI] [PubMed] [Google Scholar]
- 32.Hadzsiev K., Maasz A., Kisfali P., Kalman E., Gomori E., Pal E., Berenyi E., Komlosi K., Melegh B. Mitochondrial DNA 11777C>A mutation associated Leigh syndrome: case report with a review of the previously described pedigrees. Neruomol. Med. 2010;12:277–284. doi: 10.1007/s12017-010-8115-9. [DOI] [PubMed] [Google Scholar]
- 33.DiMauro S., Hirano M., MELAS . In: GeneReviews™ [Internet] Pagon R.A., Bird T.D., Dolan C.R., Stephens K., Adam M.P., editors. University of Washington, Seattle; Seattle (WA): 2001. [Google Scholar]
- 34.Lebre A.S., Rio M., Faivre d'Arcier L., Vernerey D., Landrieu P., Slama A., Jardel C., Laforêt P., Rodriguez D., Dorison N., Galanaud D., Chabrol B., Paquis-Flucklinger V., Grévent D., Edvardson S., Steffann J., Funalot B., Villeneuve N., Valayannopoulos V., de Lonlay P., Desguerre I., Brunelle F., Bonnefont J.P., Rötig A., Munnich A., Boddaert N. A common pattern of brain MRI imaging in mitochondrial diseases with complex I deficiency. J. Med. Genet. 2011;48:16–23. doi: 10.1136/jmg.2010.079624. [DOI] [PubMed] [Google Scholar]
- 35.Efremov R.G., Baradaran R., Sazanov L.A. The architecture of respiratory complex I. Nature. 2010;465:441–445. doi: 10.1038/nature09066. [DOI] [PubMed] [Google Scholar]
- 36.Murai M., Sekiguchi K., Nishioka T., Miyoshi H. Characterization of the inhibitor binding site in mitochondrial NADH-ubiquinone oxidoreductase by photoaffinity labeling using a quinazoline-type inhibitor. Biochemistry. 2009;48:688–698. doi: 10.1021/bi8019977. [DOI] [PubMed] [Google Scholar]
- 37.Efremov R.G., Sazanov L.A. Structure of the membrane domain of respiratory complex I. Nature. 2011;476:414–420. doi: 10.1038/nature10330. [DOI] [PubMed] [Google Scholar]
- 38.Chomyn A., Martinuzzi A., Yoneda M., Daga A., Hurko O., Johns D., Lai S.T., Nonaka I., Angelini C., Attardi G. MELAS mutation in mtDNA binding site for transcription termination factor causes defects in protein synthesis and in respiration but no change in levels of upstream and downstream mature transcripts. Proc. Natl. Acad. Sci. U. S. A. 1992;89:4221–4225. doi: 10.1073/pnas.89.10.4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruiz-Pesini E., Lott M.T., Procaccio V., Poole J., Brandon M.C., Mishmar D., Yi C., Kreuziger J., Baldi P., Wallace D.C. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 2007;35:D823–D828. doi: 10.1093/nar/gkl927. (URL: http://www.mitomap.org) [DOI] [PMC free article] [PubMed] [Google Scholar]