

Abstract
Hepatosplenic T-cell lymphoma (HSTL) is a rare entity mostly derived from γδ T cells that shows a fatal outcome. Its pathogenesis remains largely unknown. HSTL samples (7γδ, 2αβ) and the DERL2 HSTL-cell line were subject to combined gene expression profiling and array-based comparative genomic hybridization. Compared to other T-cell lymphomas, HSTL disclosed a distinct molecular signature irrespective of TCR cell lineage. Compared to PTCL,NOS and normal γδ cells, HSTL overexpressed genes encoding NK-cell associated molecules, oncogenes (FOS, VAV3), the Sphingosine-1-phosphatase receptor 5 involved in cell trafficking and the tyrosine kinase SYK, whereas the tumor suppressor gene AIM1 was among the most downexpressed. Methylation analysis of DERL2 cells demonstrated highly methylated CpG islands of AIM1 and decitabine treatment induced significant increase in AIM1 transcripts. Notably, Syk was demonstrated in HSTL cells with its phosphorylated form present in DERL2 cells by Western blot, and in vitro DERL2 cells were sensitive to a Syk inhibitor. Genomic profiles confirmed recurrent isochromosome 7q (n=6/9) without alterations at 9q22 and 6q21 containing SYK and AIM1 genes, respectively. The current study identifies a distinct molecular signature for HSTL and highlights oncogenic pathways which offer rationale for exploring new therapeutic options such as Syk inhibitors and demethylating agents.
Keywords: Adult; Aged; Base Sequence; Cell Lineage; genetics; Chromosome Aberrations; Cluster Analysis; Crystallins; metabolism; Drug Resistance, Neoplasm; genetics; Female; Gene Expression Profiling; Gene Expression Regulation, Neoplastic; Genes, Neoplasm; genetics; Humans; Intracellular Signaling Peptides and Proteins; antagonists & inhibitors; metabolism; Isochromosomes; genetics; Liver Neoplasms; drug therapy; genetics; pathology; Lymphoma, T-Cell; drug therapy; genetics; pathology; Male; Membrane Proteins; metabolism; Middle Aged; Molecular Sequence Data; Molecular Targeted Therapy; Protein-Tyrosine Kinases; antagonists & inhibitors; metabolism; Receptors, Antigen, T-Cell, alpha-beta; genetics; Receptors, Antigen, T-Cell, gamma-delta; genetics; Splenic Neoplasms; drug therapy; genetics; pathology; Tumor Markers, Biological; genetics; metabolism; Young Adult
INTRODUCTION
Hepatosplenic T-cell lymphoma (HSTL), originally described as hepatosplenic γδ T cell lymphoma, is a rare lymphoma entity with peculiar clinical presentation - hepatosplenomegaly without significant lymphadenopathy - and pathological features - intrasinusal/sinusoidal infiltration by neoplastic T cells in the bone marrow, spleen and liver[1–3]. The disease occurs predominantly in young adults, in association with a setting of long-term immunosuppression in solid organ transplant recipients or with prolonged antigenic stimulation [4]. Cases have also been reported in children treated by azathioprine and infliximab for Crohn’s disease[5]. While most HSTL are derived from the γδ subset, a few similar cases with an αβ phenotype have also been described[6,7], and the simplified designation “hepatosplenic T-cell lymphoma” was favored in the latest World Health Organization classification[8]. HSTL is associated with a recurrent isochromosome 7q and less often, trisomy 8[9], but its pathogenesis remains largely unknown.
Despite relatively innocuous cytology, the disease is highly aggressive with an almost constant fatal outcome and a median overall survival barely exceeding one year[4]. Occasional long survivors have been reported and few patients respond to cytarabine or deoxycoformycin[4,10]. Therapeutic strategies curative in a significant proportion of other aggressive subtypes of lymphoma, have proved to be ineffective in HSTL and efficient treatment modalities remain to be defined.
Over the past years, genome-wide molecular profiling studies have contributed significant insights to the pathobiology of several T-cell lymphoma entities[11–14] and brought informative data on the multiple molecular subgroups in PTCL, not otherwise specified (PTCL,NOS)[15,16]. In that respect, data on HSTL are scarce[13,17]. In the current study, we analyzed a series of HSTL samples in relation to normal γδ cells, PTCL,NOS and extranodal NK/T-cell lymphoma, nasal-type (NKTCL), another entity derived from cytotoxic lymphocytes of the innate immune system. The aim of the study was to (1) characterize the molecular signature of HSTL, (2) identify potential candidate pathways relevant to pathogenesis, and (3) search for biomarkers useful in the diagnostic purposes or in the future targeted therapies.
PATIENTS, MATERIALS AND METHODS
Patient characteristics and tumor samples
Nine HSTL patients with high quality RNA and/or DNA extracted from frozen tumor samples were selected for this study. All patients had spleen, liver and bone marrow involvement without lymphadenopathies. Three patients had been included in previous reports[4,9]. The main clinical, phenotypic and molecular characteristics are summarized in Table 1. The tumor samples, comprised six splenic tissue samples and three cell suspensions (from spleen, bone marrow and blood), two of which were enriched in tumor cells (samples HSTL_01 and HSTL_09). All cases were reviewed by three hematopathologists (L.d.L, Y.H. and P.G.) and diagnosed according to the WHO criteria[8]. The tumor cells had a CD3+, CD2+, CD5−, TiA1+, GzmB-immunophenotype and were negative for EBV. T-cell receptor (TCR )lineage was determined by immunohistochemistry and/or flow cytometry for TCRβ and TCRδ chain expression and by GC-clamp multiplex PCR for TCRγ and/or δ chain rearrangements ((PCR)-δ-DGGE procedure)[18]. In total, seven cases with a δTCR1+, βF1− immunophenotype and/or a biallelic rearrangement of the TCRδ chain[18,19], were classified as γδ HSTL and two cases with a δTCR1−, βF1+ phenotype as αβ HSTL. Four of seven investigated cases disclosed isochromosome 7q.
Table 1.
Summary of clinical, pathological, immunohistochemical, and cytogenetic features of patients enrolled in the study.
| EBI ID | Age (at dx) | Sex | Diagnosis | Organ | Type of sample | CD3 | CD2 | CD5 | CD7 | CD4 | CD8 | βF1 | δTCR | CD56 | TiA1 | GzmB | EBV ° | T clone | Cytogenet |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HSTL_02 | 41 | F | HSTL γδ | Spleen | Tissue | + | + | − | − | − | − | − | + * | + | + | − | − | + R(Jδ) G(β) | iso7q (+) |
| HSTL_03 | 73 | F | HSTL γδ | Spleen | Tissue | + | + | − | − | − | + | − | + | + | + | + | − | + | iso7q (−) |
| HSTL_04 | 38 | M | HSTL γδ | Spleen | Tissue | + | nd | − | − | − | − | + | + ** | − | + | − | − | + | iso7q (+) trisomy 8 |
| HSTL_05 | 56 | M | HSTL αβ | Spleen | Tissue | + | nd | − | nd | − | − | + | − | − | − | − | − | + | iso7q (+) |
| HSTL_06 | 53 | F | HSTL αβ | Spleen | Tissue | + | − | − | + | − | − | + | − | + | + | − | − | + | iso7q (−) |
| HSTL_08 | 24 | F | HSTL γδ | Blood | Cell suspension (no cell sorting) | + | nd | − | + | − | − | − | + | + | + | nd | nd | + | |
| HSTL_09 | 36 | M | HSTL γδ | Blood | Cell suspension (cell sorting CD56+) Purity 77% | + | + | − | + | − | − | − | + | + | + | − | nd | + | iso7q (+) |
| HSTL_01 | 86 | F | HSTL γδ | Spleen | Cell suspension (cell sorting CD3+ CD56+) Purity 84% | + | + | −/+ | nd | − | − | − | + | + | + | − | − | + | iso7q (−) |
| HSTL_10 | 67 | M | HSTL γδ | Spleen | Tissue | + | + | − | + | − | − | − | + ** | − | + | − | − | + | Iso7q (−) |
| HSTL_07 DERL2 | 30 | M | HSTL γδ | Blood | Cell line | + | + | − | + | − | + | + | + | − | Iso7q (+) |
HSTL_02 with a γδ HSTL phenotype at diagnosis disclosed a TCR-silent phenotype (βF1−, δTCR1−) at the time of analysis.
δ chain rearrangement determination using a GC-clamp multiplex polymerase chain reaction (PCR)-δ-DGGE procedure
EBV investigation by in situ hybridization with EBERs probes and/or immunohistochemistry with LMP-1 antibody
Twelve additional HSTL cases were selected for validations (10 formalin-fixed tissues for immunohistochemistry and 2 frozen samples for RT-PCR analyses).
The study was approved by the institutional review board “Comité de Protection des Personnes Ile de France IX”, Créteil, France CPP N°08-009 (06/05/08).
Cell lines and normal γδ T cells
Eight samples of normal peripheral blood activated γδ T cells (6 from Correia[20] and 2 from Zhang[21]) and 3 samples of normal resting γδ T cells (from Correia[20]) were used for comparison. Normal γδ T cells sorted using magnetic anti-TCRγ/δ microbeads kit (Miltenyi Biotec, Bergisch Gladbach, Germany) from 3 spleens removed for benign conditions were also obtained for qRT-PCR validation. DERL2, a γδ cell line derived from HSTL[22], was cultured in IMDM supplemented with 2 mM L-glutamine and 20% heat-inactivated human serum (Invitrogen, Carlsbad, CA) in the presence of recombinant human interleukin (IL)-2 (100 U/ml, Laboratoires Chiron, Amsterdam). Centrifugated pellets of DERL2 cells were fixed in ethanol to construct paraffin-embedded blocks[14].
Microarray procedures
Microarray analyses, from extraction to labeling and hybridization, were performed as previously reported[11,14].
Gene expression analyses
HG-U133-plus-2.0 Affymetrix array data were obtained for nine HSTL and the DERL-2 cell line (Table 1) and deposited to ArrayExpress under accession number E-MTAB-638. HG-U133-plus-2.0 profiles of 42 PTCL-NOS, 23 AITL, 23 NKTCL and 3 HSTL previously reported by our group (E-TABM-783, E-TABM-702, n=40)[11,14] and others (GSE19067, GSE6338, n=51)[12,13] were used for analyses. Control samples (8 activated γδ T-cells, 3 resting γδ T-cells, 18 B-cells, 6 spleens and 9 lymph nodes) were collected by us (n=6) or from public sources (E-MEX-1601, GSE13906, GSE12195, GSE15271, GSE7307)[20,21,23,24]. Affymetrix raw data of all samples were normalized in batch using RMA algorithm. The clustering analysis of the 50 tumor samples from our series was performed as already described [14]. We used moderate T-tests to identify genes differentially expressed between two groups of samples and AUC criteria to identify discriminant genes. KEGG and Biocarta pathways (and related genes) were obtained from ftp://ftp.genome.ad.jp/pub/kegg/pathways/hsa and http://www.biocarta.com. Pathways were ranked according to the scores of three different algorithms including globaltest, SAM-GS and GSA. Methodological details are given in the Supplementary Materials and Methods (Method S1).
Array-based comparative genomic hybridization
DNAs extracted from 7 HSTL and DERL2 cell line were hybridized on Agilent Sureprint CGH array 4 × 180 k covering over 170,000 coding and non-coding human sequences. CGH array raw Cy3/Cy5 intensities were quantile normalized independently for each sample; smoothing was performed using tilingArray R package, copy number status (Gain/Loss) determination in the smoothed segments was done as previously reported[14]. Details are given as supplemental (Method S1).
Quantitative reverse-transcriptase PCR analysis of candidate genes
The expression level of candidate genes (FOS, FOSB, AIM1, RHOB, ABCB1, VAV3, KIR3DS1, S1PR5) identified in gene expression analysis was determined by TaqMan® quantitative reverse-transcriptase PCR (qRT-PCR) (Applied Biosystems, Foster City, CA) in 11 primary HSTL tumors (including the 9 cases submitted to GEP analysis), the DERL2 cell line and 3 normal splenic γδ T cell samples as control. All primers and probes were purchased from Applied Biosystems and the gene expression was measured using Mastercycler® ep realplex2S system (Eppendorf, Hamburg, Germany). Quantifications were done in duplicate and mean values and standard deviation were calculated for each transcript as previously described[25].
Immunohistochemical analysis of selected candidate genes
Immunohistochemistry was performed on deparaffinized tissue sections using a standard indirect avidin-biotin immunoperoxidase method. After appropriate antigen retrieval, sections were stained for FosB (Cell Signaling Technology, Danvers, MA); CD56, CD163 and GSTP1 (Novocastra-Leica, Wetzlar, Germany); β-catenin (BD Biosciences, San Diego, USA); Bcl-10 (Zymed, San Francisco, USA), ICAM-1 (Atlas Antibodies, Stockolm, Sweden), VCAM-1 and Syk (Santa Cruz Biotechnology, Santa Cruz, USA), Blimp-1 (BioLegend UK Ltd, Cambridge, UK). For granzyme H (gzm H)(4G5) a tyramide signal amplification system was applied, as previously described[14]. Adequate controls were included.
Methylation analysis of AIM1
Two μg of genomic DNA were treated with bisulfite using the Epitect® Bisulfite Kit (Qiagen, Courtaboeuf, France) according to manufacturer’s protocol. Nested bisulfite-specific PCR for the 2 CpG islands of AIM1 was performed on DERL2 cells DNA (details, primers and PCR programs used are given in Supplemental Methods (Method S1)). PCR products were cloned in plasmid using TOPO TA cloning kit for sequencing (Invitrogen). Ten clones for each CpG islands were screened and sequenced on the ABI 3130X1 genetic analyser (Applied Biosystems).
5-Aza-2′-deoxycytidine treatment of DERL2 cells
DERL2 cells were treated with the demethylating agent 5-Aza-2′-deoxycytidine (Decitabine, Merck Chemicals, Nottingham, UK) at 10μM or 40μM for 96 hours (decitabine was added every 24 hours) alone or in combination with 500 nM histone deacetylase inhibitor trichostatin A (TSA) (Sigma Aldrich, Saint-Quentin Fallavier, France). In the combined treatment (decitabine and TSA), cells were treated with 10 μM or 40 μM of decitabine for 96 hours (every 24 hours) followed by 500 nM of TSA for the last 24 hours. Total RNA was extracted and qRT-PCR was performed to evaluate the re-expression of the demethylated gene AIM1 after decitabine treatment. In addition the number of apoptotic cells was determined by Annexin V+, 7AAD+ cells using a Cyan flow cytometer (Beckman Coulter, Villepinte, France).
Immunoblot analysis
Total cell proteins extracted from normal blood T cells [purified after CD2 magnetic beads selection and activated or not by CD3 (1μg/ml), CD28 (1μg/ml) and IL2 (100U/ml)], overnight serum deprivated DERL2 cells with or without CD3, CD28 and IL2 activation and overnight serum deprivated SUDHL4 cells with or without BCR stimulation (anti-Fab′2, 4μg/ml) were analyzed for the expression of Syk, phospho-Syk (Tyr525/526)(C87C1) (Cell Signaling technology) and β-actin (Sigma Aldrich). Immunoblotting was done as previously described[25].
Apoptosis assay with a Syk inhibitor
Normal activated γδ T cells were obtained as previously described[26], DERL2 cells were plated at 1,000,000 cells per well. They were incubated for 48 hours in the absence or presence of Syk inhibitor II (Merck Chemicals) at 2 different concentrations (40 and 60 μM). The number of apoptotic cells was determined by Annexin V+, 7AAD+ cells. For the normal activated γδ T cells, TCRVγ9 apoptotic cells were determined by measuring the pourcentage of TCRVγ9+ 7AAD+ cells.
RESULTS
1. HSTL as a molecularly distinct entity irrespective of TCR γδ or αβ lineage
Unsupervised consensus clustering of the expression profiles of 9 HSTL tumors and 41 additional T-cell lymphoma samples including PTCL,NOS, angioimmunoblastic T-cell lymphoma (AITL) and NKTCL demonstrated a clear separation between HSTL and other T-cell lymphomas entities. The HSTL cluster comprised two subgroups consisting of HSTL tissue samples and HSTL cells (sorted cells and cell line) (Figure 1). Interestingly, αβ and γδ HSTL clustered together. One non-hepatosplenic γδ T-cell lymphoma case (PTCL_20) with nodal presentation clustered within the NKTCL branch.
Figure 1. HSTL samples cluster separately to the other T-cell lymphomas, irrespective of their αβ or γδ T-cell lineage.
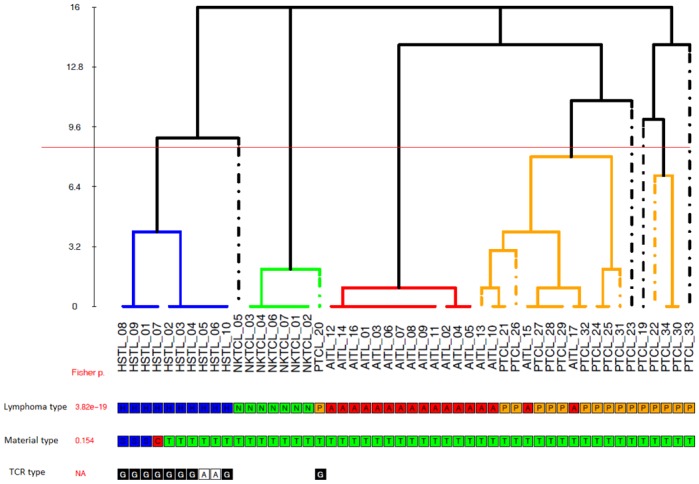
Unsupervised consensus clustering of gene expression profiles of 9 HSTL, 7 NKTCL, 16 PTCL,NOS and 17 AITL tumor samples. Core clusters (C1, C2, C3, C4) are defined as groups of at least 5 samples being co-clustered in at least 14 of the 16 initial partitions (see M&M), and are individualized by the horizontal red line. Fisher exact test p-values for association between the consensus partition C1/C2/C3/C4 and sample’s annotations (Lymphoma.type, Material.type, TCR.type) are shown in red.
Lymphoma.type : {H : Hepatosplenic T-cell lymphoma (HSTL); N : NK/T-cell lymphoma, nasal type (NKTCL); A : Angioimmunoblastic T-cell Lymphoma (AITL); P : Peripheral T-cell lymphoma, not otherwise specified (PCTL,NOS)}
Material.type { C: cell line; S : sorted tumor cells; T : tissue sample}
TCR.type : T-cell Receptor type { A: alpha-beta ; G: gamma-delta}(NA: not available)
On supervised analysis, αβ and γδ HSTL appeared to have a highly similar gene signature. As illustrated in supplemental figure S1, a similar level of transcripts of NK cell-associated molecules including killer immunoglobulin-like receptors (KIRs) were found in HSTL, irrespective of αβ or γδ lineage. Besides the genes coding for the γδ and αβ TCR which, as expected, were the most differentially expressed between both groups, only a small proportion of genes appeared overexpressed in γδ HSTL (LCK, SYT11, BCL2L11, KLRC1...) or downexpressed (CR1, TMPRSS3, ITGA9...) compared to αβ HSTL (Supplemental table S1). Comparison of HSTL gene expression profiles with that of either PTCL,NOS or NKTCL revealed similar proportions of differentially expressed genes (H1 Proportion, see Method S1) of 37% and 39% respectively), however in unsupervised clustering analysis, HSTL profiles appeared to be closer to that of NKTCL, another extranodal entity derived from cytotoxic cells, than that of PTCL,NOS and AITL.
Compared to both PTCL,NOS and NKTCL, several T-cell associated transcripts encoding the γδ T-cell receptor (TCR) subunit were among the top genes overexpressed in HSTL. Two of the interesting genes found to be upregulated in HSTL were S1PR5, a gene known to be involved in the homing of NK cells into the spleen[27] and ABCB1 encoding for the p-glycoprotein multidrug transporter (MDR1). These findings were confirmed by qRT-PCR that demonstrated high S1PR5 and ABCB1 transcripts in HSTL compared to PTCL,NOS and NKTCL (Figure 2). Genes encoding KIRs and other NK cell-associated molecules (NCAM1, CD244) were specifically overexpressed compared to PTCL,NOS (Tables 2 and 3).
Figure 2. Validation of selected genes by quantitative RT-PCR analysis.
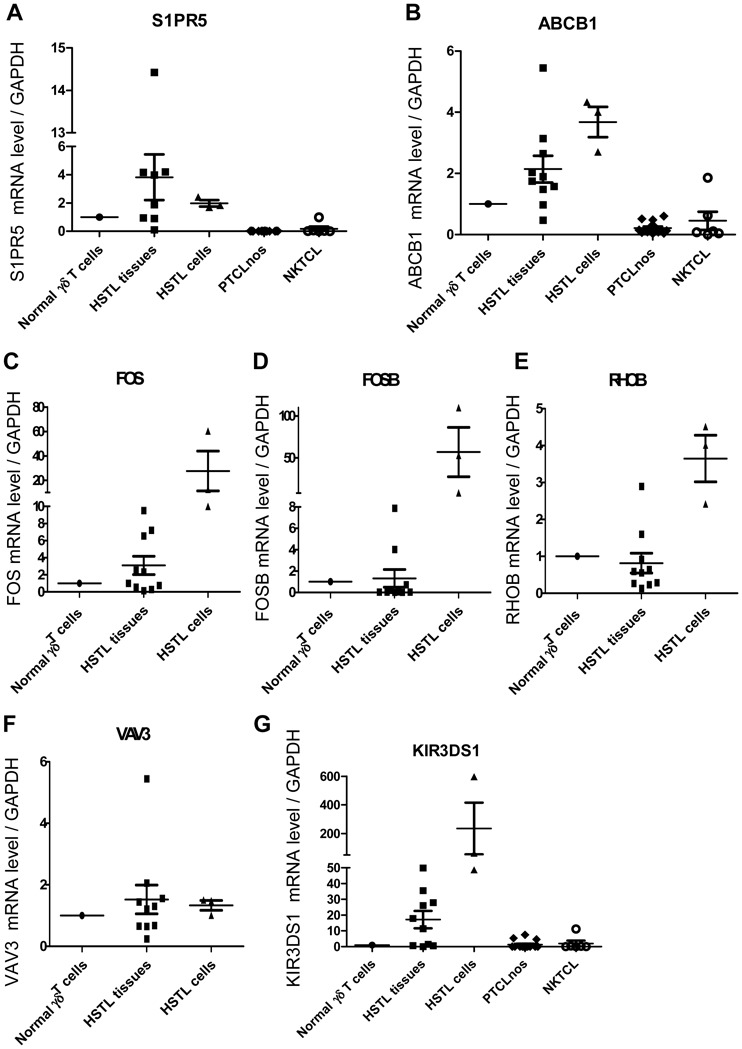
qRT-PCR analysis for S1PR5, ABCB1, FOS, FOSB, RHOB, VAV3 and KIR3DS1 in HSTL tissues (n=10), cells (n=3), PTCL,NOS (n=14) and NKTCL (n=6). The results are expressed as relative fold change compared with normal γδ T cells sorted from spleen. Values for each sample were normalized to GAPDH. Arbitrary value of 1 was assigned to normal γδ T cells.
Table 2.
Selection of genes differentially expressed in HSTL primary tumors compared to PTCL,NOS with P-value less than 0.001. A complete list of genes is given in the supplemental table S1.
| Genes overexpressed in HSTL compared to PTCL,NOS | |
|---|---|
| Gene class and specific genes | Fold change |
| NK cell associated molecules | |
| KLRC3 | 73,95 |
| KIR2DS2 | 33,03 |
| KLRD1 | 25,39 |
| CD16 | 11,83 |
| KIR3DL2 | 21,58 |
| KLRB1 | 17,52 |
| KIR3DS1 | 16,88 |
| KIR2DL2 | 20,35 |
| KIR3DL1 | 19,85 |
| KIR2DL3 | 16,35 |
| KIR3DL3 | 18,95 |
| KIR2DL1 | 17,42 |
| KLRC4 | 14,41 |
| NCAM | 18,04 |
| CD244 | 10,65 |
| KIR2DS5 | 11,59 |
| KIR2DL5A | 12,02 |
| KIR2DS1 | 9,47 |
| KIR2DS3 | 9,05 |
| KLRG1 | 9,39 |
| KLRC1 /// KLRC2 | 10,37 |
| KIR2DL3 /// KIR2DL4 /// KIR2DL5A | 6,04 |
| CTSW (Cathepsin w) | 6,76 |
| KLRK1 | 6,45 |
| Microenvironment | |
| Hemoglobin gamma | 17,62 |
| Hemoglobin beta | 15,13 |
| Hemoglobin alpha | 14,39 |
| DEFA1 /// DEFA1B /// DEFA3 | 13,64 |
| CCL3 (MIP1) | 4,44 |
| Chemokines | |
| CXCL7 (PPBP) | 21,06 |
| CXCL6 | 8,96 |
| Multi Drug Resistance | |
| ABCB1 (MDR1) | 11,72 |
| ABCB1 /// ABCB4 | 6,74 |
| Oncogenes | |
| MYBL1 | 18,81 |
| VAV3 | 3,86 |
| Cell adhesion // Cell to cell interaction | |
| CD11d | 48,35 |
| PGM5 | 6,90 |
| T cell receptor | |
| TRA@ // TRD@ | 13,22 |
| TARP // TRGC2 | 10,94 |
| Transcription factors | |
| TCF21 | 12,87 |
| PRDM16 | 7,86 |
| WT1 | 3,70 |
| Growth Factors | |
| IGFBP2 | 7,52 |
| AREG (amphiregulin) | 8,96 |
| PDGFD | 7,83 |
| Extracellular matrix interaction | |
| ADAMTS17 | 12,93 |
| Others | |
| S1PR5 | 49,99 |
| Genes downregulated in HSTL compared to PTCL,NOS | |
| Chemokines | |
| CCL19 | 0,011 |
| CXCL9 | 0,016 |
| CCL18 | 0,041 |
| CXCL10 | 0,044 |
| TFH signature | |
| CXCL13 | 0,057 |
| CD200 | 0,165 |
| ICOS | 0,158 |
| CXCR5 | 0,246 |
| Immunomodulation | |
| IDO1 | 0,074 |
| IL4I1 | 0,160 |
| Tumor suppressor | |
| AIM1 | 0,195 |
| Others | |
| TIAM1 | 0,097 |
| CD5 | 0,209 |
Table 3.
Selection of genes differentially expressed in HSTL primary tumors compared to NKTCL with P-value less than 0.001. A complete list of genes is given in the supplementary table S1.
| Genes overexpressed in HSTL compared to NKTCL | |
|---|---|
| Gene class and specific genes | Fold change |
| NK cell associated molecules | |
| KLRB1 | 17,26 |
| KLRG1 | 6,81 |
| Microenvironment | |
| Hemoglobin delta | 32,87 |
| Hemoglobin alpha | 22,79 |
| Hemoglobin beta | 19,46 |
| Hemoglobin gamma | 15,18 |
| CD36 | 14,63 |
| T cell receptor | |
| TARP//TRGC2 | 11,12 |
| CD3D | 7,34 |
| CD3G | 6,89 |
| CD247 (CD3Z) | 3,82 |
| Chemokines | |
| CXCL7 | 23,27 |
| CXCL6 | 10,73 |
| Transcription factors | |
| NR4A2 | 17,77 |
| TCF21 | 11,72 |
| GATA6 | 6,16 |
| PRDM16 | 5,99 |
| PRDM1 | 2,81 |
| Signal transduction | |
| SPRY2 | 9,87 |
| PLCB1 | 9,66 |
| MAP4K3 | 7,81 |
| RHOB | 7,61 |
| SPRY1 | 5,59 |
| MAP3K5 | 2,35 |
| Wnt signaling | |
| TCF7L2 | 6,35 |
| TLE1 | 5,55 |
| Oncogenes | |
| VAV3 | 5,25 |
| FYN | 4,29 |
| MAF | 3,46 |
| Multi Drug Resistance | |
| ABCB1 | 2,54 |
| Growth factor | |
| PDGFD | 9,26 |
| Cell adhesion/ extracellular matrix | |
| CD11d | 28,67 |
| ADAMTS17 | 9,53 |
| Apoptosis | |
| BCL2L11 | 4,59 |
| Cell cycle | |
| CCNL1 | 3,34 |
| CCND3 | 2,42 |
| Others | |
| S1PR5 | 39,64 |
| TMEM178 | 32,26 |
| TANC1 | 32,24 |
| FCRL3 | 20,27 |
| CD5L | 14,89 |
| CXXC5 | 14,83 |
| Genes downregulated in HSTL compared to NKTCL | |
| Cytotoxic molecules | |
| Granzyme B | 0,15 |
| Granzyme K | 0,21 |
| Chemokines | |
| CXCL10 | 0,05 |
| Apoptosis | |
| BIRC5 | 0,26 |
| Immunomodulation | |
| IL4I1 | 0,26 |
| Tumor suppressor | |
| AIM1 | 0,26 |
AIM1, reported as a tumor suppressor gene [28], was underexpressed in HSTL compared both to PTCL,NOS and NKTCL. The genes underexpressed in HSTL compared to PTCL,NOS included genes encoding TFH–associated molecules (CXCL13, ICOS, CD200, CXCR5), others involved in immunomodulation (IDO1, IL4I1) as well as CD5, a T-cell surface antigen known to be absent in HSTL cells.
2. Candidate genes relevant to HSTL pathogenesis
To search for candidate genes relevant to the pathogenesis of HSTL, we compared HSTL signature to that of normal γδ T cells. A selection of significantly overexpressed and underexpressed genes distinguishing HSTL from normal activated or resting γδ T cells is summarized in Table 4 (p<0.01). Among the most overexpressed genes in HSTL were those related to NK-cell associated molecules such as KIRs, KLR, CD244 and NCAM1. The other overexpressed genes were related to oncogenes (FOS, VAV3, MAF and BRAF), cell adhesion (VCAM1, CD11d, ICAM1), tyrosine kinases (SYK), signal transduction (SPRY2, RHOB, MAP4K3, SPRY1), the sonic hedgehog pathway (GLI3, PRKAR2B, PRKACB and PRKAR1A), the WNT pathway (FRZB, TCF7L2, BAMBI, TLE1, CTNNB1, APC and FZD5) and S1PR5. Interestingly, many of these genes appeared to be also overexpressed in HSTL sorted cells compared to normal activated or resting cells γδ T cells (supplemental Table S1). This was the case for genes encoding FOS, VAV3, RHOB, S1PR5 and NK-cell associated molecules indicating that their overexpression could be attributed to the neoplastic cells. These findings were confirmed by qRT-PCR analysis (Figure 2) demonstrating overexpression of FOS, RHOB, VAV3, S1PR5 and KIR3DS1 mRNAs in HSTL sorted cells, and to a lesser extent in HSTL tissues, in comparison with normal γδ T cells. Furtheremore, another oncogene FOSB (belonging to the AP1 family) was overexpressed in HSTL cells compared to activated γδ T cells. This finding was corroborated by qRT-PCR analysis (Figure 2D) and by immunohistochemistry evidencing the presence of FosB-positive cells coexpressing CD2 or TIA1 in HSTL (Figure 3A). In addition, membrane CD56 (9/17) and cytoplasmic Syk (8/9) were demonstrated in the neoplastic cells infiltrating the sinuses in the spleen or bone marrow (Figure 3C and D).
Table 4.
Selection of genes differentially expressed in HSTL primary tumors compared to normal γδ T cells with P-value less than 0.01. A complete list of genes is given in the supplementary table S1.
| Genes overexpressed in HSTL compared to normal γδ T cells with p<0.01 | ||
|---|---|---|
| Gene class and specific genes | Fold change | |
| Activated | Resting | |
| Oncogenes | ||
| FOS * | 6,50 | |
| VAV3 ° | 3,52 | 3,67 |
| MAF | 4,42 | |
| NK cell associated molecules | ||
| KIR2DS2 * ° | 13,98 | 16,63 |
| KLRC3 * ° | 11,33 | 14,87 |
| KIR3DL1//KIR3DL2 * ° | 9,56 | 11,16 |
| KIR2DL1 * ° | 8,90 | 7,57 |
| KIR2DL2 * ° | 8,60 | 8,85 |
| KIR2DS5 * ° | 7,26 | 5,19 |
| KIR3DS1 * ° | 7,03 | 6,34 |
| KIR2DL3 * ° | 6,83 | 9,06 |
| KIR2DL5A * ° | 6,40 | 6,09 |
| KIR3DL3 * ° | 5,75 | 6,51 |
| KLRC4 * ° | 5,70 | 5,17 |
| KIR2DS3 * ° | 5,69 | 6,34 |
| KIR2DS1 * ° | 5,00 | 5,17 |
| KIR2DS4 * ° | 3,23 | 3,67 |
| CD244 * | 7,23 | 3,18 |
| NCAM1 * | 6,27 | |
| Microenvironment | ||
| Hemoglobin beta | 46,31 | 34,14 |
| Hemoglobin alpha | 34,93 | 24,06 |
| CXCL12 | 31,57 | 27,39 |
| Immunoglobulin | 26,29 | 21,08 |
| CD163 | 8,86 | |
| Signal transduction | ||
| SPRY2 * ° | 13,77 | 33,98 |
| RHOB * ° | 7,03 | |
| MAP4K3 * ° | 5,96 | 9,18 |
| SPRY1* ° | 6,50 | 7,97 |
| Sonic hedgehog | ||
| GLI3 * | 8,86 | 5,94 |
| PRKAR2B * | 8,25 | 3,15 |
| PRKACB ° | 3,25 | 3,75 |
| PRKAR1A ° | 2,80 | 7,63 |
| Tyrosine kinase | ||
| SYK * | 10,59 | 2,68 |
| Cell adhesion | ||
| CD11d * ° | 37,57 | 33,11 |
| VCAM1 | 15,06 | 71,58 |
| ICAM1 | 5,71 | |
| Wnt pathways | ||
| FRZB | 14,01 | 14,19 |
| TCF7L2 * | 10,96 | |
| BAMBI | 8,58 | 7,52 |
| TLE1 * | 7,16 | 4,48 |
| CTNNB1 * | 3,99 | 11,24 |
| APC | 3,60 | 2,33 |
| FZD5 | 2,53 | 3,43 |
| Others | ||
| S1PR5 * | 8,16 | 3,51 |
| Genes downregulated in HSTL compared to normal γδ T cells with p<0.01 | ||
| Cytotoxic molecules | ||
| Granulysin * ° | 0,03 | 0,03 |
| Granzyme H * ° | 0,03 | 0,05 |
| Granzyme B * | 0,04 | 0,17 |
| Granzyme K * ° | 0,26 | 0,10 |
| Chemokines | ||
| LTA * | 0,02 | |
| TNF | 0,03 | |
| IFNG * | 0,03 | |
| Tumor suppressor | ||
| AIM1 * ° | 0,34 | 0,24 |
| Others | ||
| CD5 * ° | 0,24 | 0,20 |
genes also differentially expressed in HSTL cells compared to normal activated γδ T cells.
genes also differentially expressed in HSTL cells compared to normal resting γδ T cells.
Figure 3. Validation of genes overexpressed in gene expression profiling by immunohistochemistry.
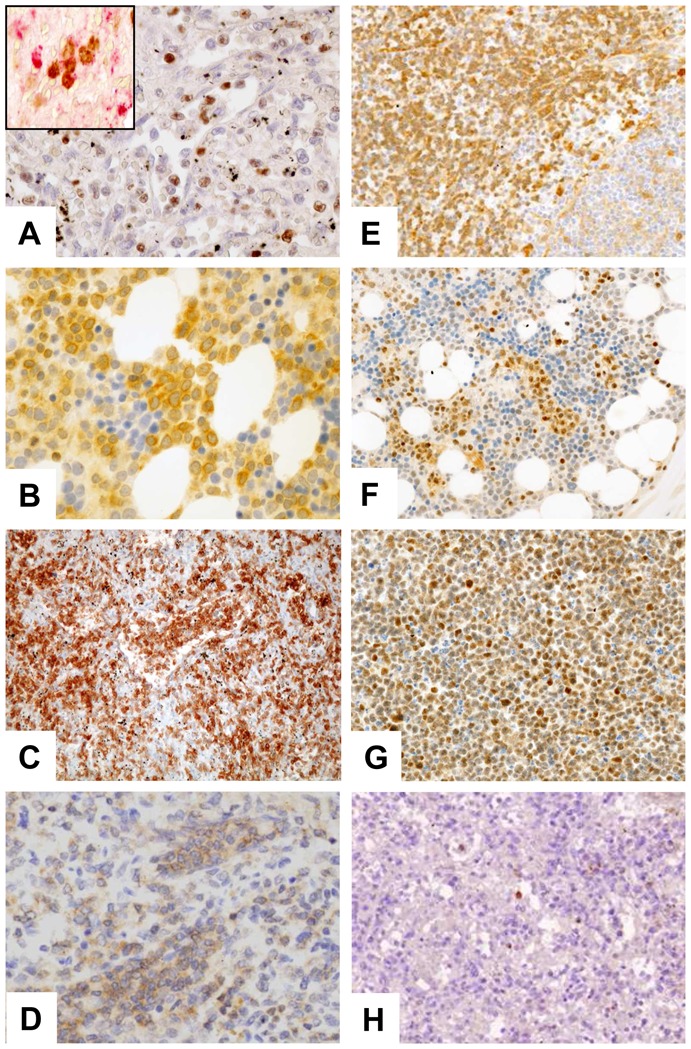
Representative HSTL disclosed staining of neoplastic cells for (A) FOSB, (B) SYK, (C) CD56, (D) BCL10, (E) GSTP1 and (F–G) PRDM1. Panel G refer to DERL2 cells. Staining of NK cells for (H) Granzyme H were observed. Images were captured with a Zeiss Axioskop2 microscope (Zeiss). Photographs were taken with an Olympus DP70 camera. Images were acquired with Olympus DP Controller 2002, and images were processed with Adobe Photoshop Version 7.0 (Adobe Systems). Original magnification, ×400 (A–L).
Genes associated with cytotoxicity (Granulysin, Granzyme H, Granzyme K, Granzyme B), cytokines (LTA, TNF, IFNG), the tumor suppressor AIM1 and CD5 were among genes those significantly underexpressed in HSTL compared to normal cell counterpart. Absence of expression of cytotoxic molecules such as Granzyme B (Table 1) and Granzyme H was shown by immunostaining (Figure 3H) whereas the dramatic reduction of AIM1 transcripts was demonstrated by qRT-PCR in both HSTL tissues and sorted cells compared to normal γδ cells (Figure 5A).
Figure 5. AIM1 is methylated in HSTL DERL2 cell line.
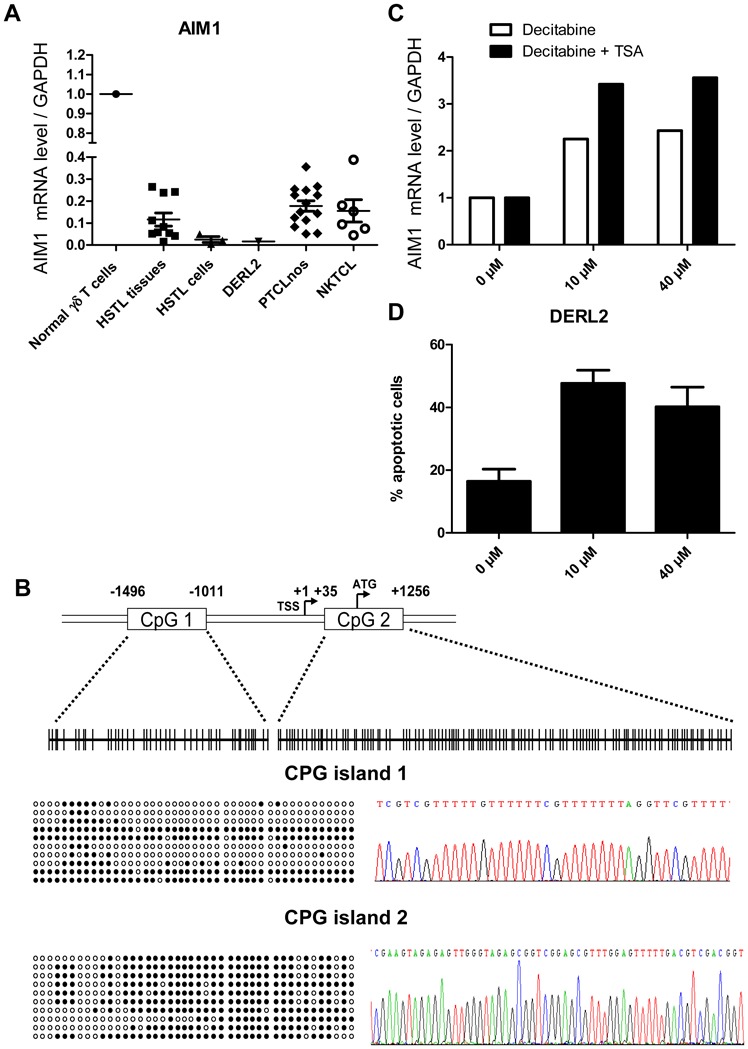
(A) Real-time PCR quantification of AIM1 was evaluated in normal γδ T cells, HSTL tissues, HSTL cells, DERL2 cells, PTCL,NOS and NKTCL. The results are expressed as relative fold change compared with normal γδ T cells sorted from spleen. Each sample was normalized to GAPDH. (B) Localization of CpG islands (CpG1 and CpG2) in the AIM1 promoter and schematic representation of the sequenced genomic fragments including all CpG sites (vertical bars). Approximate spacing of the CpG dinucleotides is shown for the 44 CpGs of island 1 and the 119 CpGs of island 2 (Top). Ten representative clones from DERL2 cells are presented, only 43 CpGs of island 2 were represented because of unmethylation of the first 76 CpGs (Left). Example of methylated cytosines after bisulfite-converted genomic DNA treatment (Right). Methylated CpG, ●; Unmethylated CpG, ○.
(C) Real-time PCR quantification of AIM1 was evaluated in DERL2 cells after 96 hours of 10μM or 40μM decitabine treatment (every 24hours) with or without 500nM TSA treatment for the last 24 hours. (D) DERL2 cell line were cultured with or without 10μM or 40μM of decitabine for 96 hours (decitabine was added every 24 hours). Cellular apoptosis was analyzed using 7AAD staining.
3. Biomarkers discriminating HSTL from other lymphomas and normal controls
Expression of several genes in the molecular signature of HSTL appeared most likely to be attributed to the normal spleen signature. This is the case of ICAM-1, VCAM-1 and CD163 gene products, whose expression was shown by immunohistochemistry to be restricted to non neoplastic cells, that is, endothelial cells (VCAM1, ICAM1) and macrophages (CD163). To build a biomarker list differentiating HSTL from other T-cell lymphoma entities and eliminating the observed microenvironmental signature, supervised tests (see Method S1 for details) were performed between HSTL and other lymphomas (i.e. AITL, PTCL,NOS and NKTCL), HSTL and control samples (i.e. normal lymph nodes, spleen, γδ T cells, B cells) as well as between HSTL obtained from tissues and cell suspensions. This yielded a list of 54 genes distinguishing HSTL from other lymphomas entities and normal controls (Supplemental Figure S2 and tables S2, S3 and S4). Among these genes were those encoding KIR molecules (KIR3DS1), CD244, DTNBP1 and S1PR5. The application of this list of biomarkers to an independent series of lymphoma samples (including 4 HSTL, 15 AITL and 11 PTCL,NOS) using a different plateform[17] allowed discrimination of HSTL from other PTCLs (supplemental Figure S3).
4. Relevant pathways and genes potentially involved in HSTL resistance to therapy
Pathways analyses showed significant enrichment of a number of pathways which are involved in cancer development or can be the target of innovative treatments, such as VEGF, MAP kinase, JAK-STAT, mTOR, Notch signaling pathways, cytokine-cytokine receptor interaction, cell adhesion (Table 6 and supplemental table S5). The Wnt signature was found deregulated but activation of the pathway was not supported by immunohistochemistry since the expression of β-catenin was restricted to endothelial cells. Consistent with previous findings[17], pathways related to NK-cell mediated cytotoxicity were among the most differentially expressed compared to other T-cell lymphoma entities and normal γδ T cells (Figure 4A). Furthermore, as illustrated in figure 4B, many genes in the TCR signaling pathway (VAV3, BCL10), and linked to the AP1 pathway (FOS) were enriched in HSTL compared to PTCL,NOS and NKTCL. These finding were supported by the high levels of VAV3, FOS, FOSB transcripts demonstrated by qRT-PCR, and by immunohistochemical detection of Bcl-10 in the neoplastic cells of all investigated HSTL (5/5) (Figure 3D). Significant enrichment of genes in the pathways of Sprouty regulation of tyrosine kinase and the Sonic Hedgehog signaling [such as genes for the composition of cAMP-dependent kinase (PKA)(PRKAR2B, PRKAR1A and PRKACB), the transcription factor Ci (GLI3), the receptor patched (PTCH1) and GSK3B] were found in HSTL (Figure 4). Interestingly, several genes in the multidrug resistance signalling pathways (ABCB1, GSTP1) were enriched in HSTL (Figure 4E) compared to PTCL,NOS and NKTCL as well as PRDM1, another gene reported to be associated to resistance to therapy in PTCLs[29]. High levels of ABCB1 transcripts were confirmed by qRT-PCR (Figure 2B) and expression of GSTP1 and Blimp-1 was demonstrated by immunohistochemistry in HSTL cells of all evaluated cases, as well as in DERL2 cells (Figures 3). As illustrated in figure 4F, HSTL were closer to activated γδ T cells than resting γδ T cells with respect to the expression of cell cycle-related genes.
Table 6.
Selection of pathways differentially expressed in HSTL compared to PTCL,NOS, NKTCL or normal γδ T cells.
| Pathway name | HSTL vs PTCL,nos | HSTL vs NKTCL | HSTL vs activated γδ T cell | HSTL vs resting γδ T cell |
|---|---|---|---|---|
| Globaltest P value | Globaltest P value | Globaltest P value | Globaltest P value | |
| Biocarta 133 - Ras-Independent in NK cell- mediated cytotoxicity | 3.E-06 | 3.E-02 | 3.E-04 | 3.E-03 |
| KEGG 04650 - Natural killer cell mediated cytotoxicity | 5.E-06 | 7.E-03 | 7.E-05 | 3.E-03 |
| KEGG 04370 - VEGF signaling pathway | 6.E-06 | 1.E-03 | 2.E-04 | 3.E-03 |
| KEGG 04060 - Cytokine-cytokine receptor interaction | 6.E-06 | 4.E-03 | 1.E-04 | 7.E-03 |
| Biocarta 131 - Phospholipase C Signaling Pathway | 7.E-06 | 3.E-04 | 3.E-03 | 8.E-02 |
| Biocarta 140 - Selective expression of chemokine receptors during T-cell polarization | 7.E-06 | 3.E-02 | 5.E-05 | 1.E-02 |
| Biocarta 506 - Sprouty regulation of tyrosine kinase signals | 8.E-06 | 2.E-04 | 2.E-04 | 3.E-03 |
| KEGG 04310 - Wnt signaling pathway | 1.E-05 | 7.E-04 | 2.E-04 | 3.E-03 |
| Biocarta 590 - Sonic Hedgehog (Shh) Pathway | 1.E-05 | 5.E-03 | 6.E-05 | 3.E-03 |
| Biocarta 137 - Regulation of BAD phosphorylation | 1.E-05 | 7.E-03 | 9.E-05 | 3.E-03 |
| KEGG 04010 - MAPK signaling pathway | 1.E-05 | 3.E-03 | 2.E-04 | 3.E-03 |
| KEGG 04340 - Hedgehog signaling pathway | 3.E-05 | 1.E-03 | 5.E-04 | 2.E-02 |
| Biocarta 586 - Multi-Drug Resistance Factors | 1.E-05 | 5.E-03 | 2.E-02 | Ns |
| Biocarta 465 - Role of MEF2D in T-cell Apoptosis | 3.E-05 | 1.E-03 | 6.E-04 | 3.E-03 |
| KEGG 04660 – T cell receptor signaling pathway | 3.E-05 | 3.E-03 | 8.E-05 | Ns |
| KEGG 04630 - Jak-STAT signaling pathway | 4.E-05 | 9.E-03 | 1.E-04 | 3.E-03 |
| KEGG 04620 - Toll-like receptor signaling pathway | 5.E-05 | 7.E-03 | 5.E-04 | 7.E-03 |
| KEGG 04514 - Cell adhesion molecules (CAMs) | 6.E-05 | 3.E-03 | 3.E-04 | 7.E-03 |
| KEGG 04210 - Apoptosis | 7.E-05 | 1.E-02 | 1.E-04 | 3.E-03 |
| KEGG 04150 - mTOR signaling pathway | 1.E-04 | 3.E-03 | 2.E-04 | 3.E-03 |
| KEGG 04330 - Notch signaling pathway | 2.E-04 | 1.E-03 | 3.E-04 | 2.E-02 |
| KEGG 04670 - Leukocyte transendothelial migration | 3.E-04 | 1.E-02 | 1.E-03 | 3.E-03 |
| KEGG 05200 - Pathways in cancer | 3.E-04 | 2.E-02 | 5.E-04 | 3.E-03 |
| KEGG 04110 - Cell cycle | 1.E-02 | 5.E-03 | 3.E-03 | 3.E-03 |
| KEGG 03050 - Proteasome | 5.E-03 | 3.E-03 | 2.E-04 | 1.E-02 |
Figure 4. Cellular programs deregulated in HSTL: * correspond to resting γδ T cells.
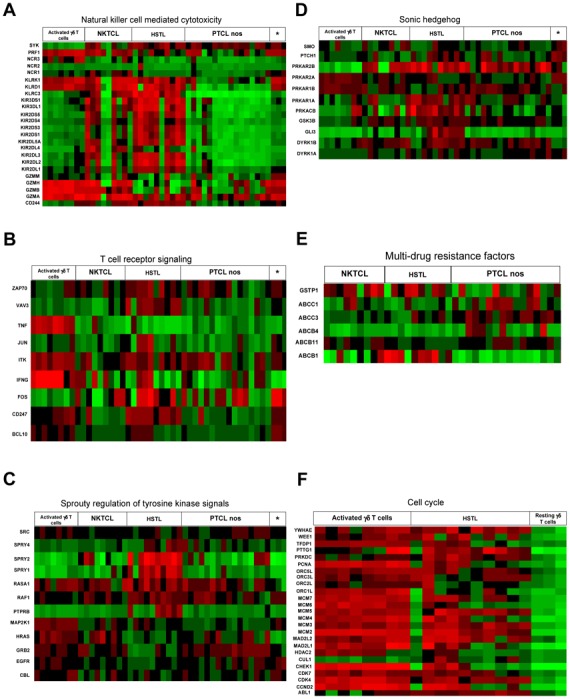
Representative molecular pathways, among those differentially expressed (as shown by enrichment analysis, see tables 6 and S5) in HSTL by comparison with NKTCL, PTCL,NOS and normal γδ T cells, are illustrated (A: Natural killer-cell mediated cytotoxicity; B: T-cell receptor signaling pathway; C: Sprouty regulation of tyrosine kinase signal pathway; D: Sonic hedgehog pathway; E: Multi-drug resistance factors and F: Cell cycle). For each line, green corresponds to the minimal intensity value (min), red corresponds to the maximal intensity value (max), and black corresponds to to (−max − min)/2.
5. Isochromosome 7 q is the main recurrent genomic aberration in HSTL
The aCGH findings confirmed recurrent isochromosome 7q in 4 of the seven tested cases (60%), with only few other alterations comprising trisomy 8 as previouly reported [9], loss of 10p arm and more focal alterations (−14q,+15q, −18q −21q, −22q) in 2 to 6 cases (30–85%)(Supplemental figure S4). In order to determine whether iso7q impacts on the molecular signature, we analysed the mean expression of genes located on the 7p and 7q arms for each sample (Supplemental figure S5). This analysis clearly showed overexpression of 7q and down expression of 7p genes in all cases with iso7q aberration as well as in 2 additional cases (HSTL_08, HSTL_09) for which the iso7q status was unknown (absence of aCGHa and FISH data), one of which could be subsequently demonstrated as iso7q by FISH (HSTL_09). To identify potential candidate genes involved in iso7q, we further compared the expression level of iso7q+ HSTL cases to normal controls (i.e. activated γδ T cells). Among the most repressed genes on 7p arm were CYCS, IKZF1 associated to regulation of apoptosis and HUS1, CBX3 involved in DNA repair, while the most overexpressed genes on 7q arm included the putative oncogene PTPN12 (Supplemental Table S1). Moreover, pathway analysis comparing iso7q+ HSTL to activated or resting γδ T cells showed a significant enrichment of apoptosis-related pathways (Supplemental Table S5).
6. AIM1 is methylated in HSTL
AIM1 (absent in melanoma 1), initially described as a tumor suppressor gene deleted in melanoma, was subsequently found to be deregulated in NKTCL[28,30]. Here we show significant downexpression of AIM1 mRNA in HSTL and DERL2 cells. Low levels of AIM1 mRNAs were confirmed by qRT-PCR in HTSL primary tumors and in DERL2 cells compared to normal γδ T cells (Figure 5A). We further searched for genetic alterations underlying AIM1 downregulation. In the absence of genomic imbalance in AIM1 locus at 6q21 in aCGH data, we asked whether AIM1 downregulation in HSTL could be the result of methylation as already reported in other hematologic malignancies such as myeloma and NKTCL[30,31]. The promoter methylation status of AIM1 was analysed in the HSTL-derived cell line, DERL-2. Sequencing of AIM1 on bisulfite-treated DNA revealed methylation in both CpG islands (Figure 5B). Furthermore, the treatment of DERL2 cells with AIM1 methylated promoter with the DNA demethylating agent decitabine with or without TSA resulted in a 2–3 fold increase in AIM1 mRNA level compared to untreated cells, supporting that AIM1 downexpression in HSTL could be reversed by demethylation (Figure 5C). Interestingly, the number of DERL2 apoptotic cells was increased after decitabine treatment (Figure 5D).
7. Syk as a candidate target for pharmacologic inhibition in HSTL
Whereas normal mature T lymphocytes lack Syk expression, Syk protein has been recently reported to be aberrantly expressed in many PTCLs[32]. GEP analysis revealed overexpression of SYK mRNA in HTSL tissues, in HSTL sorted cells and in the DERL2 cell line compared to normal γδ T cells (Figure 6A). We further demonstrated Syk expression by neoplastic HSTL cells and DERL2 cells, by immunohistochemistry and western blot (Figures 3B, 6B and 6C). Because expression of Syk does not provide information on its activity, we analyzed the phosphorylation status of Syk in DERL2 cells using a phospho-specific anti-Syk (Tyr525/526) antibody. Syk was constitutively activated in DERL2 cells (Figure 6C). The mechanism leading to Syk activation is unclear as neither SYK amplification nor ITK-SYK fusion was detected by FISH analysis in DERL2 cells (data not shown). To determine whether Syk could contribute to the growth and survival of neoplastic HSTL cells, a chemical anti-Syk agent was used to inhibit Syk activity in DERL2 cells. Syk inhibition induced an significant increase in apoptosis in a dose-dependent manner in DERL2 cell line as compared to normal activated γδ T cells (Figure 6D).
Figure 6. SYK a potential candidate target for pharmacologic inhibition.
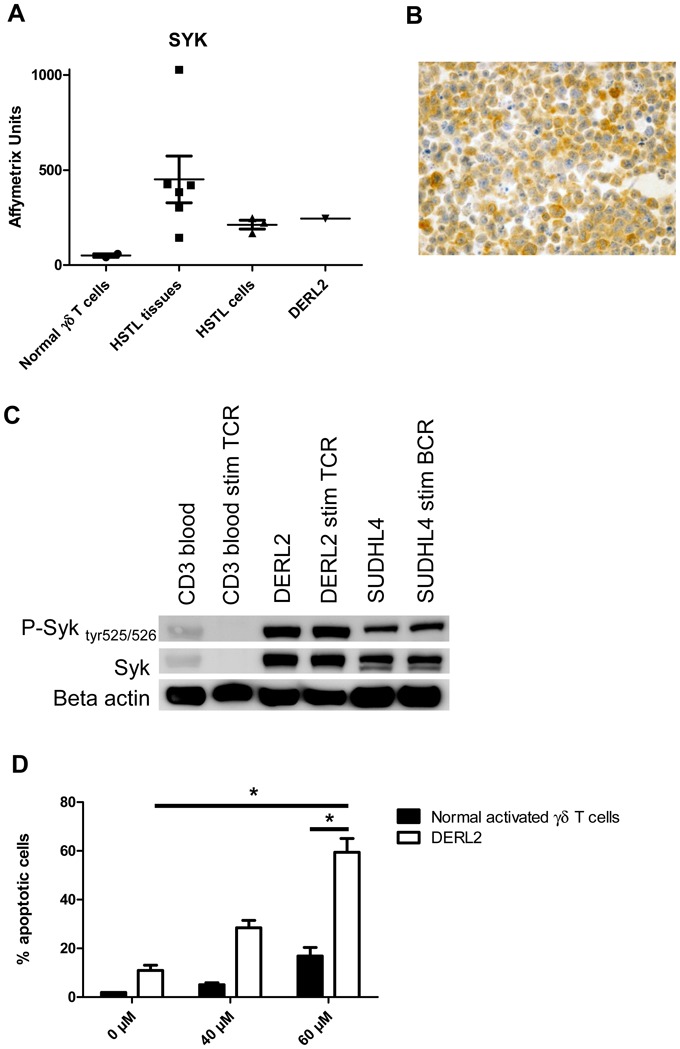
(A) Histogram representation of affymetrix datas of overexpressed SYK probe set in HSTL tissues, HSTL cells, normal γδ T cells and DERL2 cells. (B) Immunostaining of SYK in DERL2 cell line. (C) Western blot analysis of Syk, phosphorylated Syk tyr525/526 and loading control beta actin. (D) DERL2 cell line or normal activated γδ T cells were cultured with 40μM or 60μM of Syk inhibitor II or vehicle alone for 48 hours. Apoptosis was analyzed using 7AAD staining. For normal activated γδ T cells, TCRVγ9 apoptotic cells were determined by measuring the pourcentage of TCRVγ9+ 7AAD+ cells. * correspond to p<0.05.
DISCUSSION
HSTL is a rare and distinct PTCL entity of unknown pathogenesis. The disease is characterized by a peculiar distribution of the neoplastic cells, most often of γδ T-cell derivation, within sinuses of the spleen, the liver and bone marrow and by the clinical fatal outcome. Previous scarce reports showed that HSTL has a distinct gene signature[13,17]. Taking advantage of the exceptional availability of samples enriched in tumor cells, we show by consensus clustering that HSTL expression profiles clustered in a single branch independently of the spleen, marrow or blood origin of the diagnostic samples indicating that this signature is independent of the microenvironment. The distinct signature was further confirmed when a gene list was constructed by eliminating the signature of the normal spleen and lymph nodes. HSTL was initially reported as of γδ T-cell derivation but similar cases with a αβ phenotype have been later reported. In the present study, we show that two cases of HSTL with a αβ phenotype had a gene signature and also a genomic profile which were highly similar to the classical γδ HSTL variant providing strong molecular arguments for grouping γδ and αβ HSTL as a single entity as proposed in the current WHO classification[8]. The similar αβ and γδ HSTL molecular signature was largely contributed by genes implicated in cytotoxic T- and NK-cell function and development, suggesting that both αβ and γδ variants are derived from subsets of cells of the innate immune system[8,33,34]. Indeed, a subset of αβ T cells, referred as NK/T cells, share characteristics with NK cells and γδ T cells including expression of CD161 (KLRB1), CD16, CD56, KIRs molecules and participate with NK cells and γδ T cells in the innate immune system[35]. γδ T cells can accumulate in immunosuppressed patients and in the setting of chronic antigen stimulation. Expansion of γδ T cells has especially been reported in patients with kidney transplantation, systemic lupus, Hodgkin lymphoma or malaria[4] and more recently also after treatment with anti-TNF agents[36].
The elective distribution of the neoplastic cells within the sinuses and sinusoids of the bone marrow, spleen and liver is remarkable and accounts for the peculiar clinical presentation of HSTL with hepatosplenomegaly and cytopenia but without lymphadenopathy and leukemic pictures. Interestingly, one of the gene with the highest fold change of expression compared to PTCL,NOS and NKTCL and to a lesser extend in normal γδ T cells was Sphingosine-1-phosphatase receptor 5 (S1PR5). S1PR5 encodes a member of the family of S1P receptors known to be involved in T- and B-cell exit from lymphoid organs [37]. S1PR5 is preferentially expressed by mature CD56dim human NK cells[38] and is involved in NK cell trafficking in steady-state and inflammatory situations. It has been reported that S1PR5-deficient mice have defective homing of NK cells to blood and spleen [27,38]. Although the role of S1PR5 in γδ T cells is uncharacterized, the high level of S1PR5 mRNA in HSTL might explain the peculiar distribution and accumulation of neoplastic γδ cells in the spleen and bone marrow, preventing tumor cells from exiting sinusoids. Whether cell-cell interaction involving the cell adhesion molecule LFA1 (CD11a) expressed by HSTL cells and VCAM1 expressed by splenic sinusoidal cells could also play a role in the clinicopathologic picture of the disease remains to be determined.
Although our GEP findings were based on the comparison with normal blood γδ T cells and not with splenic γδ T cells, the down or overexpression of selected genes compared to normal splenic γδ T cells was validated by qRT-PCR. From the present study it appears that several candidate genes involved in distinct pathways (i.e. TCR signalling, cell cycle, sonic hedgehog, sprouty regulation of tyrosine kinase pathways and multi-drug resistance) could potentially be associated with the pathophysiology of HSTL. One of the genes we found overexpressed in HSTL is MDR-1, whose expression was significant for this lymphoma upon comparison with normal γδ T cells, PTCL,NOS and NKTCL. The P-Glycoprotein (P-gp), encoded by MDR-1, is able to export drugs out of the cytoplasm, thus reducing the intracellular accumulation of cytotoxic drugs. P-gp has been reported to be overexpressed and associated with poor outcome in various hematological malignancies including NKTCL and in DLBCL [39,40,41]. Interestingly, sensitivity to aracytine is not influenced by P-gp overexpression in multidrug resistant cell lines[42]. Although HSTL is reported as a fatal disease, recent reports have described long survivors after cytarabine-based chemotherapy[4]. The present findings might provide a rationale for the use of cytarabine containing regimen in HSTL. In addition to P-gp expression we demonstrated that Glutathione-S-transferases (GSTs), another molecule involved with resistance to alkylating agents and anthracyclines[43] was also expressed by the neoplastic HSTL cells (as shown by immunohistochemistry). One could hypothesise that the combination of P-gp and GST-pi expression might contribute to the resistance to anthracyclin-based chemotherapy regimen of HSTL and its poor outcome. High levels of GST-pi are reported in neoplastic cells of mantle cell lymphoma and DLBCL[44,45] and are correlated with poor prognosis in DLBCL[44]. The role of transcription factor Blimp-1 in PTCL is unclear. We show here that PRDM1 is expressed at mRNA and protein level in HSTL cells. While inactivation of PRDM1 through epigenetic mechanisms has been recently reported in NKTCL suggesting a role as tumor suppressor gene[30,46], its expression has been associated with chemoresistance in T-cell lymphoma[29].
Tumor development is a multistep process involving deregulation of oncogenes and tumor suppressor genes (TSG). To date, no oncogene or TSG has been identified in HSTL. AIM1 is a gene whose expression is altered in association with tumor suppression in a model of human melanoma[28]. It is frequently deleted or methylated in several solid tumors and in myeloma and more interestingly NK cell lines[14,30,31]. Altogether, these data and our findings that AIM1 is dramatically reduced in HSTL most likely due to promoter methylation suggest that AIM1 might play a role as a TSG in HSTL oncogenesis and provide rationale for testing demethylating agents in this disease. Our expression data also suggests that the recurrent iso7q aberration in HSTL could target putative TSG and oncogenes, including IKZF1 and PTPN12.
With respect to potential target for novel therapies in HSTL, we also show here Syk expression by neoplastic HSTL cells with constitutive activation in DERL2 cells compared to normal T cells. Syk is a protein tyrosine kinase involved in B-cell receptor signaling and its activation is associated with cell growth and survival in B-cell lymphomas [47,48]. Normal mature T lymphocytes lack Syk expression [49]. However, Syk protein has been recently reported as a feature common to most PTCLs, which potentially represents a novel therapeutic target, since, in addition to its aberrant expression in many PTCLs, inhibition of Syk induces apoptosis and blocks proliferation in T-cell lymphoma cell lines[32,49]. An orally available Syk inhibitor is currently being tested for B-cell lymphomas in a phase I/II clinical trial, with encouraging results[50]. In view of the findings that Syk inhibition induced apoptosis in DERL2 cells in a dose-dependent manner, Syk could also be a potential candidate target for pharmacologic inhibition in HSTL.
In conclusion, we show that the distinctive molecular signature of HSTL partly reflects the ontogeny and might explain several clinicopathologic features of the disease including the homing to sinusoids of the spleen and bone marrow. The study highlights emerging oncogenic pathways, potentially implicated in the pathophysiology of the disease. It may offer rationale for exploring new therapeutic options such as tyrosine kinase inhibitors and demethylating agents in these patients. Finally, this study also provides additional molecular arguments for grouping αβ and γδ HSTL as a single distinct entity.
Table 5.
Summary of immunohistochemical results
| HSTL (n=20) | DERL2 | |
|---|---|---|
|
| ||
| FosB | 5*/14 | − |
| CD163 | 0/3 | ND |
| ICAM1 | 0/7 | ND |
| VCAM1 | 4°/12 | ND |
| Granzyme H | 0/7 | − |
| Beta-catenin | 0/5 | ND |
| Syk | 8/9 | + |
| Blimp-1 | 7/7 | + |
| Bcl10 | 5/5 | ND |
| GSTP1 | 4/4 | ND |
| CD56 | 9/17 | + |
2 with weak (equivocal) staining in TC.
Double immunostaining showing presence of FosB-positive lymphocytes coexpressing CD2 or TIA1 in 3 investigated cases
ND : Not done
Acknowledgments
This work is part of the Carte d’Identité des Tumeurs (CIT) program (http://cit.ligue-cancer.net/index.php/en) from the Ligue Nationale Contre le Cancer and of the Tenomic project supported by a Programme Hospitalier de Recherche Clinique and the Institut National du Cancer (INCa). This work was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM), the Institut National du Cancer (INCa), the Plan cancer of the Belgian government, and the Association pour la Recherche Thérapeutique, Génétique et Immunologique dans les Lymphomes (ARTGIL). M.T. was supported by the Fondation pour la Recherche Médicale (DEQ 2010/0318253). We are extremely thankful for the contributions made by Christelle Thibault from the IGBMC platform (Affymetrix : Philippe Kastner). We also thank Joe A. Trapani for providing antibodies, L. Del Vecchio for providing DERL2 cell line, Francois Radvanyi for critical opinion in data interpretation, Jérôme Cuvillier and Nadine Vailhen from GELAP, and Virginie Fataccioli and Maryse Baia for their technical assistance
Appendix : List of contributors to the TENOMIC project
A. Martin, Hôpital Avicenne, Bobigny ; I. Soubeyran, P. Soubeyran, Institut Bergonié, Bordeaux; P. Dechelotte, A. Pilon, Pr O.Tournilhac, Hôtel-Dieu, Clermont Ferrand ; P. Gaulard, MH Delfau, A Plonquet, C. Haïoun, Hôpital H Mondor, Créteil; T. Petrella, L. Martin, JN Bastié, O Casasnovas CHU, Dijon; B. Fabre, D. Salameire, R. Gressin, D. Leroux, MC Jacob CHU, Grenoble ; L. De Leval, B. Bisig, G. Fillet, C. Bonnet, CHU of Liège ; M.C. Copin, B. Bouchindhomme, F. Morschhauser, CHU, Lille ; B. Petit, A. Jaccard, Hôpital Dupuytren, Limoges, F. Berger, B. Coiffier, CHU Sud, Lyon ; T. Rousset, P. Quittet, G. Cartron, Hôpital Gui de Chauliac-St Eloi, Montpellier ; S. Thiebault, B. Drenou, Hôpital E. Muller, Mulhouse ; K. Montagne, S. Bologna, CHU de Brabois, Nancy ; C. Bossard, S. Le Gouill, Hôtel-Dieu, Nantes ; T. Molina, Hôtel-Dieu, Paris ; J. Brière, C. Gisselbrecht, Hôpital St Louis, Paris ; B. Fabiani, A Aline-Fardin, P. Coppo, Hôpital Saint-Antoine, Paris ; F. Charlotte, J. Gabarre, Hôpital Pitié-Salpétrière, Paris ; J. Bruneau, D. Canioni, V. Verkarre, E Macintyre, V. Asnafi, O. Hermine, R. Delarue, F Suarez, D. Sibon, JP Jaïs, Hôpital Necker, Paris ; M. Parrens, JP Merlio, E Laharanne, K. Bouabdallah, Hôpital Haut Lévêque, Bordeaux ; S.Maugendre-Caulet, P. Tas, T. Lamy, CHU Pontchaillou, Rennes ; JM Picquenot, F. Jardin, C. Bastard, Centre H Becquerel, Rouen ; M. Peoc’h, J. Cornillon, CHU, Saint Etienne ; L. Lamant, G. Laurent, Lucie Oberic, Hôpital Purpan, Toulouse ; J.Bosq, P. Dartigues, V. Ribrag, Institut G Roussy, Villejuif ; M. Patey, A. Delmer, Hôpital R. Debré, Reims ; JF Emile, K. Jondeau, Hôpital Ambroise Paré, Boulogne ; MC Rousselet, Mathilde Hunault, CHU, Angers ; C. Badoual, Hôpital européen Georges Pompidou, Paris ; C. Legendre, S. Castaigne, AL Taksin, CH Versailles, Le Chesnay ; J. Vadrot, A. Devidas, CH Sud francilien, Corbeil ; Dr Gandhi DAMAJ, CHU Amiens, Hématologie Clinique. F Lemonnier, M Travert, INSERM, Créteil ;P Dessen, G Meurice, Institut G Roussy, Villejuif ; M Delorenzi, E Missiaglia, N Houhou, Swiss Institut of Bioinformatics, Lausanne ; F Radvanyi, E Chapeaublanc, Institut Curie, Paris ; S Spicuglia, CIML, Marseille ; J Soulier, Hôpital St Louis, Paris ; C Thibault, IGBMC, Illkirsch
GELA/GOELAMS
Footnotes
AUTHORSHIP:
Contribution: P.G. L.d.L and M.T. designed research; M.T., Y.H., A.d.R., N.M-G., Ja.B. and T.M. performed research; M.T., A.d.R., and P.G. analyzed and interpreted the data; L.d.L., Ja.B., J.B., B.P., and P.G. collected data; PG and A.d.R. supervised bioinformatics analyses, T.M. contributed vital analytical tools; M.T., A.d.R., L.d.L. and P.G. drafted the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
References
- 1.Gaulard P, Bourquelot P, Kanavaros P, et al. Expression of the alpha/beta and gamma/delta T-cell receptors in 57 cases of peripheral T-cell lymphomas. Identification of a subset of gamma/delta T-cell lymphomas. Am J Pathol. 1990;137:617–628. [PMC free article] [PubMed] [Google Scholar]
- 2.Farcet JP, Gaulard P, Marolleau JP, et al. Hepatosplenic T-cell lymphoma: sinusal/sinusoidal localization of malignant cells expressing the T-cell receptor gamma delta. Blood. 1990;75:2213–2219. [PubMed] [Google Scholar]
- 3.Cooke CB, Krenacs L, Stetler-Stevenson M, et al. Hepatosplenic T-cell lymphoma: a distinct clinicopathologic entity of cytotoxic gamma delta T-cell origin. Blood. 1996;88:4265–4274. [PubMed] [Google Scholar]
- 4.Belhadj K, Reyes F, Farcet JP, et al. Hepatosplenic gammadelta T-cell lymphoma is a rare clinicopathologic entity with poor outcome: report on a series of 21 patients. Blood. 2003;102:4261–4269. doi: 10.1182/blood-2003-05-1675. [DOI] [PubMed] [Google Scholar]
- 5.Mackey AC, Green L, Liang LC, Dinndorf P, Avigan M. Hepatosplenic T cell lymphoma associated with infliximab use in young patients treated for inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2007;44:265–267. doi: 10.1097/MPG.0b013e31802f6424. [DOI] [PubMed] [Google Scholar]
- 6.Macon WR, Williams ME, Greer JP, Cousar JB. Paracortical nodular T-cell lymphoma. Identification of an unusual variant of peripheral T-cell lymphoma. Am J Surg Pathol. 1995;19:297–303. doi: 10.1097/00000478-199503000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Suarez F, Wlodarska I, Rigal-Huguet F, et al. Hepatosplenic alphabeta T-cell lymphoma: an unusual case with clinical, histologic, and cytogenetic features of gammadelta hepatosplenic T-cell lymphoma. Am J Surg Pathol. 2000;24:1027–1032. doi: 10.1097/00000478-200007000-00016. [DOI] [PubMed] [Google Scholar]
- 8.Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. IARC; Lyon: 2008. [Google Scholar]
- 9.Wlodarska I, Martin-Garcia N, Achten R, et al. Fluorescence in situ hybridization study of chromosome 7 aberrations in hepatosplenic T-cell lymphoma: isochromosome 7q as a common abnormality accumulating in forms with features of cytologic progression. Genes Chromosomes Cancer. 2002;33:243–251. doi: 10.1002/gcc.10021. [DOI] [PubMed] [Google Scholar]
- 10.Bennett M, Matutes E, Gaulard P. Hepatosplenic T cell lymphoma responsive to 2′-deoxycoformycin therapy. Am J Hematol. 2010;85:727–729. doi: 10.1002/ajh.21774. [DOI] [PubMed] [Google Scholar]
- 11.de Leval L, Rickman DS, Thielen C, et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood. 2007;109:4952–4963. doi: 10.1182/blood-2006-10-055145. [DOI] [PubMed] [Google Scholar]
- 12.Piccaluga PP, Agostinelli C, Califano A, et al. Gene expression analysis of peripheral T cell lymphoma, unspecified, reveals distinct profiles and new potential therapeutic targets. J Clin Invest. 2007;117:823–834. doi: 10.1172/JCI26833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iqbal J, Weisenburger DD, Chowdhury A, et al. Natural killer cell lymphoma shares strikingly similar molecular features with a group of non-hepatosplenic gammadelta T-cell lymphoma and is highly sensitive to a novel aurora kinase A inhibitor in vitro. Leukemia. 2011;25:348–358. doi: 10.1038/leu.2010.255. [DOI] [PubMed] [Google Scholar]
- 14.Huang Y, de Reynies A, de Leval L, et al. Gene expression profiling identifies emerging oncogenic pathways operating in extranodal NK/T-cell lymphoma, nasal type. Blood. 2010;115:1226–1237. doi: 10.1182/blood-2009-05-221275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iqbal J, Weisenburger DD, Greiner TC, et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood. 2010;115:1026–1036. doi: 10.1182/blood-2009-06-227579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ballester B, Ramuz O, Gisselbrecht C, et al. Gene expression profiling identifies molecular subgroups among nodal peripheral T-cell lymphomas. Oncogene. 2006;25:1560–1570. doi: 10.1038/sj.onc.1209178. [DOI] [PubMed] [Google Scholar]
- 17.Miyazaki K, Yamaguchi M, Imai H, et al. Gene expression profiling of peripheral T-cell lymphoma including gammadelta T-cell lymphoma. Blood. 2009;113:1071–1074. doi: 10.1182/blood-2008-07-166363. [DOI] [PubMed] [Google Scholar]
- 18.Delfau-Larue MH, Dalac S, Lepage E, et al. Prognostic significance of a polymerase chain reaction-detectable dominant T-lymphocyte clone in cutaneous lesions of patients with mycosis fungoides. Blood. 1998;92:3376–3380. [PubMed] [Google Scholar]
- 19.Kanavaros P, Farcet JP, Gaulard P, et al. Recombinative events of the T cell antigen receptor delta gene in peripheral T cell lymphomas. J Clin Invest. 1991;87:666–672. doi: 10.1172/JCI115044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Correia DV, d’Orey F, Cardoso BA, et al. Highly active microbial phosphoantigen induces rapid yet sustained MEK/Erk- and PI-3K/Akt-mediated signal transduction in anti-tumor human gammadelta T-cells. PLoS One. 2009;4:e5657. doi: 10.1371/journal.pone.0005657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Ohyashiki JH, Shimizu N, Ohyashiki K. Aberrant expression of NK cell receptors in Epstein-Barr virus-positive gammadelta T-cell lymphoproliferative disorders. Hematology. 2010;15:43–47. doi: 10.1179/102453310X12583347009450. [DOI] [PubMed] [Google Scholar]
- 22.Di Noto R, Pane F, Camera A, et al. Characterization of two novel cell lines, DERL-2 (CD56+/CD3+/Tcry5+) and DERL-7 (CD56+/CD3-/TCRgammadelta-), derived from a single patient with CD56+ non-Hodgkin’s lymphoma. Leukemia. 2001;15:1641–1649. doi: 10.1038/sj.leu.2402239. [DOI] [PubMed] [Google Scholar]
- 23.Compagno M, Lim WK, Grunn A, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459:717–721. doi: 10.1038/nature07968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caron G, Le Gallou S, Lamy T, Tarte K, Fest T. CXCR4 expression functionally discriminates centroblasts versus centrocytes within human germinal center B cells. J Immunol. 2009;182:7595–7602. doi: 10.4049/jimmunol.0804272. [DOI] [PubMed] [Google Scholar]
- 25.Travert M, Ame-Thomas P, Pangault C, et al. CD40 ligand protects from TRAIL-induced apoptosis in follicular lymphomas through NF-kappaB activation and up-regulation of c-FLIP and Bcl-xL. J Immunol. 2008;181:1001–1011. doi: 10.4049/jimmunol.181.2.1001. [DOI] [PubMed] [Google Scholar]
- 26.Capietto AH, Martinet L, Fournie JJ. Stimulated gammadelta T cells increase the in vivo efficacy of trastuzumab in HER-2+ breast cancer. J Immunol. 2011;187:1031–1038. doi: 10.4049/jimmunol.1100681. [DOI] [PubMed] [Google Scholar]
- 27.Jenne CN, Enders A, Rivera R, et al. T-bet-dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. J Exp Med. 2009;206:2469–2481. doi: 10.1084/jem.20090525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ray ME, Su YA, Meltzer PS, Trent JM. Isolation and characterization of genes associated with chromosome-6 mediated tumor suppression in human malignant melanoma. Oncogene. 1996;12:2527–2533. [PubMed] [Google Scholar]
- 29.Zhao WL, Liu YY, Zhang QL, et al. PRDM1 is involved in chemoresistance of T-cell lymphoma and down-regulated by the proteasome inhibitor. Blood. 2008;111:3867–3871. doi: 10.1182/blood-2007-08-108654. [DOI] [PubMed] [Google Scholar]
- 30.Iqbal J, Kucuk C, Deleeuw RJ, et al. Genomic analyses reveal global functional alterations that promote tumor growth and novel tumor suppressor genes in natural killer-cell malignancies. Leukemia. 2009;23:1139–1151. doi: 10.1038/leu.2009.3. [DOI] [PubMed] [Google Scholar]
- 31.de Carvalho F, Colleoni GW, Almeida MS, Carvalho AL, Vettore AL. TGFbetaR2 aberrant methylation is a potential prognostic marker and therapeutic target in multiple myeloma. Int J Cancer. 2009;125:1985–1991. doi: 10.1002/ijc.24431. [DOI] [PubMed] [Google Scholar]
- 32.Feldman AL, Sun DX, Law ME, et al. Overexpression of Syk tyrosine kinase in peripheral T-cell lymphomas. Leukemia. 2008;22:1139–1143. doi: 10.1038/leu.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krenacs L, Smyth MJ, Bagdi E, et al. The serine protease granzyme M is preferentially expressed in NK-cell, gamma delta T-cell, and intestinal T-cell lymphomas: evidence of origin from lymphocytes involved in innate immunity. Blood. 2003;101:3590–3593. doi: 10.1182/blood-2002-09-2908. [DOI] [PubMed] [Google Scholar]
- 34.Morice WG, Macon WR, Dogan A, Hanson CA, Kurtin PJ. NK-cell-associated receptor expression in hepatosplenic T-cell lymphoma, insights into pathogenesis. Leukemia. 2006;20:883–886. doi: 10.1038/sj.leu.2404168. [DOI] [PubMed] [Google Scholar]
- 35.Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007;25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- 36.Kelsen J, Dige A, Schwindt H, et al. Infliximab induces clonal expansion of gammadelta-T cells in Crohn’s disease: a predictor of lymphoma risk? PLoS One. 2011;6:e17890. doi: 10.1371/journal.pone.0017890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pappu R, Schwab SR, Cornelissen I, et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science. 2007;316:295–298. doi: 10.1126/science.1139221. [DOI] [PubMed] [Google Scholar]
- 38.Walzer T, Chiossone L, Chaix J, et al. Natural killer cell trafficking in vivo requires a dedicated sphingosine 1-phosphate receptor. Nat Immunol. 2007;8:1337–1344. doi: 10.1038/ni1523. [DOI] [PubMed] [Google Scholar]
- 39.Marie JP. Drug resistance in hematologic malignancies. Curr Opin Oncol. 2001;13:463–469. doi: 10.1097/00001622-200111000-00008. [DOI] [PubMed] [Google Scholar]
- 40.Andreadis C, Gimotty PA, Wahl P, et al. Members of the glutathione and ABC-transporter families are associated with clinical outcome in patients with diffuse large B-cell lymphoma. Blood. 2007;109:3409–3416. doi: 10.1182/blood-2006-09-047621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang B, Li XQ, Ma X, Hong X, Lu H, Guo Y. Immunohistochemical expression and clinical significance of P-glycoprotein in previously untreated extranodal NK/T-cell lymphoma, nasal type. Am J Hematol. 2008;83:795–799. doi: 10.1002/ajh.21256. [DOI] [PubMed] [Google Scholar]
- 42.Michelutti A, Michieli M, Damiani D, et al. Effect of fludarabine and arabinosylcytosine on multidrug resistant cells. Haematologica. 1997;82:143–147. [PubMed] [Google Scholar]
- 43.Tew KD. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res . 1994;54:4313–4320. [PubMed] [Google Scholar]
- 44.Ribrag V, Koscielny S, Carpiuc I, et al. Prognostic value of GST-pi expression in diffuse large B-cell lymphomas. Leukemia. 2003;17:972–977. doi: 10.1038/sj.leu.2402930. [DOI] [PubMed] [Google Scholar]
- 45.Bennaceur-Griscelli A, Bosq J, Koscielny S, et al. High level of glutathione-S-transferase pi expression in mantle cell lymphomas. Clin Cancer Res. 2004;10:3029–3034. doi: 10.1158/1078-0432.ccr-03-0554. [DOI] [PubMed] [Google Scholar]
- 46.Kucuk C, Iqbal J, Hu X, et al. PRDM1 is a tumor suppressor gene in natural killer cell malignancies. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1115128108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pogue SL, Kurosaki T, Bolen J, Herbst R. B cell antigen receptor-induced activation of Akt promotes B cell survival and is dependent on Syk kinase. J Immunol. 2000;165:1300–1306. doi: 10.4049/jimmunol.165.3.1300. [DOI] [PubMed] [Google Scholar]
- 48.Gururajan M, Dasu T, Shahidain S, et al. Spleen tyrosine kinase (Syk), a novel target of curcumin, is required for B lymphoma growth. J Immunol. 2007;178:111–121. doi: 10.4049/jimmunol.178.1.111. [DOI] [PubMed] [Google Scholar]
- 49.Wilcox RA, Sun DX, Novak A, Dogan A, Ansell SM, Feldman AL. Inhibition of Syk protein tyrosine kinase induces apoptosis and blocks proliferation in T-cell non-Hodgkin’s lymphoma cell lines. Leukemia. 2010;24:229–232. doi: 10.1038/leu.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Friedberg JW, Sharman J, Sweetenham J, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. 2010;115:2578–2585. doi: 10.1182/blood-2009-08-236471. [DOI] [PMC free article] [PubMed] [Google Scholar]