

Abstract
A specific and sensitive LC-MS/MS method for analysis of F2-isoprostanes (F2-IsoPs) and prostaglandins (PGs) in urine was developed and validated to examine the levels of F2-IsoPs and prostaglandin F2α (PGF2α), in human urine in patients undergoing cardiac surgery. The rapid extraction for F2-IsoPs and PGs from urine was achieved using a polymeric weak anion solid phase extraction cartridge. The base-line separation of 8-iso-PGF2α, 8-iso-15(R)-PGF2α, PGF2α, and 15(R)-PGF2α was carried out on a Hydro-RP column (250 × 2.0 mm i.d, Phenomenex, CA) using a linear gradient of methanol:acetonitrile (1:1 v/v) in 0.1% formic acid at a flow rate of 0.2 mL/min. The method was proved to be accurate and precise for simultaneous quantification of each analyte over a linear dynamic range of 0.05–50 ng/mL with correlation coefficients greater than 0.99. The intra-day and inter-day assay precision at the lowest quality control (0.07 ng/mL) level were less than 17%. The mean extraction recoveries of F2-IsoPs and PGs were in a range of 79–100%. In applications of this method to patients undergoing cardiac surgery, post-surgery urinary concentrations of 8-iso-PGF2α increased significantly in patients (n=14) who did not develop acute kidney (AKI) (pre-surgery 0.344±0.039 vs post-surgery 0.682±0.094 ng/mg creatinine, p< 0.01), whereas there was no significant change in this isoprostane in the patients (n=4) who developed AKI (pre-surgery 0.298±0.062 vs post-surgery 0.383±0.117 ng/mg creatinine, NS). Therefore, the method is suitable for the analysis of individual F2-IsoPs and PGF2αs in both clinical and research studies.
Keywords: F2-isoprostanes, Prostaglandins, LC-MS/MS, analysis
Introduction
Assessment of the critical role oxidative stress plays in the pathophysiology of many human diseases, such as cardiovascular and kidney diseases, diabetes, and cancer [1–6] is important in pre-clinical and clinical research. F2-IsoPs, a unique series of prostaglandin-like compounds (Fig. 1) formed in vivo via a non-enzymatic mechanism involving the free radical-initiated peroxidation of arachidonic acid, are considered biomarkers of oxidative stress. There is growing acceptance that measurement of the relatively high levels of F2-IsoPs, and their metabolites in urine may be useful biomarkers to evaluate the effectiveness of clinical interventions to diminish oxidant stress and associated inflammation [7,8]. Prostaglandin F2α (PGF2α), an inflammatory mediator, and the isoprostane 8-iso-PGF2α, another reliable indicator of oxidative stress, and cytokine-related inflammatory mediators are closely associated with several inflammatory diseases [3]. This investigation aimed to develop an improved method for the measurement of F2-IsoPs and related isomers by LC-MS/MS in urine samples from patients before and after cardiac surgery. It underscored the need for precise determination of both F2-IsoPs and PGs which are structurally similar and exist in different isomeric forms at physiological concentrations.
Figure 1.
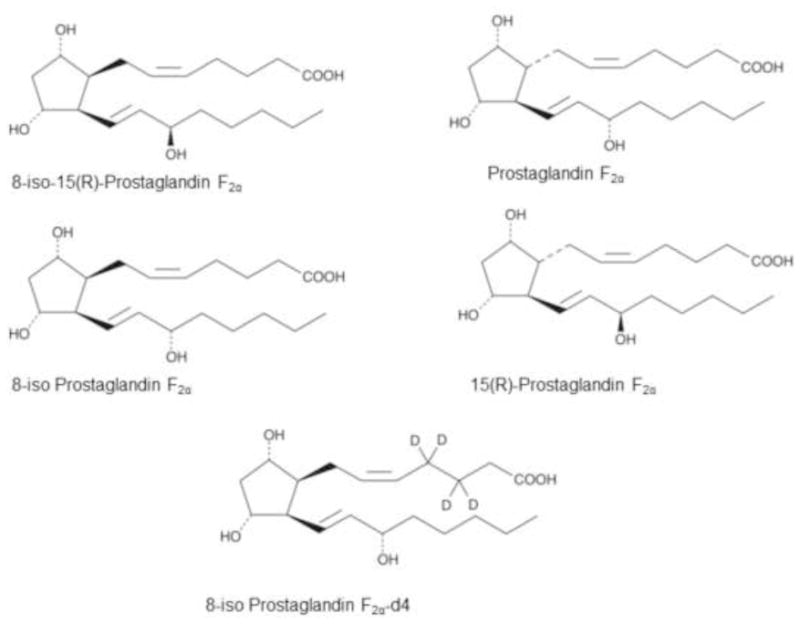
Chemical structures of standard 8-iso-PGF2α, 8-iso-15(R)-PGF2α, PGF2α, 15(R)-PGF2α and 8-iso-PGF2α-d4.
Quantification of F2-IsoPs in biological samples has been carried out using different analytical methods such as gas chromatography/mass spectrometry (GC–MS), liquid chromatography tandem mass spectrometry (LC–MS/MS), radioimmunoassay (RIA) and enzyme immunoassay (EIA) [9–15]. Radioimmunoassay is sensitive and easier to use than the other methods. However, it is less specific and only one isoprostane can be analyzed per assay [15]. In general, LC–MS/MS is a sensitive and specific analytical method. Compared to GC-MS, it can require less sample preparation steps. There are several previously reported methods available to measure F2-IsoPs by LC-MS/MS in biological samples [16,17]. However, most of these methods involve multi-step sample preparation and only deal with quantification of the most abundant F2-IsoPs. Moreover, existence of different F2-IsoPs and PGs in several isomeric forms has been a major analytical challenge for quantification of individual compounds. Recently, Langhorst and co-workers reported the determination of isomeric F2-IsoPs in urine using LC-MS/MS [13]. However, the method lacks the required sensitivity for the analysis of very low levels of F2-IsoPs. To the best of our knowledge, there exists no validated LC-MS/MS method that enables us to quantify stereo isomeric isoprostanes such as 8-iso-PGF2α, 8-iso-15-PGF2α, PGs such as PGF2α, and 15(R)-PGF2α in urine. Therefore, we describe herein a LC/MS/MS method using selected reaction monitoring (SRM) that allows sensitive detection and simultaneous quantification of isomeric F2-IsoPs and PGs in human urine using a solid phase extraction method.
Material and methods
Chemicals
8-iso-PGF2α, 8-iso-15(R)-PGF2α, PGF2α, 15(R)-PGF2α and 8-iso-PGF2α-d4 were purchased from Cayman Chemical Company (Ann Arbor, MI, USA). All of standards were dissolved or diluted into adequate volumes of methanol: water (1:1 v/v containing 1% acetic acid) to generate stock solutions, which were aliquoted into small vials and stored at 20°C. Creatinine and creatinine-d3 were obtained from Sigma-Aldrich, Milwaukee, WI and Cambridge Isotope Laboratory, Cambridge, MA. All HPLC solvents and reagents were purchased from Fisher Scientific Co. (Norcross, GA) and were of HPLC grade.
Sample preparation
Quality control samples and calibration standards
Stock solutions of individual F2-IsoPs and PGs were prepared in methanol and then diluted with methanol-water (1:1 v/v containing 1% acetic acid) to obtain appropriate working solutions containing all analytes and the internal standard (8-iso-PGF2α-d4).
Human urine used in calibration and quality control was from internal remnant pool in our laboratory. Since F2-IsoPs and PGs are endogenous compounds, the urine used to make standards and quality control samples should be free from these compounds. To evaluate this, the unspiked urine samples (200 μL) were loaded on to Strata X-AW 33u polymeric weak anion exchange cartridges (Phenomenex, Torrance, CA) which had previously been equilibrated with methanol and water. The extraction solvent, methanol-water (80/20, v/v containing 1% acetic acid), was used to elute endogenous F2-IsoPs and PGs of interest. The extracted samples were evaporated to dryness under a stream of nitrogen gas and reconstituted in 100 μL of methanol:water (1:1 v/v containing 1% acetic acid) for LC-MS/MS analysis.
For calibration standards and quality control (QC) samples, the urine pool sample (200 μL) serving as “blank urine” was spiked (10 μL) with appropriate concentrations of working solution of F2-IsoPs, PGs and IS to obtain calibration standards (50, 25, 10, 1, 2, 0.1 and 0.05 ng/mL) and QC samples (5, 0.5 and 0.07 ng/mL). After gently mixing, the samples were extracted and reconstituted as described above for LC-MS/MS analysis.
Validation study
The analytical method was validated to demonstrate the specificity, recovery, lower limit of quantification (LLOQ), accuracy and precision of measurements. Linearity was tested at 7 levels of concentrations covering a range from 0.05–50 ng/mL. The regression parameters of slope, intercept and correlation coefficient were calculated by linear least-square regression (1/x2 weighting).
The percent recovery of the method was determined by comparing the mean of peak areas obtained from the urine samples spiked prior to extraction with the peak areas obtained from spiked post-extraction urine samples. The matrix effect (ME) was evaluated for all analytes and the internal standard. For this, the urine samples were processed and then spiked with analytes and IS after extraction. The ME for each analyte was calculated by comparison of mean peak area obtained for blank urine samples (n = 4–5) spiked with F2-IsoPs, PGs and IS after extraction (X) and peak area of standards in methanol-water (1:1) at concentrations 0.07, 0.5 and 5 ng/mL (Y). The MEs were calculated as follows.
ME values >0 and <0 indicate ionization suppression and ionization enhancement, respectively.
The intra-day accuracy (%bias) and precision (presented as %CV) of this analytical method was determined using quality control (QC) samples in 5 replicates of 0.07, 0.5, and 5 ng/mL of F2-IsoPs and PGs in urine within a day. The inter-day assay precision was determined by analyzing the QC samples during three consecutive days (n =15 at each level). The % bias was used as an important tool for the accuracy and calculated with the following equation:
The limit of quantification (LOQ) was defined as the smallest amount of the analyte that could be measured in a sample with sufficient precision and accuracy (within 20% for both parameters) and was chosen as the lowest concentration on the calibration curve.
Selectivity
In selectivity experiments, urine samples from different persons were analyzed for any residual F2-IsoPs, and PGs. Furthermore, a zero sample that was only spiked with 8-iso-PGF2α-d4 was tested for any undeuterated 8-iso-PGF2α in this deuterated IS.
Stability
The QC samples were investigated thoroughly to evaluate their stability under different conditions (room temperature, freeze/thaw cycles, autosampler and long term storage stability). The room temperature stability was evaluated at ambient temperature (~23°C) over 24 h using QC samples in five replicates. The stability of F2-IsoPs, and PGs in human urine following repeated freeze-thaw (three cycles) was assessed using QC samples. The autosampler stability was evaluated at 4°C over 48 h. The long-term storage stability at −20°C for over 30 days was also evaluated.
Liquid chromatography-mass spectrometry
LC-MS/MS analyses of urine samples was performed using a system consisting of a Shimadzu Prominence HPLC with a refrigerated auto sampler (Shimadzu Scientific Instruments, Inc. Columbia, MD), and an API 4000 (Applied Biosystems/MDS Sciex, Concord, Ontario, Canada) triple quadrupole mass spectrometer.
Chromatography was performed using a Hydro-RP column (250 × 2.0 mm i.d, Phenomenex, CA). The mobile phase consisted of water [A] and methanol:acetonitrile (1:1 v/v) [B] (both containing 0.1% formic acid) with a flow rate of 0.2 mL/min. The gradient started at 55% B and went up to 65% B over 12 min). It was, followed an increase to 100% B at 13 min and this was maintained for a further 1 min. At 15 min there was a linear decrease from 100 to 55%. The column effluent was introduced into the mass spectrometer using electrospray ionization (ESI) in the negative ion mode.
Nitrogen was used as nebulizer, gas 1 and curtain gas. The SRM analysis was conducted by monitoring the precursor ion to product ion transitions from m/z 353/193 and 353/309 (8-iso-PGF2α, 8-iso-15(R)-PGF2α and PGF2α,15(R)-PGF2α), and m/z 357/197 and 357/313 (8-iso-PGF2α-d4). MSMS parameters were optimized to obtain best sensitivity. The declustering potential, temperature, collision energy and ion source gas (GS1) were optimized and set at −80 V, 600 °C, −35 eV and 40 psi, respectively. The dewll time was 50 ms. The LC-MS/MS system was controlled by Analyst software version 1.4.2.
Clinical application
Patients undergoing cardiac surgery (either coronary bypass surgery or valve surgery) were recruited from the San Diego VA Hospital and their urine samples before surgery and 2, 6, 12, 24 and 48 hours after surgery were collected (Tables 1 and 2). For this analysis the pre-operative sample was compared with the post-operative sample in which patients had the highest creatinine level (mg/dL). Of the 15 patients analyzed, four developed AKI as defined as a 0.3 mg/dL elevation in creatinine (Table 2).
Table 1.
Characteristics of patients undergoing surgery for cardiac problems who did not develop acute kidney injury
| Patient # | Age (yr) | Sex | Pre-operative Creatinine (mg/mL) | Post-operative Creatinine (mg/mL) | DM | HTN | HL | HbA1C (%) | Surgery Type |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 68 | M | 0.96 | 0.87 | Yes | Yes | Yes | 6.0 | CABG |
| 2 | 62 | M | 0.84 | 0.80 | No | No | No | 7.5 | CABG |
| 3 | 66 | M | 0.76 | 1.03 | No | No | Yes | 5.2 | CABG |
| 4 | 59 | M | 0.94 | 0.84 | No | No | Yes | 5.3 | CABG AVR |
| 5 | 70 | M | 1.03 | 0.88 | No | Yes | No | 5.3 | CABG AVR |
| 6 | 64 | M | 0.93 | 0.93 | No | Yes | No | 5.9 | CABG MVR |
| 7 | 64 | M | 2.18 | 2.25 | No | Yes | No | 5.1 | MVR |
| 8 | 61 | M | 1.15 | 1.09 | No | Yes | Yes | 5.5 | AVR |
| 9 | 66 | M | 0.85 | 0.88 | Yes | Yes | Yes | 6.4 | CABG |
| 10 | 71 | M | 2.09 | 2.11 | No | No | Yes | 5.7 | CABG |
| 11 | 67 | M | 1.06 | 0.97 | Yes | Yes | No | 5.5 | CABG |
DM = Diabetes mellitus; HTN = hypertension; HL = hyperlipidemia; CABG = coronary arterial by-pass grafting; AVR = aortic valve replacement; MVR = mitral valve replacement
Table 2.
Characteristics of patients undergoing surgery for cardiac problems who developed acute kidney injury
| Patient # | Age (yr) | Sex | Pre-operative Creatinine (mg/mL) | Post-operative Creatinine (mg/mL) | DM | HTN | HL | HbA1C (%) | Surgery Type |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 63 | M | 1.33 | 1.70 | Yes | Yes | Yes | 6.6 | CABG |
| 2 | 72 | M | 0.77 | 1.21 | No | Yes | Yes | 5.5 | AVR |
| 3 | 60 | M | 1.46 | 1.85 | No | Yes | No | 5.5 | AVR |
| 4 | 68 | M | 0.96 | 1.31 | No | Yes | Yes | 5.6 | AVR |
DM = Diabetes mellitus; HTN = hypertension; HL = ; CABG = coronary arterial by-pass grafting; AVR = aortic valve replacement
At UAB, urines were collected from a group of healthy human volunteers (n=18), who gave informed consent, both prior to and immediately following taking part in mild exercise (45 min of walking). The study was approved by the UAB Institutional Review Board. Urine collected from the middle third of their urine void was immediately placed on ice and stored at −20°C prior to analysis. Urine creatinine was analyzed following the published method (18).
Analysis of F2-IsoP and PGF2α
Sample preparation and analysis
First, 1 mL of each urine obtained from the clinical studies was added to 8-iso-PGF2α-d4 (2 ng/mL final concentration) and the solution then was processed and analyzed by the methods as described above for method development and validation. Each sample was analyzed in duplicate.
Statistical analysis
Data are presented as means ± standard deviation and the unpaired t-test was used for statistical comparison. A p value < 0.05 was considered significant.
RESULTS
LC-MS/MS
Due to the presence of a carboxylic group in both F2-IsoPs and PGs, ESI operating in negative ion mode provided the best sensitivity. Deprotonated molecular ions [M-H]− m/z 353 of standards (8-iso-PGF2α, 8-iso-15(R)-PGF2α, PGF2α and 15(R)-PGF2α) were induced to fragment in the collision cell, and after optimization, the most abundant product ions m/z 193 and 309 were chosen for quantitative SRM analysis. For SRM analysis, the mass transitions m/z 353/193 and 353/309 were selected for F2-IsoPs and PGs and m/z 357/197 and 357/313 for 8-iso-PGF2α-d4.
In order to obtain both baseline separation and optimized sensitivity of these analytes, variables such as column type, gradient, and MS/MS operating parameters were investigated. Several columns and different solvents were tested to obtain a high degree of sensitivity, good separation and a short analysis time. Chromatographic conditions were optimized to provide baseline separation of isomeric F2-IsoPs and PGs with adequate peak shape. A Hydro-Rp column (250 × 2.0 mm i.d) with water and methanol:acetonitrile (1:1 v/v containing 0.1% formic acid) provided the best separation and sensitivity. Use of acetonitrile in the mobile phase led to the best chromatographic separation, but at the expense of lowered sensitivity. While use of methanol alone gave the highest sensitivity, it failed to resolve the four F2-IsoPs and PG isomers. As shown in Fig. 3A, a 20 min run time with acidified methanol:acetonitrile (1:1 v/v) provided efficient separation of all isomers.
Figure 3.
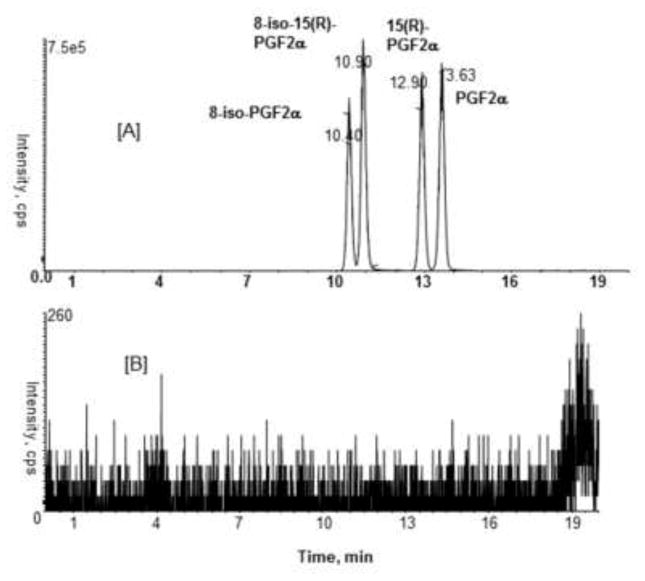
Representative SRM chromatograms for F2-IsoPs and PGs in urine (10 ng/mL) [A]; 10 μL methanol blank [B].
Sample preparation was carried out using Strata X-AW 33u polymeric weak anion cartridge (Phenomenex, Torrance, CA) and provided excellent recoveries of analytes from urine after eluting with methanol-water (80/20, v/v containing 1% acetic acid).
Method validation
The methods were validated in accordance with the FDA guidance for bioanalytical method validation and also based on the paper of Shah et al. [19,20]. A full validation was performed for human urine based on the following criteria.
Linearity
The calibration curves for standards were generated at different concentrations ranging from 0.05 to 50 ng/mL (50, 25, 10, 1, 2, 0.1, and 0.05 ng/mL). The standard curve was linear over this 1000-fold concentration range. Table 3 shows the summary of the individual standard data obtained in the five replicates. LOQ was determined to be 0.05 ng/mL, demonstrating the precision (CV%) each analyte in a range of 4.90–19.39 (Table 3). These results indicated a linear relationship between the peak areas and concentrations of all analytes in this assay.
Table 3.
Summary of calibration curves (n =5)
| Nominal concentration (ng/mL) | Measured mean concentration (ng/mL) ± S.D | CV(%) | |
|---|---|---|---|
| 8-Iso-PGF2α | 50 | 49.56 ± 2.11 | 4.26 |
| 25 | 25.54 ± 1.01 | 3.96 | |
| 10 | 9.70 ± 0.57 | 5.88 | |
| 2 | 1.97 ± 0.14 | 7.38 | |
| 1 | 0.90 ± 0.00 | 6.42 | |
| 0.1 | 0.09 ± 0.01 | 7.44 | |
| 0.05 | 0.04 ± 0.00 | 6.43 | |
| 8-Iso-15R-PGF2α | 50 | 47.24 ± 1.2 | 2.68 |
| 25 | 24.22 ± 1.13 | 4.67 | |
| 10 | 9.68 ± 0.22 | 2.32 | |
| 2 | 1.93 ± 0.09 | 5.16 | |
| 1 | 0.94 ± 0.02 | 2.92 | |
| 0.1 | 0.09 ± 0.00 | 5.55 | |
| 0.05 | 0.04 ± 0.00 | 4.90 | |
| PGF2α | 50 | 51.48 ± 1.69 | 3.28 |
| 25 | 25.04 ± 0.88 | 3.53 | |
| 10 | 10.02 ± 0.51 | 5.15 | |
| 2 | 1.86 ± 0.08 | 4.59 | |
| 1 | 0.89 ± 0.06 | 6.89 | |
| 0.1 | 0.09 ± 0.01 | 12.67 | |
| 0.05 | 0.04 ± 0.00 | 19.39 | |
| 15R-PGF2α | 50 | 50.15 ± 1.62 | 3.25 |
| 25 | 25.47 ± 0.35 | 1.41 | |
| 10 | 9.43 ± 0.14 | 1.54 | |
| 2 | 1.79 ± 0.09 | 5.29 | |
| 1 | 0.89 ± 0.02 | 3.05 | |
| 0.1 | 0.09 ± 0.00 | 4.44 | |
| 0.05 | 0.04 ± 0.00 | 14.15 | |
%CV = coefficient of variation (SD/mean × 100)
Accuracy and precision
The accuracy and precision of this assay for all analytes were determined at various concentrations (5, 0.5, 0.07 ng/mL) of the QC samples. The performance characteristics of the method were established by validation procedures employing assays with standard solutions, sample blanks and spiked samples. The intra-day accuracy (%bias) and inter-day precision (%CV) were found to be well within acceptable limits as described in Table 4. For the lowest quality control (0.07 ng/mL), the intra-day and inter-day precisions for the analytes were less than 17% and the intra-day bias were less than 20%. These results indicated that the method is sensitive, reproducible, and reliable.
Table 4.
Precision and accuracy of quality control samples
| Analyte | Nominal concentration (ng/mL) | Accuracy (%bias) | Precision (%CV) | Inter-day | ||||
|---|---|---|---|---|---|---|---|---|
| Day 1 | Day 2 | Day 3 | Day 1 | Day 2 | Day 3 | |||
| 8-iso-PGF2α | 5 | 0.68 | −7.55 | 0.74 | 1.94 | 3.94 | 5.76 | 1.59 |
| 0.5 | 9.72 | −5.40 | −1.11 | 4.74 | 4.78 | 6.29 | 0.90 | |
| 0.07 | 14.46 | −0.77 | 3.43 | 6.16 | 6.49 | 9.13 | 4.15 | |
| 8-iso-15(R)-PGF2α | 5 | −2.56 | −3.33 | −1.90 | 2.98 | 0.92 | 4.67 | 3.22 |
| 0.5 | 2.40 | −2.23 | 3.27 | 6.45 | 8.49 | 7.53 | 1.15 | |
| 0.07 | −3.68 | −6.04 | 3.07 | 16.59 | 14.59 | 3.88 | 8.29 | |
| PGF2α | 5 | −1.28 | −6.48 | −1.67 | 1.56 | 6.49 | 2.84 | 2.40 |
| 0.5 | 8.28 | 6.06 | −1.67 | 5.98 | 6.75 | 8.95 | 1.10 | |
| 0.07 | 0.10 | −12.02 | 16.24 | 12.18 | 12.71 | 15.41 | 8.88 | |
| 15(R)-PGF2α | 5 | −0.32 | −2.89 | −9.47 | 2.45 | 5.79 | 4.84 | 1.67 |
| 0.5 | 4.16 | 12.25 | 0.78 | 4.64 | 6.11 | 3.93 | 1.90 | |
| 0.07 | −11.54 | 19.46 | −8.23 | 8.97 | 9.68 | 3.12 | 9.12 | |
Inter-day variation (15 replicates at each concentration)
Specificity and selectivity
Representative SRM chromatograms of a zero sample (urine spiked with IS) and unspiked urine sample after extraction are shown in Fig. 2. The LC-MS/MS method demonstrated high specificity because only ions derived from IS (20 ng/mL) was observed (Fig. 2A and B). Furthermore, no quantifiable amount of F2-IsoPs and PGs were found in an extracted unspiked urine sample (Fig. 2C). These data indicate that there were no detectable endogenous substances in urine interfering in the analysis of F2-IsoPs and PGs.
Figure 2.
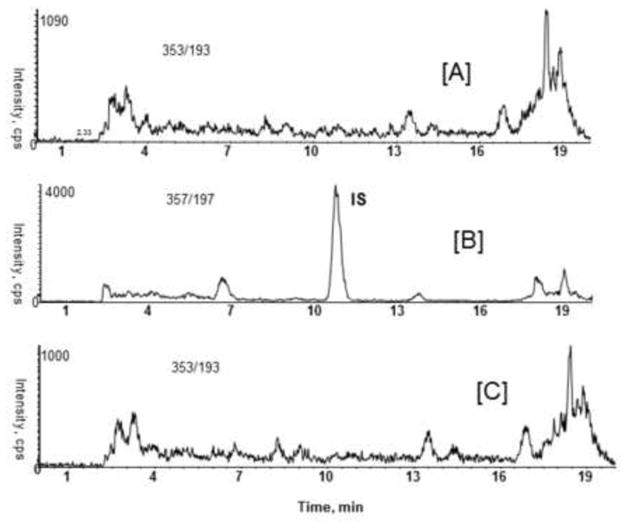
Representative SRM chromatograms of human urine samples processed with or without IS. Matrix samples with IS (20 ng/mL) [A] and [B]; without IS [C].
Carry over
Carry-over is one of the most commonly encountered problems of LC/MS/MS method development. It can affect the accuracy and precision of a method and should be evaluated during method validation. As can be seen in Fig. 3, when methanol blank was injected right after the analyte spiked urine sample (10 ng/mL), no detectable carry over was observed in the current LC-MS/MS method.
Matrix effect and recovery
The adverse consequences of matrix effects on the results of quantitative LC-MS/MS analyses have been fully recognized and the assessment of matrix effects is becoming an integral part of method development and validation. Human urine samples were extracted, then spiked and analyzed for potential interferences by endogenous matrix components. The MEs from endogenous urinary metabolites on each analyte are listed in Table 5. The MEs observed for 8-iso-PGF2α at 0.5 and 5 ng/mL were 21.48 and 17.0%, respectively, indicating endogenous interference from urine matrix. However, the use of deuterated internal standard (8-iso-PGF2α-d4) can neutralize the effect of matrix and the results for linearity, precision and recovery are acceptable for all analytes [21]. These results indicated that there were minor matrix effects for these compounds in human urine.
Table 5.
Calculated recovery and matrix effects of urine on the peak area response of analytes
| Analyte | Concentration (ng/mL) | Mean recovery (%)± SD | Mean matrix effect (%) |
|---|---|---|---|
| 8-iso PGF2α | 5 | 85.3±8.8 | 17.0 |
| 0.5 | 83.8±8.5 | 21.5 | |
| 0.07 | 87.4±10.3 | 14.5 | |
| 8-iso-15(R) | 5 | 81.5±6.8 | 2.0 |
| 0.5 | 80.7±7.4 | 12.3 | |
| 0.07 | 100.2±7.7 | 0.4 | |
| PGF2α | 5 | 85.7±6.0 | −1.0 |
| 0.5 | 82.1±8.6 | 0.9 | |
| 0.07 | 99.5±24.2 | −13.1 | |
| 15(R)-PGF2α | 5 | 79.4±7.9 | 6.1 |
| 0.5 | 83.0±10.0 | 4.3 | |
| 0.07 | 90.1±13.1 | −9.4 |
Recovery studies of each analyte were performed at concentrations of 5, 0.5 and 0.07 ng/mL by comparing the peak areas of the extracted urine samples with those of post spiked samples. Overall mean extraction recoveries at these concentrations were in a range of 79–100%.
Stability
The stability of QC samples was investigated thoroughly to evaluate their stability in urine samples under different conditions (autosampler, room temperature, freeze-thaw cycles and long term storage stability). The autosampler stability was evaluated at 4°C over 48 h and the mean measured concentrations showed that the analytes are stable for at least 48 h when stored at 4°C. All stability data are summarized in Table 6.
Table 6.
Stability of quality control samples
| Compound | Nominal Concentration (ng/mL) | Mean measured concentration (ng/mL) ±SD | |||
|---|---|---|---|---|---|
| room temp | long term | autosampler | Freeze-thaw cycles | ||
| 8-iso-PGF2α | 5 | 4.92±0.11 | 4.79±0.10 | 4.62±0.18 | 4.84±0.71 |
| 0.5 | 0.49±0.08 | 0.46±0.03 | 0.47±0.02 | 0.52±0.04 | |
| 0.07 | 0.07±0.00 | 0.07±0.00 | 0.06±0.00 | 0.07±0.00 | |
| 8-iso-15(R)-PGF2α | 5 | 4.68±0.05 | 4.77±0.07 | 4.83±0.04 | 4.70±0.11 |
| 0.5 | 0.51±0.03 | 0.05±0.04 | 0.48±0.04 | 0.50±0.01 | |
| 0.07 | 0.07±0.00 | 0.07±0.01 | 0.06±0.01 | 0.07±0.00 | |
| PGF2α | 5 | 4.69±0.23 | 4.84±0.26 | 4.68±0.30 | 4.48±0.07 |
| 0.5 | 0.43±0.17 | 0.50±0.04 | 0.53±0.04 | 0.52±0.01 | |
| 0.07 | 0.07±0.00 | 0.07±0.00 | 0.06±0.01 | 0.06±0.00 | |
| 15(R)-PGF2α | 5 | 5.11±0.19 | 4.87±0.29 | 4.85±0.28 | 4.49±0.07 |
| 0.5 | 0.53±0.012 | 0.53±0.03 | 0.56±0.03 | 0.51±0.02 | |
| 0.07 | 0.07±0.00 | 0.08±0.01 | 0.07±0.00 | 0.06±0.00 | |
Repeated freeze and thawing of urine samples spiked with analytes did not affect their stability (Table 6). The analytes were stable in the frozen urine for at least three freeze-thaw cycles. The longer-term (over 1 month) stability in human urine was evaluated by analyzing frozen QC samples in five replicates. The mean measured concentrations shown in Table 6 indicated that 8-iso-PGF2α can undergo isomerization during long storage.
Measurement of F2-IsoPs and PGF2α in clinical samples
The LC-MS/MS method was applied to quantification of F2-IsoPs and PGs in urine samples obtained from fifteen patients before and after cardiac surgery from the UAB-UCSD O’Brien Acute Kidney Injury Research Center. The samples were extracted after spiking with IS as described for QC and calibration standards in sample preparation section. The urinary levels of F2-IsoPs but not PGs changed significantly following cardiac surgery (Fig. 4). The levels of 8-iso-PGF2α, - the most abundant form of F2-IsoP and an indicator of in vivo oxidative stress - increased 85% from baseline (0.333±0.030 to 0.617±0.103 ng/mg creatinine, p<0.01). A 62% increase was observed for 8-iso-15(R)-PGF2α (0.363±0.050 vs 0.589±0.090 ng/mg creatinine, p<0.05). Non-significant changes occurred in PGF2α (1.460±0.270 vs 1.068±0.251 ng/mg creatinine) and its 15R isomer (0.227±0.0.074 vs 0.349±0.138 ng/mg creatinine) in urine samples collected after surgery. Interestingly, the increase in the two F2-IsoPs was confined to those patients (5 of the 15) who did not experience acute kidney injury. In the four AKI patients, the 8-iso-PGF2α and 8-iso-15(R)-PGF2α concentrations were not significantly changed (0.298±0.062 to 0.383±0.117 ng/mg creatinine, P=0.54, and 0.245±0.070 to 0.336±0.097, P=0.48, respectively). A representative SRM chromatogram demonstrating detection of F2-IsoPs and PGs in human urine samples from an AKI patient is shown in Fig. 5.
Figure 4.
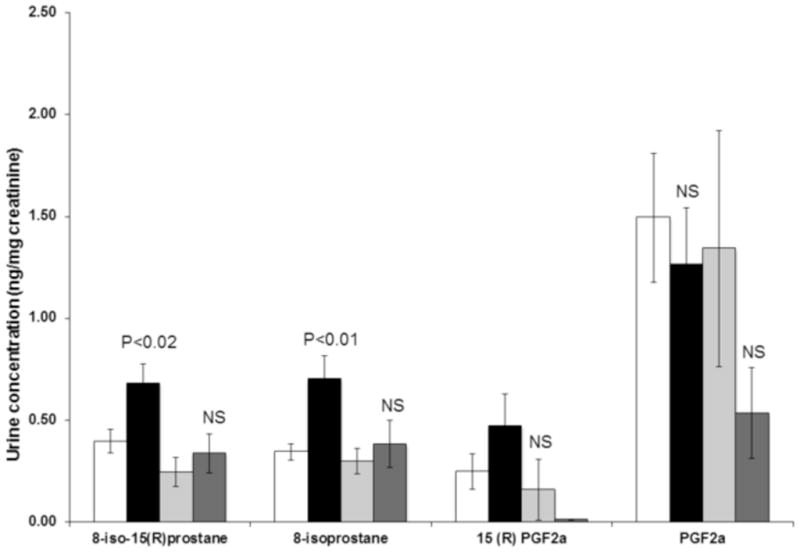
Isoprostane and PGF2α levels before and after cardiac surgery. Values are expressed as means ± SEM and p-values indicate the significance of differences between values before and after surgery. The white boxes are values for patients without acute kidney injury prior to surgery; the black boxes are values after surgery in these patients. The light gray boxes are patients with acute kidney injury prior to surgery; the dark gray boxes are values after surgery in these patients. The statistical significances are for differences between pre- and post-surgery values.
Figure 5.
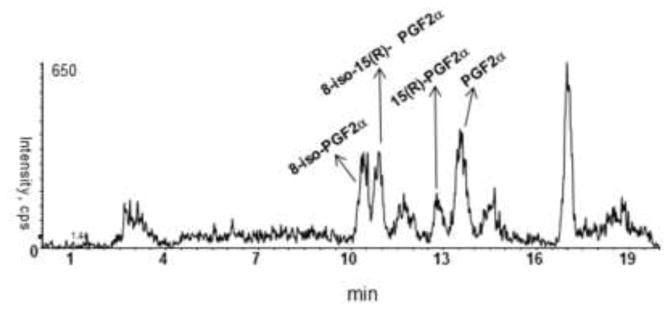
Representative SRM chromatogram for F2-IsoPs and PGs in a urine sample from an AKI patient.
In a study in 18 healthy volunteers, the pre-exercise urinary concentrations of 8-iso-PGF2α, 8-iso-15(R)-PGF2α, PGF2α and 15R-PGF2α were 0.395±0.051, 0.477±0.081, 1.618±0.216 and 0.291±0.112 ng/mg creatinine, respectively. Following exercise, the urinary concentrations of 8-iso-PGF2α, 8-iso-15(R)-PGF2α, PGF2α and 15R-PGF2α were not significantly changed (0.464±0.072, 0.374±0.060, 2.369±0.555 and 0.231±0.057 ng/mg creatinine, respectively).
Reproducibility in clinical samples
The volumes of the urine samples that were available in this study allowed replicate analyses to be performed. The mean reproducibility (coefficient of variation) of duplicates in these samples was 8.59%. For those with isoprostane concentrations >1 ng/creatinine, the mean reproducibility was 5.48%. In only six of the 113 analytical runs where there was a measurable peak were the reproducibilities of duplicates outside of the acceptable range (20%).
DISCUSSION
An accurate, reproducible LC-MS/MS assay for the simultaneous quantification of F2-IsoPs and PGs in human urine has been developed and validated. The assay permits analysis over a range of 0.05–50 ng/mL of analytes in 1 mL of aliquots of human urine with precise and accurate quantification. The main advantages of this LC-MS/MS method are: (1) simplicity, 2) excellent recovery; (3) absence of a significant matrix effect; (4) absence of carry over; (5) base line separation of the isomers measured; and (6) validation over a large dynamic range (calibrators from 0.05 to 50 ng/mL).
The analysis of F2-IsoPs and PGs is of great interest in assessing quantitative stress clinically in acute and chronic disease and in corresponding experimental models. Their measurement nonetheless represents several analytical challenges. First, they are typically present in urine and plasma at concentrations close to 1 ng/mL. For many mass spectrometry methods this is only an order of magnitude above the LOQ of the F2-IsoPs and PGs. However, while 1 mL or more volumes of urine can be obtained in clinical situations, in experimental rodent models of oxidative stress, particularly in mice, these volumes can be harder to come by. Future solutions to the issue of sensitivity may include the use of a lower solution flow rate nano-ESI to reduce the size of the charged droplet initially produced in the spraying process, thereby increasing MS sensitivity [22,23]. Other approaches such as formation of derivatives containing positively charged, quaternary nitrogen atoms can be utilized to enhance the detection limit in positive ion mode [24].
The concentrations of F2-IsoPs and PGs in normal healthy subjects as measured using the method described in this study are comparable to those in other reports using LC-MS/MS methods [9, 25, 26]. As noted by Klawitter et al. [25], LC-MS/MS methods are superior to enzyme-linked immunosorbent assay (ELISA) for F2-isoPs since the latter gave values for urinary concentrations that were 0.4–61.9-fold higher than those obtained by LC-MS/MS and were highly dependent on the manufacturer of the ELISA kit.
Using the LC-MS/MS method, elevated levels of F2-IsoPs were found in urine samples collected from patients following cardiac surgery, indicating a potential involvement of isoprostanes in vascular physiology and pathogenesis. In contrast, in those patients who subsequently developed an acute kidney injury (AKI), the increases in F2-IsoPs were not observed. It is possible that since the AKI patients were defined as those who had elevated levels of creatinine, the origin(s) of creatinine and the F2-IsoPs are independent, thereby causing a reduction in the F2-IsoPs/creatinine ratio.
In normal healthy subjects, mild exercise caused no significant changes in urinary F2-IsoPs and PGF2a and is consistent with previous studies [27–29]. It is noteworthy that chronic mild exercise lowers F2-IsoPs [25].
We conclude that this method provides a simple procedure for assaying urinary F2-IsoPs together with PGs and has reliable reproducibility.
Highlights.
Measurement of F2-isoprostanes and prostaglandins in human urine by LC-MS/MS.
Base line separation of the isomers measured.
Quantification of analytes over a linear dynamic range (0.05–50 ng/mL).
Excellent recoveries (79–100%), no major matrix effects and absence of carry over.
Urinary F2-isoprostanes and PGs analysis in patients undergoing cardiac surgery.
Acknowledgments
We appreciate the advice given during this study by Dr. Ginger Milne, Vanderbilt University and Dr. Alan Taylor, Oregon State University. Dr. Molly Bray, UAB, kindly allowed us access to the participants in the TIGER study. Research on the LC-MS analysis of urinary isoprostanes and prostaglandins is supported by a grant (P30 DK079337; Anupam Agarwal, PI) from the National Institute of Diabetes, Digestive and Kidney Diseases to the UAB-UCSD O’Brien Center for Acute Kidney Injury. Dr. Taub is supported by a Pilot and Feasibility Grant from the O’Brien Center for Acute Kidney Injury. Funds for the purchase of the mass spectrometer used in this study were provided by a grant (SB, PI) from the UAB Health Services Foundation General Endowment Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kashyap P, Farrugia G. Neurogastroenterol Motil. 2011;23:111. doi: 10.1111/j.1365-2982.2010.01659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dobrain AD, Davies JJ, Schriver SD, Lauterio TJ, Prewitt RL. Hypertension. 2001;37:554. doi: 10.1161/01.hyp.37.2.554. [DOI] [PubMed] [Google Scholar]
- 3.Davi G, Ciabattoni G, Consoli A, Mezzetti A, Falco A, Santarone S, Pennese E, Vitacolonna E, Bucciarelli T, Costantini F, Capani F, Patrono C. Circulation. 1999;99:224. doi: 10.1161/01.cir.99.2.224. [DOI] [PubMed] [Google Scholar]
- 4.Belli R, Amerio P, Brunetti L, Orlando G, Toto P, Proietto G, Vacca M, Tulli A. Int J Immunopathol Pharmacol. 2005;18:497. doi: 10.1177/039463200501800309. [DOI] [PubMed] [Google Scholar]
- 5.Chrissobolis S, Miller AA, Drummond GR, Kemp-Harper BK, Sobey CG. Front Biosci. 2011;16:1733. doi: 10.2741/3816. [DOI] [PubMed] [Google Scholar]
- 6.Morrow JD, Roberts LJ., II Methods Enzymol. 1994;223:163. doi: 10.1016/s0076-6879(94)33019-0. [DOI] [PubMed] [Google Scholar]
- 7.Il’yasova D, Spasojevic I, Wang F, Tolun AA, Base K, Young SP, Marcom PK, Marks J, Mixon G, DiGiulio R, Millington DS. Cancer Epidemiol Biomarkers Prev. 2010;19:1506. doi: 10.1158/1055-9965.EPI-10-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wedes SH, Khatri SB, Zhang R, Wu W, Comhair SA, Wenzel S, Teague WG, Israel E, Erzurum SC, Hazen SL. Clin Transl Sci. 2009;2:112–117. doi: 10.1111/j.1752-8062.2009.00095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan W, Byrd GD, Ogden MW. J Lipid Res. 2007;48:1607. doi: 10.1194/jlr.M700097-JLR200. [DOI] [PubMed] [Google Scholar]
- 10.Tsikas D, Schwedhelm E, Fauler J, Gutzki FM, Mayatepeck E, Frolich JC. J Chromatogr B. 1998;716:7. doi: 10.1016/s0378-4347(98)00275-8. [DOI] [PubMed] [Google Scholar]
- 11.Schwedhelm E, Tsikas D, Durand T, Gutzki FM, Guy A, Rossi JC, Frolich JC. J Chromatogr B. 2000;744:99. doi: 10.1016/s0378-4347(00)00236-x. [DOI] [PubMed] [Google Scholar]
- 12.Ferretti A, Flanagan VP. J Chromatogr B. 1997;694:271. doi: 10.1016/s0378-4347(97)00142-4. [DOI] [PubMed] [Google Scholar]
- 13.Langhorst ML, Hastings MJ, Yokoyama WH, Hung SC, Cellar N, Kuppannan K, Young SA. J Agric Food Chem. 2010;58:6614. doi: 10.1021/jf101146q. [DOI] [PubMed] [Google Scholar]
- 14.Tsikas D. J Chromatogr B. 1998;717:201. doi: 10.1016/s0378-4347(98)00210-2. [DOI] [PubMed] [Google Scholar]
- 15.Proudfoot J, Barden A, Mori TA, Burke V, Croft KD, Beilin LJ, Puddey IB. Anal Biochem. 1999;272:209. doi: 10.1006/abio.1999.4187. [DOI] [PubMed] [Google Scholar]
- 16.Wagner RS, Weare C, Jin N, Mohler ER, Rhoades RA. Prostaglandins. 1997;54:581. doi: 10.1016/s0090-6980(97)00127-5. [DOI] [PubMed] [Google Scholar]
- 17.Davies SS, Zackert W, Luo Y, Cunningham CC, Frisard M, Roberts LJ., 2nd Anal Biochem. 2006;348:185. doi: 10.1016/j.ab.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi N, Boysen G, Li F, Li Y, Swenberg JA. Kidney Int. 2007;71:266. doi: 10.1038/sj.ki.5002033. [DOI] [PubMed] [Google Scholar]
- 19.U.S. Food and Drug Administration: Centre for Drug Evaluation and Research. Guidance for Industry. Bioanalytical Method Validation. 2001 www.fda.gov/cder/quidance/4252fnl.htm.
- 20.Shah VP, Midha KK, Findlay JWA, Hill HM, Hulse JD, McGilveray IJ, McKay G, Miller KJ, Patnaik RN, Powell ML, Tonelli A, Viswanathan CT, Yacobi A. Pharm Res. 2000;17:1551. doi: 10.1023/a:1007669411738. [DOI] [PubMed] [Google Scholar]
- 21.Badawi N, Simonsen KW, Steentoft A, Bernhoft IM, Linnet K. Clinical Chemistry. 2009;55:2004. doi: 10.1373/clinchem.2008.122341. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt A, Karas M. J Am Soc Mass Spectrom. 2003;14:492–500. doi: 10.1016/S1044-0305(03)00128-4. [DOI] [PubMed] [Google Scholar]
- 23.Tang K, Page JS, Smith RD. J Am Soc Mass Spectrom. 2004;15:1416–1423. doi: 10.1016/j.jasms.2004.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kojima M, Kamada-Nobusada T, Komatsu H, Takei K, Kuroha T, Mizutani M, Ashikari M, Ueguchi-Tanaka M, Matsuoka M, Suzuki K, Sakakibara H. Plant Cell Physiol. 2009;50:1201. doi: 10.1093/pcp/pcp057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klawitter J, Haschke M, Shokati T, Klawitter J, Christians U. Rapid Commun Mass Spectrom. 2011;25:463. doi: 10.1002/rcm.4871. [DOI] [PubMed] [Google Scholar]
- 26.Taylor AW, Bruno RS, Traber MG. Lipids. 2008;43:925. doi: 10.1007/s11745-008-3222-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campbell PT, Gross MD, Potter JD, Schmitz KH, Duggan C, McTiernan A, Ulrich CM. Med Sci Sports Exerc. 2010;42:1448. doi: 10.1249/MSS.0b013e3181cfc908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nikolaidis MG, Kyparos A, Vrabas IS. Prog Lipid Res. 2011;50:89. doi: 10.1016/j.plipres.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 29.Medina S, Domínguez-Perles R, Cejuela-Anta R, Martínez-Sanz JM, Gil P, García-Viguera C, Ferreres F, Gil JI, Gil-Izquierdo A. Prostaglandins Other Lipid Mediat. 2012 Jul 21; doi: 10.1016/j.prostaglandins.2012.07.002. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]