

Abstract
New antiarrhythmic drugs for treatment of atrial fibrillation should ideally be atrial selective in order to avoid pro-arrhythmic effects in the ventricles. Currently recognized atrial selective targets include atrial Nav1.5 channels, Kv1.5 channels and constitutively active Kir3.1/3.4 channels, each of which confers atrial selectivity by different mechanisms. Na+ channel blockers with potential- and frequency-dependent action preferentially suppress atrial fibrillation because of the high excitation rate and less negative atrial resting potential, which promote drug binding in atria. Kv1.5 channels are truly atrial selective because they do not conduct repolarizing current IKur in ventricles. Constitutively active IK,ACh is predominantly observed in remodelled atria from patients in permanent atrial fibrillation (AF). A lot of effort has been invested to detect compounds which will selectively block Kir3.1/Kir3.4 in their remodelled constitutively active form. Novel drugs which have been and are being developed aim at atrial-selective targets. Vernakalant and ranolazine which mainly block atrial Na+ channels are clinically effective. Newly designed selective IKur blockers and IK,ACh blockers are effective in animal models; however, clinical benefit in converting AF into sinus rhythm (SR) or reducing AF burden remains to be demonstrated. In conclusion, atrial-selective antiarrhythmic agents have a lot of potential, but a long way to go.
 |
Prof. Ursula Ravens studied medicine followed by a postdoc position in pharmacology. After some years as Professor of Cardiovascular Pharmacology in Essen she became director of the Department of Pharmacology and Toxicology of the Medical Faculty of TU Dresden, Germany. Her research interest include pharmacological aspects of cardiac cellular electrophysiology with a strong focus on atrial fibrillation, differentiation and reprogramming of stem cells for regenerative medicine, and pharmacology of the urinary tract. Dr. Claire Poulet studied biology at the University Paris XII in France and joined the Dresden International Graduate School for Biomedicine and Bioengineering to write her PhD thesis on “Cardiomyogenic potential of skeletal muscle-derived progenitor cells”. She continued in Ursula Ravens' lab as a post-doc and has since contributed to the characterization of various ion channels in human atrial cardiomyocytes and fibroblasts. Prof. Erich Wettwer studied biology and did a PhD in Physiology investigating Ca homeostasis in skeletal muscle cells. He joined Ursula Ravens' lab in Essen and continued to work with her in Dresden. His scientific Interests are the role of auxiliary subunits in the regulation of ion channel expression and function in human cardiac tissue and expression systems. PD Dr. Michael Knaut studied medicine and became a cardiac surgeon at the Heart Centre Dresden. Scientifically, he has conducted and supervised numerous clinical studies required for the introduction of new energy sources for ablation of atrial fibrillation in cardiac surgery. With the pharmacology group in Dresden he shares the interest in atrial fibrillation, which has led to a long standing collaboration.
Introduction
The most prevalent cardiac rhythm disorder atrial fibrillation (AF) is responsible for significant morbidity and mortality, especially in the elderly. Current therapeutic options consist of anticoagulation for stroke prevention, and of rate or rhythm control for ameliorating symptoms and preventing tachycardia-induced deterioration of ventricular function (Camm et al. 2012). Rhythm control can be achieved with antiarrhythmic drugs, electrical cardioversion, and ablation strategies. All these treatment modalities have their advantages and limitations. Drug treatment of atrial fibrillation is limited by low efficacy and side effects of currently available antiarrhythmic agents combined with their propensity to induce life-threatening ventricular arrhythmias. Therefore, the ongoing search for new agents against AF with a more favourable benefit-to-harm relation has led to the development of atrial-selective antiarrhythmic drugs (Burashnikov et al. 2007; Ehrlich et al. 2007).
Though this review focuses on ion channels as targets for new antiarrhythmic drugs, any process involved in underlying cardiac disease may also prove a useful candidate target for new potential therapeutic agents in AF, and innovative strategies in this field have been recently reviewed (Savelieva & Camm, 2008). However, here, we will give a brief outline of the mechanisms of arrhythmia and our current understanding of antiarrhythmic action before we discuss novel antiarrhythmic drugs that have recently been developed or conceived as being atrial selective.
Cardiac action potentials (APs)
At the cellular level, the excitation of a cardiomyocyte is initiated by depolarization beyond the threshold for activation of Na+ current (INa). The large inward current surge depolarizes the cell membrane to the potential range of L-type Ca2+ channel activation, and the resulting inward current contributes to the plateau of the action potential. Repolarizing outward currents are conducted via various K+ channels, regulate the plateau, and shape the final repolarization phase. Cellular ionic homeostasis is maintained by pumps and transporters. The Na+–Ca2+ exchanger (NCX) utilizes the transmembrane Na+ concentration gradient to remove Ca2+ from the cell. Because of its transport stoichiometry of 3 Na+ to 1 Ca2+, the NCX is electrogenic, i.e. it reverses direction of Ca2+ transport in dependence of membrane potential, contributing depolarizing current in its Ca2+-extruding ‘forward’ mode at resting potential and repolarizing current in its Ca2+-influx ‘reverse’ mode in the plateau potential range.
Cardiac cells remain refractory as long as the membrane is depolarized, and, after repolarization, until enough Na+ channels have recovered from inactivation for re-excitation. Thus, effective refractory period (ERP) is governed by action potential duration (APD) and by the rate of recovery of Na+ channels from inactivation. Action potentials propagate between adjacent cardiomyocytes via gap junctions formed by connexins. These low resistance pathways must pass a current large enough to depolarize neighbouring cells beyond the threshold of a propagated action potential. Therefore, conduction velocity is a function of INa and of cell-to-cell coupling via gap junctions.
Mechanisms of arrhythmia
In general, arrhythmias develop when abnormal impulse formation and or abnormal automaticity encounter pathological conduction. A simple extrasystole may deteriorate into fibrillation when the excitatory wave front travels around an anatomical or functional obstacle back to its site of origin and encounters myocardium that is no longer refractory. The concept of reentry of the excitation wave front for maintenance of arrhythmias implies that shortening of effective refractory period and/or impaired conduction will shorten the wavelength of a reentry circuit thereby allowing more reentry circuits to be situated in a certain area of heart tissue. The ‘leading circle’ theory of reentry with multiple wavelets causing fibrillation (Allessie et al. 2002) is now supplemented by the idea of rotors. In this concept a number of driving sources (‘drivers’) give rise to excitatory wave fronts. The wave front and the tail of the wave are curved and meet each other in a central core around which the rotor ‘spins’ (see Pandit & Jalife, 2013 for recent review). With the help of extensive computer-based modelling and mapping these concepts help to guide ablation (Narayan et al. 2012).
Atrial fibrillation
Despite an enormous body of research on the pathophysiology of atrial fibrillation (AF), expertly reviewed by Schotten et al. (2011) and Wakili et al. (2011), the exact mechanism of AF remains unknown. Atrial fibrillation is initiated when abnormal excitation (abnormal automaticity, ectopic focus, drivers) triggers reentry in a vulnerable substrate. Typical sources of ectopic activity are early and late after-depolarizations caused and/or maintained by compromised cellular Ca2+ handling (Nattel & Dobrev, 2012). Abnormal automaticity is often found in intrinsically rapidly firing atrial cardiomyocytes located within the sleeves of myocardium extending into the pulmonary veins (Spach et al. 1972). Indeed, ablation of excitatory activity in the pulmonary veins can successfully terminate AF (Haissaguerre et al. 1998). Conversely, many pathological conditions such as ischaemia, infarction or heart failure maintain an arrhythmogenic substrate. A common denominator of these conditions is that they cause inflammation, extracellular matrix remodelling and fatty infiltrations. In fact, fibrosis, atrial dilatation and uncoupled gap junctions contribute to the pathological substrate in AF (Yue et al. 2011; Dobrev et al. 2012).
In an experimental setting and under many clinical circumstances, AF tends to become more resistant to conversion into sinus rhythm the longer the arrhythmia persists (Wijffels et al. 1995). Prolonged duration of AF leads to functional and structural changes in the atrial myocardium, that are considered to support maintenance of AF (‘remodelling’; for review see Allessie et al. 2002). Compelling evidence emerges that fibrosis is a major component of structural remodelling and by disrupting normal conduction patterns contributes to maintenance of AF (Burstein & Nattel, 2008; Schotten et al. 2011).
Electrical remodelling including altered regulation of many ion channels and transporters is responsible for the change of the characteristic ‘spike-and-dome’ shape of atrial action potentials into a triangular form (for review, see Dobrev & Ravens, 2003). Remodelling of Na+ channels in atrial fibrillation is modest: mRNA and maximum current density are between 10 to 20% lower in atrial myocardium from patients in permanent AF than in sinus rhythm (SR; Bosch et al. 1999; Wettwer et al. 2013). In our hands, AF does not alter Na+ channel availability (Toussaint et al. 2011). Most K+ channels are down-regulated both functionally and at the expression level, with exception of the inward rectifier IK1 which is in fact up-regulated (Dobrev & Ravens, 2003). Acetylcholine-activated K+ channels become constitutively active during AF-induced remodelling (Dobrev et al. 2005). Atrial fibrillation is also associated with remodelling of gap junctions (Polontchouk et al. 2001).
Last but not least, genetic factors appear to play a role in ‘lone’ atrial fibrillation, i.e. AF in the absence of any heart disease (for reviews see Lubitz et al. 2010 and Magnani et al. 2011). Mutations in ion channels that associate with familial AF are reported to induce both loss and gain of function. Some examples are: loss-of-function (Chen et al. 2007) but also gain-of-function mutations in Na+ channels as evidenced by association between LQT-3 syndrome and familial AF (Benito et al. 2008); loss or gain of function for Kv1.5 channels (Olson et al. 2006; Christophersen et al. 2012); gain of function in Kir2.1 channels (Xia et al. 2005; Deo et al. 2013); and loss of function for gap junction protein connexin 40 (Cx40) linked to intercellular communication (Firouzi et al. 2004; Sun et al. 2013), to name but a few.
Antiarrhythmic versus pro-arrhythmic drug effects
Conventional antiarrhythmic drugs have been classified into four groups according to their mechanism of action (Vaughan Williams, 1975): (1) Na+ channel blockers, (2) β-adrenoceptor (β-AR) blockers, (3) APD prolonging drugs, and (4) Ca2+ channel blockers. The principles of antiarrhythmic drug action include suppression of excitability and prolongation of ERP. Excitability is reduced by blocking Na+ channels (class 1), reducing β-adrenergic drive (class 2), and reversal of sinus tachycardia (class 2, class 4). Effective refractory period can be prolonged either by lengthening of APs (class 3), or by enhancing post-excitatory refractoriness (class 1). Though reduction in excitability and prologation of refractoriness are usually associated with antiarrhythmic outcome, each of these mechanisms also includes a certain propensity for becoming pro-arrhythmic. Na+ channel block slows conduction thereby promoting reentry; β-AR blockers and Ca2+ channel blockers cause bradycardia and atrioventricular (AV) block; extensive APD prolongation induced for instance by selective hERG (K+) channel blockers increases the risk for early afterdepolarizations leading to torsade de pointes, a polymorphic ventricular tachyarrhythmia that can easily turn into ventricular fibrillation (Curran et al. 1995; Mitcheson et al. 2000).
Atrial selective drug targets
New antiarrhythmic drugs for treatment of atrial fibrillation should be atrial selective in order to avoid pro-arrhythmic effects in the ventricles. Currently recognized atrial selective targets include atrial Nav1.5 channels, Kv1.5 channels and constitutively active Kir3.1/3.4 channels, each of which confers atrial selectivity by different mechanisms.
Na+ channels
In atrial tissue, a smaller fraction of Na+ channels is available for excitation at physiological heart rates than in ventricular tissue because of differences in resting membrane potential and steady-state inactivation of Na+ channels, at least in dog atria (Burashnikov et al. 2007). In man, voltage values for half-maximum Na+ channel inactivation are similar in atria and ventricles (Sakakibara et al. 1992, 1993), nevertheless the more depolarized membrane potential in atria is sufficient for lower Na+ channel availability in comparison with ventricle.
Na+ channels have a higher affinity for blocking drugs in their activated/inactivated than in their closed state (Hondeghem & Katzung, 1984; Starmer et al. 1984). Therefore, high frequencies favour drug binding during the APs and restrict drug dissociation during the short diastolic intervals. Because of the high activation rate in the fibrillating atria, rate-dependent Na+ channel blockers more strongly suppress atrial than ventricular Na+ current, because Na+ channels recover more completely during the long ventricular diastolic interval (Burashnikov & Antzelevitch, 2009).
When atrial excitation rate is normalized after effective conversion to sinus rhythm, diastolic interval increases so that drug dissociates from the channels, relieves Na+ channel block and at the same time reduces pro-arrhythmic side effects. The atrial-selective action of class 1 drugs is particularly prominent in rapidly dissociating, strongly potential-dependent drugs, because of the relatively depolarized atrial resting potential.
It has recently been proposed that permanent AF is associated with development of ‘late’INa (INa,late; Sossalla et al. 2010) caused by atrial Na+ channels which do not inactivate completely. This current was originally detected in ventricular cells of ischaemic, hypertrophic, or failing hearts from animals and humans (Undrovinas et al. 1999; Maltsev et al. 2007; Maltsev & Undrovinas, 2008; Zaza et al. 2008) or in congenital long QT syndrome (LQT-3; Bennett et al. 1995). Moreover, the antianginal drug ranolazine is supposed to block INa,late with higher potency than peak INa (Antzelevitch et al. 2004), suggesting atrial selectivity of the compound in atrial fibrillation. It must be noted, however, that this concept has been challenged (Toussaint et al. 2011) and therefore, the existence of INa,late in atrial cardiomyocytes requires confirmation.
Comparison of drug effects on canine and human atrial tissue
Despite their negative record in the cardiac arrhythmia suppression trial (CAST, Echt et al. 1991), class 1 drugs were found to be safe in patients without a history of heart disease for conversion of AF and maintenance of SR, which led to the concept of atrial selectivity (Burashnikov et al. 2007). Clinical class 1 antiarrhythmic drugs loose their efficacy as the arrhythmia proceeds from first episodes to long standing AF, possibly because of conduction disturbances due to fibrosis. Nevertheless, Na+ channels do not alter their sensitivity to block by vernakalant (Wettwer et al. 2013) or ranolazine (Toussaint et al. 2011).
Vernakalant was registered in 2011 as a mixed ion channel blocker for intravenous conversion of AF into SR. In a canine coronary perfused left atrial preparation, the drug exihibits frequency-dependent block of Na+ channels which causes post-excitation refractoriness. Vernakalant also elevates the plateau phase and increases action potential duration of canine atrial APs. In right atrial trabeculae from patients in SR, vernakalant shortens rather than prolongs APD at 90% repolarisation (APD90) and does not elevate the plateau (Fig. 1A and B). Ranolazine also exhibits frequency-dependent Na+ channel block. In canine and human atrial preparations the drug prolongs late repolarization and slightly shortens APD at 20% repolarization (APD20). The selective hERG channel blocker d,l-sotalol only prolongs APD90 in a reverse use-dependent manner and, again, the effects were similar in canine and human atrial tissues (Fig. 1A and B, unpublished observations).
Figure 1. Typical action potentials recorded from canine left (A) and human right atrial tissue (B) before and after exposure to various antiarrhythmic drugs.
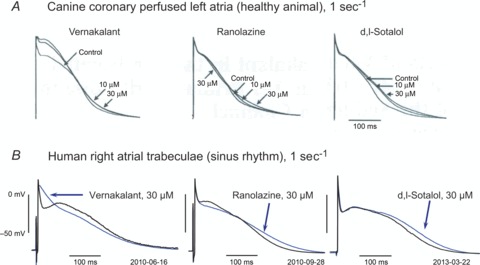
A, in healthy canine coronary perfused left atria stimulated at a rate of 2 s−1, vernakalant, ranolazine and the hERG channel blocker d,l-sotalol prolonged APD during late repolarization phases. Note that vernaklant elevated the plateau, ranolazine slightly shortened the plateau phase, and d,l-sotalol did not affect it at all. Reproduced from Burashnikov et al. (2012), with permission of the publisher. B, similar effects of ranolazine and dofetilide (as an alternative example of a selective hERG channel blocker) in isolated right atrial trabeculae from patients in SR (stimulation rate 1 s−1), but no plateau elevation with vernakalant (unpublished observations).
Despite its documented multi-ion channel actions, vernakalant was promoted as the first IKur blocker available for treatment of AF. Indeed, elevation of the plateau phase in healthy canine atria, is consistent with IKur block. In human atrial trabeculae from SR patients, however, vernakalant fails to elevate the plateau phase as in the canine preparation (Fig. 1A; Burashnikov et al. 2012; Wettwer et al. 2013). This difference suggests that, despite obvious similarity in shape of canine and human atrial APs, distinct K+ channel subtypes must contribute in each species. In fact, there is some controversy in the literature about whether IKur in dog is conducted via Kv3.1 or Kv1.2 rather than via Kv1.5 as in man (Nattel et al. 1999; Fedida et al. 2003).
In any case, although development of new drugs for AF cannot do without animal models (for review see Nishida et al. 2010), these do have their limitations concerning similarity to and reproducibility of human AF pathophysiology (Kirchhof et al. 2009) and uncritical extrapolation may lead to erroneous drug classification.
Kv1.5 channels
In 1991, Kv1.5 channels (HK2) encoded by KCNA5 were cloned from the human heart and found to be much more abundant in atria than in ventricles (Tamkun et al. 1991). When stably expressed in a mouse cell line, this channel conducts an ultrarapidly activating, outwardly rectifying current IKur (Snyders et al. 1993). In the same year, IKur was discovered in human atrial cardiomyocytes (Fedida et al. 1993; Wang et al. 1993), where it contributes to early repolarization of the action potential (AP). Since the current is absent in the human ventricle, IKur is considered as an atrial-selective drug target with antiarrhythmic potential. The anticipated antiarrhythmic mechanism of IKur blockers is prolongation of atrial action potential duration (APD) and effective refractory period (ERP) without any effect on QT-interval (Amos et al. 1996; Li et al. 1996). For these reasons, IKur has received a lot of interest as a potential drug target for the treatment of AF (for review see Ford & Milnes, 2008; Tamargo et al. 2009; Ravens, 2010; Ravens & Wettwer, 2011).
Block of repolarizing current is expected to prolong APD, and hence to terminate reentry by prolonging ERP. However, low concentrations of 4-aminopyridine (4-AP) which selectively block IKur elevate the AP plateau in human atrial tissue from patients in sinus rhythm, but shorten rather than prolong APD90. There is compelling evidence that the elevated plateau phase induced by the mixed Ito/IKur blocker AVE0118 activates NCX in its reverse mode contributing repolarizing current for abbreviation of APD90 (Schotten et al. 2007). Furthermore, increased activation of rapidly activated outward rectifying current IKr at more positive plateau potentials could also accelerate final repolarization (Gintant, 2000).
In contrast, 4-AP prolongs APD90 in tissue from patients in permanent AF (>6 months), and the same holds true for the IKur blockers AVE0118 and XEN-D0101 (Fig. 2; Wettwer et al. 2004; Schotten et al. 2007; Christ et al. 2008; Ford et al. 2013). Moreover, similar findings have been reported for dog atrial preparations (Burashnikov & Antzelevitch, 2008), whereas pig atrial APs were prolonged by 4-AP (Ehrlich et al. 2006). The differences in responses between SR and AF tissue are most likely due to profound AF-induced electrical remodelling (see Schotten et al. 2011 for review). In particular, down-regulation of IKur is expected to reduce the efficacy of IKur blockers in long-lasting AF.
Figure 2. Effects of various IKur blockers on plateau elevation and action potential duration in human right atrial trabeculae from patients in sinus rhythm (SR) and atrial fibrillation (AF).
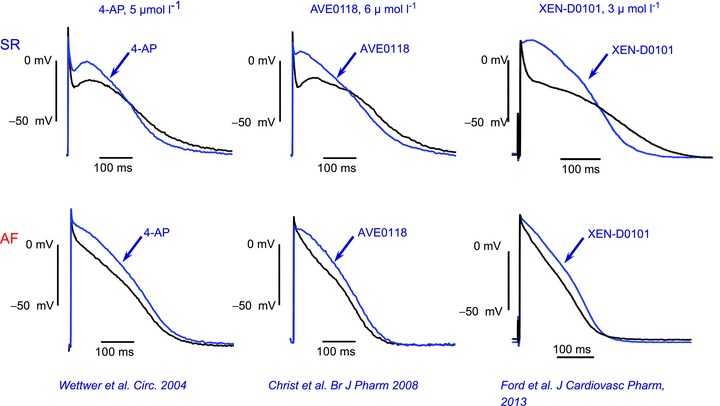
Stimulation rate 1 s−1, temperature 37°C. Note that all IKur blockers shorten action potential during the final phase of repolarization in SR preparations, whereas they prolong the action potential duration in AF preparations (unpublished observations). For experimental details see Christ et al. (2008), Ford et al. (2013) and Wettwer et al. (2013).
Numerous compounds have been screened for high Kv1.5 selectivity against all major cardiac ion channels, characterized electrophysiologically in isolated cardiomyocytes and cardiac tissue, and tested for their antiarrhythmic activity in various animal models of AF. Despite these efforts, proof-of-concept of antiarrhythmic efficacy in human is still lacking. In order to fill this gap, a ‘first-in-human’ study was recently reported with the highly selective IKur blocker MK-0448 (N-{6-[(1S)-1-(4-fluorophenyl)-2,2-di(pyridine-3-l)ethyl]pyridine2yl}methane sulphonamide; Pavri et al. 2012). Despite promising pre-clinical results with the compound, an invasive electrophysiological trial in healthy young male volunteers did not reveal any increase in atrial effective refractory period. Therefore the authors suggested that IKur block was likely to have limited value in the prevention of atrial fibrillation. However, it must be emphasized, that all electrophysiological testing was in a frequency range much below that of AF so that the question of whether or not IKur block is effective in pharmacological conversion of recent onset AF into SR and/or reducing AF burden by maintenance of SR remains unanswered.
IK,ACh (Kir 3.1/Kir 3.4 channels)
Acetylcholine (ACh)-regulated potassium current (IK,ACh) is conducted through G-protein activated inwardly rectifying K channels (GIRK1 and GIRK4) whose α-subunits are encoded by Kir3.1/Kir3.4 (for review see Yamada et al. 1998). The channels are more abundant in atrial than in ventricular muscle (Krapivinsky et al. 1995). They mediate AF induced by vagal stimulation via activation of muscarinic M2 receptors. In knockout mice lacking this channel, vagal stimulation is no longer able to trigger AF (Kovoor et al. 2001). Activation of IK,ACh hyperpolarizes the membrane and shortens atrial action potentials, thereby contributing to maintenance of AF by promoting reentry (reduced wavelength) and/or stabilizing rotors (negative membrane potential). Although GIRK channels are down regulated in long-term AF, (Dobrev et al. 2001), they can contribute to basal inward rectification due to development of constitutive activity, i.e. channel activation in the absence of any M2 receptor ligand (Dobrev et al. 2005). For these reasons, block of IK,ACh is considered an interesting target for AF therapy.
Tertiapin-Q (TQ) is a derivative of the 21-amino acid large peptide toxin tertiapin of honey bee venom, in which the original methionine residue is replaced with a glutamine residue (Jin & Lu, 1999). In the atrial tissue, TQ inhibits Kir3 channels with high selectivity over background inward rectifier Kir2 channels (Jin & Lu, 1999) and also suppresses constitutively active IK,ACh at low-nanomolar concentrations (Cha et al. 2006), as the congener tertiapin does in human cells (Dobrev et al. 2005). Moreover, vagally induced AF in dog is terminated by intravenous application of TQ (Hashimoto et al. 2006). In our hands, TQ has little effect in multicellular preparations (Fig. 3): TQ (100 nmol l−1) slightly reversed the effect of 2 μmol l−1 carbachol, but did not prevent the AP shortening with 10 μmol l−1 carbachol. However, in isolated cardiomyocytes from SR patients, TQ (100 nmol l−1) fully reversed the effect of carbachol (2 μmol l−1) on APD. Possibly, the difference in responses to TQ between tissue and single cardiomyocytes can be accounted for by impaired diffusion of the peptide in intact tissue, although even larger peptides, like the sea anemone toxin ATX II which modulates Na+ channel inactivation, are perfectly effective in multicellular preparations (Ravens, 1976).
Figure 3. Effects of carbachol and tertiapin-Q (TQ) on human atrial action potentials and IK,ACh.
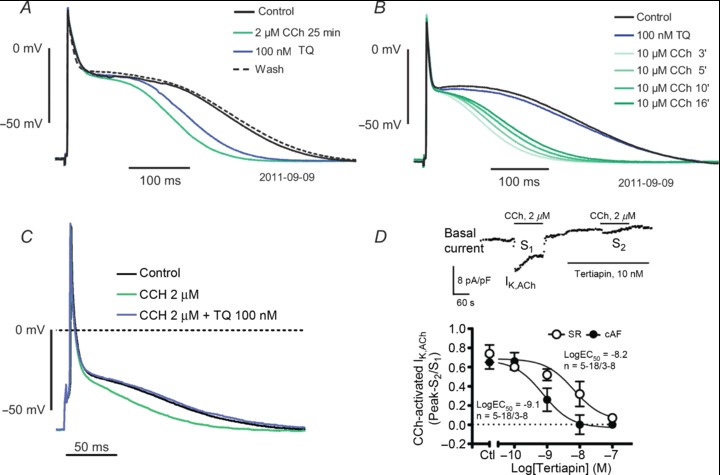
A and B, action potentials (stimulation rate 1 s−1) recorded in trabeculae. C, action potentials recorded in an isolated cardiomyocytes from a SR patient. Note, that TQ (100 nmol l−1) hardly reversed or prevented the APD shortening induced by carbachol in multicellular preparations, but fully reversed the carbachol effect in isolated cells (unpublished observations). D, inhibitory effect of tertiapin on actelcholine-activated current in atrial cardiomyocytes from patients in SR and AF. Inset; inward rectifier current (basal current) activated by ramp clamp steps was analysed at −100 mV. The carbachol (2 μmol l−1) stimulated current increase (IK,ACh) which was fully reversible upon washout, was suppressed after exposure to tertiapin in a concentration-dependent manner. Reproduced from Dobrev et al. (2005), with permission of the publisher.
Several antiarrhythmic agents including azimilide, dofetilide, dronedarone, ibutilide, sotalol and terikalant are known to block IK,ACh (Mori et al. 1995; Altomare et al. 2000; Nishida et al. 2007), and hence this effect may contribute to their efficacy in AF. In dogs, TQ prolongs action potential duration and suppresses inducibility of AF episodes (Cha et al. 2006). In addition to their major antiarrhythmic action, propafenone (class 1), dofetilide (class 3), flecainide (class 1) and AVE0118 (non-selective IKur blocker) impair IK,ACh, yet only flecainide and AVE0118 appear to also suppress constitutive activity of ACh-activated K+ channels (Voigt et al. 2010).
The benzopyrane derivative NIP-142 selectively blocks IK,ACh, thereby reversing the shortening effect of carbachol or adenosine on guinea-pig action potentials, and these effects are confined to the atria (Matsuda et al. 2006). Indeed, NIP-142 inhibits vagally induced atrial fibrillation. The congener NIP-151 blocks IK,ACh with a potency that is more than 4 orders of magnitude higher than its block of IKr and is highly effective in two canine AF models, i.e. aconitine- or vagal nerve stimulation-induced AF (Hashimoto et al. 2008). The authors conclude that because it is less likely to induce pro-arrhythmia than IKr blockers NIP-151 might be useful for the treatment of AF.
Although many drugs have IK,ACh blocking properties, selective IK,ACh blockade has only recently been reported using the compound NTC-801 which was suggested to exert antifibrillatory action by atrial-selective prolongation of effective refractory period (Machida et al. 2011). This new compound has been tested in expression systems and is effective against AF induced by vagal nerve stimulation, and also in aconitine- and rapid atrial pacing-induced AF. No data are as yet available for native human tissue.
‘Novel’ ion channels
Increasing evidence suggests that various additional ion channels might contribute to the cardiac action potential, e.g. two-pore-domain potassium (K2P) channels, small conductance Ca2+-activated K+ channels, mechano-sensitive channels could serve as drug targets in AF (see Ravens, 2010), however, little is known about their atrial selectivity and whether or not their pharmacological modulation is indeed effective in AF. Recently, some members of the family of transient receptor potential (TRP) channels have been shown to relate to the pathology of AF. In particular, TRPM7 and TRPC3 channels are involved in controlling atrial fibroblasts activation by regulating Ca2+ entry (Du et al. 2010; Harada et al. 2012; Yue et al. 2013). Drugs targeting these channels may be beneficial in AF because of their anti-fibrotic potential.
Gap junctions, connexins
Facilitating conduction by improving intercellular communication via gap junctions is an important new antiarrhythmic principle (Dhein et al. 2010). Discovery of antiarrhythmic peptides (Aonuma et al. 1980) and their synthetic modulation has spurred the development of the stable hexapeptide rotagatipe (ZP123: Kjolbye et al. 2003).
AF induced enhanced lateral expression of connexin 43 (Cx43) and Cx40, together with enhanced transverse conduction velocity in left atrial tissue. Alterations in localization of Cx43 and conduction changes were both antagonized by metoprolol, showing that pharmacological modulation of gap junction remodelling seems, in principle, possible.
Conclusion
While this review focuses on ion channels as targets for new drugs in AF therapy, future directions have to take a more holistic approach, with the main goal of prevention of AF onset by effective treatment of underlying cardiovascular diseases. In this context biomarkers indicating risk of developing AF may prove of great benefit. Various atrial-selective targets for antiarrhythmic drugs against AF have been identified and novel compounds are being developed. Although these drugs are effective in animal models, clinical benefit in converting AF into SR or reducing AF burden remains to be demonstrated for selective IKur blockers and IK,ACh blockers. In conclusion, we would like to adapt and restate the title of the classic publication about antiarrhythmic drugs (Hondeghem & Snyders, 1990): atrial-selective drugs have a lot of potential, but a long way to go.
Acknowledgments
We wish to thank Konstanze Fischer and Annegret Häntzschel for expert technical assistance.
Glossary
- ACh
acetylcholine
- AF
atrial fibrillation
- APD
action potential duration
- Cx
connexin
- ERP
effective refractory period
- GIRK
G-protein activated, inward rectifying K+ channel
- IK,ACh
acetylcholine-regulated potassium current
- IKur
ultrarapidly activating, outwardly rectifying current
- Ito
transient outward current
- INa,late
late sodium current
- LQT-3
long QT syndrome 3
- NCX
Na+–Ca2+ exchanger
- SR
sinus rhythm
- TQ
tertiapin-Q
- TRP
transient receptor potential (channels)
Additional information
Competing interests
None.
Author contributions
U.R., conception, design, and writing of the review; C.P., analysis of data, writing and critical reading; E.W., conception, design and critical revision; M.K., conception, design and critical reading. All authors approved the final version of the manuscript.
Funding
U.R. gratefully acknowledges funding by Fondation Leducq (07 CVD 03, ‘Leducq European-North American Atrial Fibrillation Research Alliance’), the German Federal Ministry of Education and Research (Atrial Fibrillation Competence Network, member of the steering committee); and the EU FP7-Health-2010-single-stage ‘EUTRAF’ (European Network for Translational Research in Atrial Fibrillation; no. 216057).
References
- Allessie M, Ausma J, Schotten U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc Res. 2002;54:230–246. doi: 10.1016/s0008-6363(02)00258-4. [DOI] [PubMed] [Google Scholar]
- Altomare C, Barbuti A, Viscomi C, Baruscotti M, DiFrancesco D. Effects of dronedarone on acetylcholine-activated current in rabbit SAN cells. Br J Pharmacol. 2000;130:1315–1320. doi: 10.1038/sj.bjp.0703446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amos GJ, Wettwer E, Metzger F, Li Q, Himmel HM, Ravens U. Differences between outward currents of human atrial and subepicardial ventricular myocytes. J Physiol. 1996;491:31–50. doi: 10.1113/jphysiol.1996.sp021194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, Di Diego JM, Fish JM, Cordeiro JM, Thomas G. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation. 2004;110:904–910. doi: 10.1161/01.CIR.0000139333.83620.5D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aonuma S, Kohama Y, Akai K, Komiyama Y, Nakajima S, Wakabayashi M, Makino T. Studies on heart. XIX. Isolation of an atrial peptide that improves the rhythmicity of cultured myocardial cell clusters. Chem Pharm Bull (Tokyo) 1980;28:3332–3339. doi: 10.1248/cpb.28.3332. [DOI] [PubMed] [Google Scholar]
- Benito B, Brugada R, Perich RM, Lizotte E, Cinca J, Mont L, Berruezo A, Tolosana JM, Freixa X, Brugada P, Brugada J. A mutation in the sodium channel is responsible for the association of long QT syndrome and familial atrial fibrillation. Heart Rhythm. 2008;5:1434–1440. doi: 10.1016/j.hrthm.2008.07.013. [DOI] [PubMed] [Google Scholar]
- Bennett PB, Yazawa K, Makita N, George AL., Jr Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- Bosch RF, Zeng X, Grammer JB, Popovic K, Mewis C, Kuhlkamp V. Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc Res. 1999;44:121–131. doi: 10.1016/s0008-6363(99)00178-9. [DOI] [PubMed] [Google Scholar]
- Burashnikov A, Antzelevitch C. Can inhibition of IKur promote atrial fibrillation. Heart Rhythm. 2008;5:1304–1309. doi: 10.1016/j.hrthm.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burashnikov A, Antzelevitch C. Atrial-selective sodium channel block for the treatment of atrial fibrillation. Expert Opin Emerg Drugs. 2009;14:233–249. doi: 10.1517/14728210902997939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burashnikov A, Di Diego JM, Zygmunt AC, Belardinelli L, Antzelevitch C. Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation. 2007;116:1449–1457. doi: 10.1161/CIRCULATIONAHA.107.704890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burashnikov A, Pourrier M, Gibson JK, Lynch JJ, Antzelevitch C. Rate-dependent effects of vernakalant in the isolated non-remodeled canine left atria are primarily due to block of the sodium channel: comparison with ranolazine and DL-sotalol. Circ Arrhythm Electrophysiol. 2012;5:400–408. doi: 10.1161/CIRCEP.111.968305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol. 2008;51:802–809. doi: 10.1016/j.jacc.2007.09.064. [DOI] [PubMed] [Google Scholar]
- Camm AJ, Lip GY, De CR, Savelieva I, Atar D, Hohnloser SH, Hindricks G, Kirchhof P. 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation – developed with the special contribution of the European Heart Rhythm Association. Europace. 2012;14:1385–1413. doi: 10.1093/europace/eus305. [DOI] [PubMed] [Google Scholar]
- Cha TJ, Ehrlich JR, Chartier D, Qi XY, Xiao L, Nattel S. Kir3-based inward rectifier potassium current: potential role in atrial tachycardia remodeling effects on atrial repolarization and arrhythmias. Circulation. 2006;113:1730–1737. doi: 10.1161/CIRCULATIONAHA.105.561738. [DOI] [PubMed] [Google Scholar]
- Chen LY, Ballew JD, Herron KJ, Rodeheffer RJ, Olson TM. A common polymorphism in SCN5A is associated with lone atrial fibrillation. Clin Pharmacol Ther. 2007;81:35–41. doi: 10.1038/sj.clpt.6100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ T, Wettwer E, Voigt N, Hala O, Radicke S, Matschke K, Varro A, Dobrev D, Ravens U. Pathology-specific effects of the IKurItoIK,ACh blocker AVE0118 on ion channels in human chronic atrial fibrillation. Br J Pharmacol. 2008;154:1619–1630. doi: 10.1038/bjp.2008.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christophersen IE, Olesen MS, Liang B, Andersen MN, Larsen AP, Nielsen JB, Haunso S, Olesen SP, Tveit A, Svendsen JH, Schmitt N. Genetic variation in KCNA5: impact on the atrial-specific potassium current IKur in patients with lone atrial fibrillation. Eur Heart J. 2012;34:1517–1525. doi: 10.1093/eurheartj/ehs442. [DOI] [PubMed] [Google Scholar]
- Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- Deo M, Ruan Y, Pandit SV, Shah K, Berenfeld O, Blaufox A, Cerrone M, Noujaim SF, Denegri M, Jalife J, Priori SG. KCNJ2 mutation in short QT syndrome 3 results in atrial fibrillation and ventricular proarrhythmia. Proc Natl Acad Sci U S A. 2013;110:4291–4296. doi: 10.1073/pnas.1218154110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhein S, Hagen A, Jozwiak J, Dietze A, Garbade J, Barten M, Kostelka M, Mohr FW. Improving cardiac gap junction communication as a new antiarrhythmic mechanism: the action of antiarrhythmic peptides. Naunyn Schmiedebergs Arch Pharmacol. 2010;381:221–234. doi: 10.1007/s00210-009-0473-1. [DOI] [PubMed] [Google Scholar]
- Dobrev D, Carlsson L, Nattel S. Novel molecular targets for atrial fibrillation therapy. Nat Rev Drug Discov. 2012;11:275–291. doi: 10.1038/nrd3682. [DOI] [PubMed] [Google Scholar]
- Dobrev D, Friedrich A, Voigt N, Jost N, Wettwer E, Christ T, Knaut M, Ravens U. The G protein-gated potassium current IK,ACh is constitutively active in patients with chronic atrial fibrillation. Circulation. 2005;112:3697–3706. doi: 10.1161/CIRCULATIONAHA.105.575332. [DOI] [PubMed] [Google Scholar]
- Dobrev D, Graf E, Wettwer E, Himmel HM, Hala O, Doerfel C, Christ T, Schuler S, Ravens U. Molecular basis of downregulation of G-protein-coupled inward rectifying K+ current (IK,ACh) in chronic human atrial fibrillation: decrease in GIRK4 mRNA correlates with reduced IK,ACh and muscarinic receptor-mediated shortening of action potentials. Circulation. 2001;104:2551–2557. doi: 10.1161/hc4601.099466. [DOI] [PubMed] [Google Scholar]
- Dobrev D, Ravens U. Remodeling of cardiomyocyte ion channels in human atrial fibrillation. Basic Res Cardiol. 2003;98:137–148. doi: 10.1007/s00395-003-0409-8. [DOI] [PubMed] [Google Scholar]
- Du J, Xie J, Zhang Z, Tsujikawa H, Fusco D, Silverman D, Liang B, Yue L. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ Res. 2010;106:992–1003. doi: 10.1161/CIRCRESAHA.109.206771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echt DS, Liebson PR, Mitchell LB, Peters RW, Obias-Manno D, Barker AH, Arensberg D, Baker A, Friedman L, Greene HL, et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med. 1991;324:781–788. doi: 10.1056/NEJM199103213241201. [DOI] [PubMed] [Google Scholar]
- Ehrlich JR, Hoche C, Coutu P, Metz-Weidmann C, Dittrich W, Hohnloser SH, Nattel S, Gögelein H. Properties of a time-dependent potassium current in pig atrium: evidence for a role of Kv1.5 in repolarization. J Pharmacol Exp Ther. 2006;319:898–906. doi: 10.1124/jpet.106.110080. [DOI] [PubMed] [Google Scholar]
- Ehrlich JR, Nattel S, Hohnloser SH. Novel anti-arrhythmic drugs for atrial fibrillation management. Curr Vasc Pharmacol. 2007;5:185–195. doi: 10.2174/157016107781024073. [DOI] [PubMed] [Google Scholar]
- Fedida D, Eldstrom J, Hesketh JC, Lamorgese M, Castel L, Steele DF, Van Wagoner DR. Kv1.5 is an important component of repolarizing K+ current in canine atrial myocytes. Circ Res. 2003;93:744–751. doi: 10.1161/01.RES.0000096362.60730.AE. [DOI] [PubMed] [Google Scholar]
- Fedida D, Wible B, Wang Z, Fermini B, Faust F, Nattel S, Brown AM. Identity of a novel delayed rectifier current from human heart with a cloned K+ channel current. Circ Res. 1993;73:210–216. doi: 10.1161/01.res.73.1.210. [DOI] [PubMed] [Google Scholar]
- Firouzi M, Ramanna H, Kok B, Jongsma HJ, Koeleman BP, Doevendans PA, Groenewegen WA, Hauer RN. Association of human connexin40 gene polymorphisms with atrial vulnerability as a risk factor for idiopathic atrial fibrillation. Circ Res. 2004;95:e29–e33. doi: 10.1161/01.RES.0000141134.64811.0a. [DOI] [PubMed] [Google Scholar]
- Ford J, Milnes J, Wettwer E, Christ T, Rogers M, Sutton K, Madge D, Virag L, Jost N, Horvath Z, Matschke K, Varro A, Ravens U. Human electrophysiological and pharmacological properties of XEN-D0101: A novel atrial selective Kv1.5/IKur inhibitor. J Cardiovasc Pharmacol. 2013;61:408–419. doi: 10.1097/FJC.0b013e31828780eb. [DOI] [PubMed] [Google Scholar]
- Ford JW, Milnes JT. New drugs targeting the ardiac ultra-rapid delayed-rectifier current (IKur): rationale, pharmacology and evidence for potential therapeutic value. J Cardiovasc Pharmacol. 2008;52:105–120. doi: 10.1097/FJC.0b013e3181719b0c. [DOI] [PubMed] [Google Scholar]
- Gintant GA. Characterization and functional consequences of delayed rectifier current transient in ventricular repolarization. Am J Physiol Heart Circ Physiol. 2000;278:H806–H817. doi: 10.1152/ajpheart.2000.278.3.H806. [DOI] [PubMed] [Google Scholar]
- Haissaguerre M, Jais P, Shah DC, Takahashi A, Hocini M, Quiniou G, Garrigue S, Le Mouroux A, Le Metayer P, Clementy J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. 1998;339:659–666. doi: 10.1056/NEJM199809033391003. [DOI] [PubMed] [Google Scholar]
- Harada M, Luo X, Qi XY, Tadevosyan A, Maguy A, Ordog B, Ledoux J, Kato T, Naud P, Voigt N, Shi Y, Kamiya K, Murohara T, Kodama I, Tardif JC, Schotten U, Van Wagoner DR, Dobrev D, Nattel S. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation. 2012;126:2051–2064. doi: 10.1161/CIRCULATIONAHA.112.121830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto N, Yamashita T, Tsuruzoe N. Tertiapin, a selective IKACh blocker, terminates atrial fibrillation with selective atrial effective refractory period prolongation. Pharmacol Res. 2006;54:136–141. doi: 10.1016/j.phrs.2006.03.021. [DOI] [PubMed] [Google Scholar]
- Hashimoto N, Yamashita T, Tsuruzoe N. Characterization of in vivo and in vitro electrophysiological and antiarrhythmic effects of a novel IKACh blocker, NIP-151: a comparison with an IKr-blocker dofetilide. J Cardiovasc Pharmacol. 2008;51:162–169. doi: 10.1097/FJC.0b013e31815e854c. [DOI] [PubMed] [Google Scholar]
- Hondeghem LM, Katzung BG. Antiarrhythmic agents: the modulated receptor mechanism of action of sodium and calcium channel-blocking drugs. Annu Rev Pharmacol Toxicol. 1984;24:387–423. doi: 10.1146/annurev.pa.24.040184.002131. [DOI] [PubMed] [Google Scholar]
- Hondeghem LM, Snyders DJ. Class III antiarrhythmic agents have a lot of potential but a long way to go. Reduced effectiveness and dangers of reverse use dependence. Circulation. 1990;81:686–690. doi: 10.1161/01.cir.81.2.686. [DOI] [PubMed] [Google Scholar]
- Jin W, Lu Z. Synthesis of a stable form of tertiapin: a high-affinity inhibitor for inward-rectifier K+ channels. Biochemistry. 1999;38:14286–14293. doi: 10.1021/bi991205r. [DOI] [PubMed] [Google Scholar]
- Kirchhof P, Bax J, Blomstrom-Lundquist C, Calkins H, Camm AJ, Cappato R, Cosio F, Crijns H, Diener HC, Goette A, Israel CW, Kuck KH, Lip GY, Nattel S, Page RL, Ravens U, Schotten U, Steinbeck G, Vardas P, Waldo A, Wegscheider K, Willems S, Breithardt G. Early and comprehensive management of atrial fibrillation: executive summary of the proceedings from the 2nd AFNET-EHRA consensus conference ‘research perspectives in AF’. Eur Heart J. 2009;30:2969–77c. doi: 10.1093/eurheartj/ehp235. [DOI] [PubMed] [Google Scholar]
- Kjolbye AL, Knudsen CB, Jepsen T, Larsen BD, Petersen JS. Pharmacological characterization of the new stable antiarrhythmic peptide analog Ac-D-Tyr-D-Pro-D-Hyp-Gly-D-Ala-Gly-NH2 (ZP123): in vivo and in vitro studies. J Pharmacol Exp Ther. 2003;306:1191–1199. doi: 10.1124/jpet.103.052258. [DOI] [PubMed] [Google Scholar]
- Kovoor P, Wickman K, Maguire CT, Pu W, Gehrmann J, Berul CI, Clapham DE. Evaluation of the role of IKACh in atrial fibrillation using a mouse knockout model. J Am Coll Cardiol. 2001;37:2136–2143. doi: 10.1016/s0735-1097(01)01304-3. [DOI] [PubMed] [Google Scholar]
- Krapivinsky G, Gordon EA, Wickman K, Velimirovic B, Krapivinsky L, Clapham DE. The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature. 1995;374:135–141. doi: 10.1038/374135a0. [DOI] [PubMed] [Google Scholar]
- Li GR, Feng J, Yue L, Carrier M, Nattel S. Evidence for two components of delayed rectifier K+ current in human ventricular myocytes. Circ Res. 1996;78:689–696. doi: 10.1161/01.res.78.4.689. [DOI] [PubMed] [Google Scholar]
- Lubitz SA, Ozcan C, Magnani JW, Kaab S, Benjamin EJ, Ellinor PT. Genetics of atrial fibrillation: implications for future research directions and personalized medicine. Circ Arrhythm Electrophysiol. 2010;3:291–299. doi: 10.1161/CIRCEP.110.942441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida T, Hashimoto N, Kuwahara I, Ogino Y, Matsuura J, Yamamoto W, Itano Y, Zamma A, Matsumoto R, Kamon J, Kobayashi T, Ishiwata N, Yamashita T, Ogura T, Nakaya H. Effects of a highly selective acetylcholine-activated K+ channel blocker on experimental atrial fibrillation. Circ Arrhythm Electrophysiol. 2011;4:94–102. doi: 10.1161/CIRCEP.110.951608. [DOI] [PubMed] [Google Scholar]
- Magnani JW, Rienstra M, Lin H, Sinner MF, Lubitz SA, McManus DD, Dupuis J, Ellinor PT, Benjamin EJ. Atrial fibrillation: current knowledge and future directions in epidemiology and genomics. Circulation. 2011;124:1982–1993. doi: 10.1161/CIRCULATIONAHA.111.039677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA, Silverman N, Sabbah HN, Undrovinas AI. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: implications for repolarization variability. Eur J Heart Fail. 2007;9:219–227. doi: 10.1016/j.ejheart.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA, Undrovinas A. Late sodium current in failing heart: friend or foe. Prog Biophys Mol Biol. 2008;96:421–451. doi: 10.1016/j.pbiomolbio.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, Ito M, Ishimaru S, Tsuruoka N, Saito T, Iida-Tanaka N, Hashimoto N, Yamashita T, Tsuruzoe N, Tanaka H, Shigenobu K. Blockade by NIP-142, an antiarrhythmic agent, of carbachol-induced atrial action potential shortening and GIRK1/4 channel. J Pharmacol Sci. 2006;101:303–310. doi: 10.1254/jphs.fp0060324. [DOI] [PubMed] [Google Scholar]
- Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci U S A. 2000;97:12329–12333. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Hara Y, Saito T, Masuda Y, Nakaya H. Anticholinergic effects of class III antiarrhythmic drugs in guinea pig atrial cells. Different molecular mechanisms. Circulation. 1995;91:2834–2843. doi: 10.1161/01.cir.91.11.2834. [DOI] [PubMed] [Google Scholar]
- Narayan SM, Krummen DE, Enyeart MW, Rappel WJ. Computational mapping identifies localized mechanisms for ablation of atrial fibrillation. PLoS One. 2012;7:e46034. doi: 10.1371/journal.pone.0046034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattel S, Dobrev D. The multidimensional role of calcium in atrial fibrillation pathophysiology: mechanistic insights and therapeutic opportunities. Eur Heart J. 2012;33:1870–1877. doi: 10.1093/eurheartj/ehs079. [DOI] [PubMed] [Google Scholar]
- Nattel S, Yue L, Wang Z. Cardiac ultrarapid delayed rectifiers: a novel potassium current family of functional similarity and molecular diversity. Cell Physiol Biochem. 1999;9:217–226. doi: 10.1159/000016318. [DOI] [PubMed] [Google Scholar]
- Nishida A, Reien Y, Ogura T, Uemura H, Tamagawa M, Yabana H, Nakaya H. Effects of azimilide on the muscarinic acetylcholine receptor-operated K+ current and experimental atrial fibrillation in guinea-pig hearts. J Pharmacol Sci. 2007;105:229–239. doi: 10.1254/jphs.fp0070940. [DOI] [PubMed] [Google Scholar]
- Nishida K, Michael G, Dobrev D, Nattel S. Animal models for atrial fibrillation: clinical insights and scientific opportunities. Europace. 2010;12:160–172. doi: 10.1093/europace/eup328. [DOI] [PubMed] [Google Scholar]
- Olson TM, Alekseev AE, Liu XK, Park S, Zingman LV, Bienengraeber M, Sattiraju S, Ballew JD, Jahangir A, Terzic A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet. 2006;15:2185–2191. doi: 10.1093/hmg/ddl143. [DOI] [PubMed] [Google Scholar]
- Pandit SV, Jalife J. Rotors and the dynamics of cardiac fibrillation. Circ Res. 2013;112:849–862. doi: 10.1161/CIRCRESAHA.111.300158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavri BB, Greenberg HE, Kraft WK, Lazarus N, Lynch JJ, Salata JJ, Bilodeau MT, Regan CP, Stump G, Fan L, Mehta A, Wagner JA, Gutstein DE, Bloomfield D. MK-0448, a specific Kv1.5 inhibitor: Safety, pharmacokinetics and pharmacodynamic electrophysiology in experimental animal models and in humans. Circ Arrhythm Electrophysiol. 2012;5:1193–1201. doi: 10.1161/CIRCEP.111.969782. [DOI] [PubMed] [Google Scholar]
- Polontchouk L, Haefliger JA, Ebelt B, Schaefer T, Stuhlmann D, Mehlhorn U, Kuhn-Regnier F, De Vivie ER, Dhein S. Effects of chronic atrial fibrillation on gap junction distribution in human and rat atria. J Am Coll Cardiol. 2001;38:883–891. doi: 10.1016/s0735-1097(01)01443-7. [DOI] [PubMed] [Google Scholar]
- Ravens U. Electromechanical studies of an Anemonia sulcata toxin in mammalian cardiac muscle. Naunyn Schmiedebergs Arch Pharmacol. 1976;296:73–78. doi: 10.1007/BF00498842. [DOI] [PubMed] [Google Scholar]
- Ravens U. Antiarrhythmic therapy in atrial fibrillation. Pharmacol Ther. 2010;128:129–145. doi: 10.1016/j.pharmthera.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Ravens U, Wettwer E. Ultra-rapid delayed rectifier channels: molecular basis and therapeutic implications. Cardiovasc Res. 2011;89:776–785. doi: 10.1093/cvr/cvq398. [DOI] [PubMed] [Google Scholar]
- Sakakibara Y, Furukawa T, Singer DH, Jia H, Backer CL, Arentzen CE, Wasserstrom JA. Sodium current in isolated human ventricular myocytes. Am J Physiol Heart Circ Physiol. 1993;265:H1301–H1309. doi: 10.1152/ajpheart.1993.265.4.H1301. [DOI] [PubMed] [Google Scholar]
- Sakakibara Y, Wasserstrom JA, Furukawa T, Jia H, Arentzen CE, Hartz RS, Singer DH. Characterization of the sodium current in single human atrial myocytes. Circ Res. 1992;71:535–546. doi: 10.1161/01.res.71.3.535. [DOI] [PubMed] [Google Scholar]
- Savelieva I, Camm J. Anti-arrhythmic drug therapy for atrial fibrillation: current anti-arrhythmic drugs, investigational agents, and innovative approaches. Europace. 2008;10:647–665. doi: 10.1093/europace/eun130. [DOI] [PubMed] [Google Scholar]
- Schotten U, de Haan S, Verheule S, Harks EG, Frechen D, Bodewig E, Greiser M, Ram R, Maessen J, Kelm M, Allessie M, Van Wagoner DR. Blockade of atrial-specific K+-currents increases atrial but not ventricular contractility by enhancing reverse mode Na+/Ca2+-exchange. Cardiovasc Res. 2007;73:37–47. doi: 10.1016/j.cardiores.2006.11.024. [DOI] [PubMed] [Google Scholar]
- Schotten U, Verheule S, Kirchhof P, Goette A. Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol Rev. 2011;91:265–325. doi: 10.1152/physrev.00031.2009. [DOI] [PubMed] [Google Scholar]
- Snyders DJ, Tamkun MM, Bennett PB. A rapidly activating and slowly inactivating potassium channel cloned from human heart. Functional analysis after stable mammalian cell culture expression. J Gen Physiol. 1993;101:513–543. doi: 10.1085/jgp.101.4.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossalla S, Kallmeyer B, Wagner S, Mazur M, Maurer U, Toischer K, Schmitto JD, Seipelt R, Schondube FA, Hasenfuss G, Belardinelli L, Maier LS. Altered Na+ currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J Am Coll Cardiol. 2010;55:2330–2342. doi: 10.1016/j.jacc.2009.12.055. [DOI] [PubMed] [Google Scholar]
- Spach MS, Barr RC, Jewett PH. Spread of excitation from the atrium into thoracic veins in human beings and dogs. Am J Cardiol. 1972;30:844–854. doi: 10.1016/0002-9149(72)90009-4. [DOI] [PubMed] [Google Scholar]
- Starmer CF, Grant AO, Strauss HC. Mechanisms of use-dependent block of sodium channels in excitable membranes by local anaesthetics. Biophys J. 1984;46:15–27. doi: 10.1016/S0006-3495(84)83994-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Yang YQ, Gong XQ, Wang XH, Li RG, Tan HW, Liu X, Fang WY, Bai D. Novel germline GJA5/Connexin40 mutations associated with lone atrial fibrillation impair gap junctional intercellular communication. Hum Mutat. 2013;34:603–609. doi: 10.1002/humu.22278. [DOI] [PubMed] [Google Scholar]
- Tamargo J, Caballero R, Gomez R, Delpon E. IKur/Kv1.5 channel blockers for the treatment of atrial fibrillation. Expert Opin Investig Drugs. 2009;18:399–416. doi: 10.1517/13543780902762850. [DOI] [PubMed] [Google Scholar]
- Tamkun MM, Knoth KM, Walbridge JA, Kroemer H, Roden DM, Glover DM. Molecular cloning and characterization of two voltage-gated K+ channel cDNAs from human ventricle. FASEB J. 1991;5:331–337. doi: 10.1096/fasebj.5.3.2001794. [DOI] [PubMed] [Google Scholar]
- Toussaint D, Christ T, Wettwer E, Ravens U. Late sodium current as a promising antiarrhythmic drug target for treatment of atrial fibrillation. Naunyn Schmiedebergs Arch Pharmacol. 2011;383:61. [Google Scholar]
- Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci. 1999;55:494–505. doi: 10.1007/s000180050306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan Williams EM. Classification of antidysrhythmic drugs. Pharmacol Ther B. 1975;1:115–138. doi: 10.1016/0306-039x(75)90019-7. [DOI] [PubMed] [Google Scholar]
- Voigt N, Rozmaritsa N, Trausch A, Zimniak T, Christ T, Wettwer E, Matschke K, Dobrev D, Ravens U. Inhibition of IK,ACh current may contribute to clinical efficacy of class I and class III antiarrhythmic drugs in patients with atrial fibrillation. Naunyn Schmiedebergs Arch Pharmacol. 2010;381:251–259. doi: 10.1007/s00210-009-0452-6. [DOI] [PubMed] [Google Scholar]
- Wakili R, Voigt N, Kaab S, Dobrev D, Nattel S. Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Invest. 2011;121:2955–2968. doi: 10.1172/JCI46315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Fermini B, Nattel S. Sustained depolarization-induced outward current in human atrial myocytes. Evidence for a novel delayed rectifier K+ current similar to Kv1.5 cloned channel currents. Circ Res. 1993;73:1061–1076. doi: 10.1161/01.res.73.6.1061. [DOI] [PubMed] [Google Scholar]
- Wettwer E, Christ T, Endig S, Rozmaritsa N, Matschke K, Lynch JJ, Pourrier M, Gibson JK, Fedida D, Knaut M, Ravens U. The new antiarrhythmic drug vernakalant: ex vivo study of human atrial tissue from sinus rhythm and chronic atrial fibrillation. Cardiovasc Res. 2013;98:145–154. doi: 10.1093/cvr/cvt006. [DOI] [PubMed] [Google Scholar]
- Wettwer E, Hala O, Christ T, Heubach JF, Dobrev D, Knaut M, Varro A, Ravens U. Role of IKur in controlling action potential shape and contractility in the human atrium: influence of chronic atrial fibrillation. Circulation. 2004;110:2299–2306. doi: 10.1161/01.CIR.0000145155.60288.71. [DOI] [PubMed] [Google Scholar]
- Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation. 1995;92:1954–1968. doi: 10.1161/01.cir.92.7.1954. [DOI] [PubMed] [Google Scholar]
- Xia M, Jin Q, Bendahhou S, He Y, Larroque MM, Chen Y, Zhou Q, Yang Y, Liu Y, Liu B, Zhu Q, Zhou Y, Lin J, Liang B, Li L, Dong X, Pan Z, Wang R, Wan H, Qiu W, Xu W, Eurlings P, Barhanin J, Chen Y. A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem Biophys Res Commun. 2005;332:1012–1019. doi: 10.1016/j.bbrc.2005.05.054. [DOI] [PubMed] [Google Scholar]
- Yamada M, Inanobe A, Kurachi Y. G protein regulation of potassium ion channels. Pharmacol Rev. 1998;50:723–760. [PubMed] [Google Scholar]
- Yue L, Xie J, Nattel S. Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovasc Res. 2011;89:744–753. doi: 10.1093/cvr/cvq329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z, Zhang Y, Xie J, Jiang J, Yue L. Transient receptor potential (TRP) channels and cardiac fibrosis. Curr Top Med Chem. 2013;13:270–282. doi: 10.2174/1568026611313030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaza A, Belardinelli L, Shryock JC. Pathophysiology and pharmacology of the cardiac “late sodium current”. Pharmacol Ther. 2008;119:326–339. doi: 10.1016/j.pharmthera.2008.06.001. [DOI] [PubMed] [Google Scholar]