

Abstract
Many diverse data support a role of the funny current (If) in pacemaking and heart rate control. Among them, clinically relevant applications have special impact, since they translate the concept of funny channel-based pacemaking into practical developments of potential therapeutic value. For example, the If role in pacemaking is the basis for a pharmacological approach to heart rate control. Ivabradine, a specific f-channel blocker acting as a ‘pure heart rate reducing’ agent, is used today as a therapeutic tool in chronic stable angina and heart failure. Also, the pacemaking capability of funny channels has been the main rationale behind the development of ‘biological’ pacemakers, whose aim is to eventually replace the electronic pacemakers implanted today. Finally, the role of If in pacemaking implies that dysfunctional funny channels can contribute to rhythm disorders. This consideration has led to the search for inheritable forms of cardiac arrhythmias potentially linked to functional changes of HCN4 channels, the molecular correlates of funny channels. This review addresses recent reports where investigation of families with arrhythmias has allowed the identification of specific HCN4 channel mutations associated with different types of rhythm disorders, specifically with sinus bradycardia. The perspective of future research in this field is also addressed.
 |
Dario DiFrancesco is Professor of Physiology at the University of Milano where he heads The PaceLab in the Department of Biosciences. His research focuses on basic and clinically-relevant properties of the cardiac pacemaker (funny) current, which he and coworkers first described in 1979. Decades of investigation have established the role of f-channels in cardiac pacemaking and heart rate control. Recent interest in f-channels has grown because of their relevance to development of “biological” pacemakers, pharmacological control of heart rate by rate-reducing agents such as ivabradine, marketed as therapeutic tool against angina and heart failure, and familial types of f-channel-dependent arrhythmias.
Introduction
It is well established that the role played by the funny current in the heart relates principally to the generation and control of pacemaker activity in the sinoatrial node (SAN) region, although the precise extent to which f-channels contribute to determine pacemaking and rate control within the framework of the more complex set of participating mechanisms is still fiercely debated (Lakatta & DiFrancesco, 2009).
Among a whole variety of experimental results and clinically relevant applications demonstrating the contribution of If to pacemaking (Barbuti et al. 2007; DiFrancesco, 2010; DiFrancesco & Noble, 2012), genetic evidence has grown rapidly in the last few years that mutations in HCN4, the hyperpolarization-activated, cyclic-nucleotide-gated (HCN) channel isoform expressed in pacemaker tissue, are associated with rhythm disorders.
It also appears from experimental data that the role of f-channels need not be confined to the control of normal spontaneous activity in the primary pacemaker region of the heart. For example, progressive reduction of f-channel expression in pacemaker cells of cardiac-specific, inducible HCN4 knockout mice eventually leads to lethal atrioventricular block, as well as the expected slowing of SAN rate, suggesting a previously unsuspected role of HCN4 in atrioventricular node function (Baruscotti et al. 2011). Also, overexpression of If in ventricular cells from human failing hearts in dilated or ischaemic cardiomyopathy represents a potentially important pro-arrhythmogenic mechanism (Cerbai et al. 2001; Stillitano et al. 2008). Finally, it is known that ectopic beats originating in pulmonary veins are important focal sources that can initiate atrial fibrillation (Haissaguerre et al. 1998; Chen et al. 1999); pulmonary veins contain f-channel-expressing pacemaker cells which contribute to ectopic beat generation and arrhythmogenesis (Chen et al. 2009; Suenari et al. 2012), suggesting that dysfunctional f-channels can be involved in atrial fibrillation.
It is therefore important to investigate by candidate- gene screening if mutations of the HCN4 gene affecting channel function are associated with arrhythmias. Here I discuss recent reports showing that this association exists, and that identified HCN4 mutations may indeed contribute to rhythm disorders such as asymptomatic and symptomatic sinus bradycardia, tachycardia–bradycardia syndrome, atrial fibrillation, chronotropic incompetence and more complex arrhythmias.
Dysfunctional HCN4 mutations and rhythm disorders
L573X
Early evidence indicating a potential association between HCN4 mutations and irregular cardiac rhythm involved a 66-year-old woman with severe sinus bradycardia, intermittent atrial fibrillation (AF) and chronotropic incompetence (Schulze-Bahr et al. 2003). Genetic screening identified a heterozygous mutation in HCN4 consisting of a single base deletion (1631 delC), which generated a truncated protein (HCN4-573X). Residue 573 is in the C-linker (Fig. 1), implying that mutated proteins lack the cyclic nucleotide-binding domain (CNBD). As expected, mutated channels did not respond to cAMP (Fig. 2). Surprisingly, heteromeric wild-type-mutant channels were also cAMP insensitive. This result contrasts with previous data indicating that cotransfection of cAMP-responding and cAMP-non-responding subunits leads to an intermediate cAMP response; this has been shown for example with the cotransfection of HCN4 and HCN1 isoforms (Altomare et al. 2003) or of wild-type and cAMP-insensitive, R591E mutant HCN2 channel subunits (Ulens & Siegelbaum, 2003).
Figure 1.
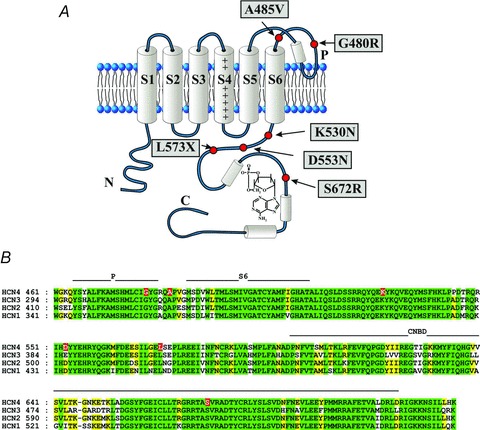
A, schematic diagram of HCN topology showing one of the four channel subunits. The six transmembrane domains (S1–S6) and the intracellular N- and C-termini are shown. The C-terminus incorporates the cyclic nucleotide binding domain (CNBD) and a bound cAMP molecule. Also shown are approximate positions of HCN4 mutations reported to be linked to rhythm disorders. B, sequence alignment of the four human HCN isoforms from the pore (P) domain through to the CNBD. The position of the mutated HCN4 residues are also indicated (red background).
Figure 2.
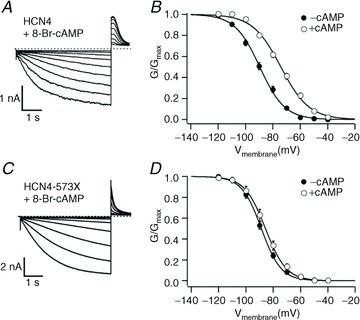
Sample traces (left) and plots of IHCN4 activation curves (right) measured in COS-7 cells showing that 573X mutant HCN4 channels (C and D), contrary to wild-type channels (A and B), did not respond to cAMP. From Schulze-Bahr et al. 2003. Reproduced with permission of the American Society for Clinical Investigation.
A limit of this study is that an extensive pedigree investigation of the family of this patient was not performed. With data referring to a single patient, familial inheritance could not be examined and it was not possible to positively correlate the arrhythmic phenotype with the HCN4 mutation.
D553N
A second report investigated a small family with a complex set of cardiac disorders (Ueda et al. 2004). Three members of this family were found to carry the HCN4 heterozygous mutation D553N (Fig. 1). The proband suffered from recurrent syncope, severe bradycardia, long QT (LQT), polymorphic ventricular tachycardia and torsade de pointes. Two other family members with the mutation had LQT, but whether they also shared other symptoms among the array of disorders manifest in the proband is not clear. Functional studies showed that mutant channels have a reduced membrane expression relative to wild-type channels, suggesting that D553N is a loss-of-function mutation interfering in a dominant-negative manner with the correct protein trafficking to the plasma membrane. This interpretation was supported by the decreased current recorded when transfecting heterozygous wild-type/mutant channels (Fig. 3A). Surprisingly, the mutation caused a rightward shift in the time constant curve, but did not cause any shift in the voltage dependence of activation (Fig. 3C).
Figure 3.
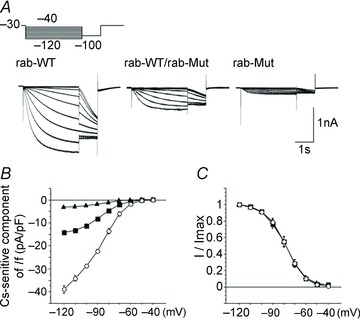
Sample traces (A) and mean current–voltage relations of IHCN4 currents (B) recorded from COS-7 cells expressing wild-type (open circles), homomeric D553N mutant (filled triangles) and heterozygous wild-type/mutant channels (filled squares), showing the reduction of current associated with the mutation. The mutation did not affect the position of the activation curve (C), although it did modify the time constant curve (not shown). From Ueda et al. 2004. Reproduced with permission of the American Society for Biochemistry and Molecular Biology.
A limit of this study is that in the presence of the complex array of symptoms of the proband and in the absence of a clear indication of which specific aspect was common to all mutation carriers, it is difficult to establish a direct correlation between the D553N mutation and the severity and complexity of the rhythm disorders described. Thus, while the loss-of-function change attributed to a trafficking defect can rationally be linked to bradycardia, it is not obviously linked to other symptoms involving ventricular features like LQT, polymorphic ventricular tachycardia or torsade de pointes. These more complex arrhythmias might arise, for example, from a mutation in an additional ion channel.
S672R
According to Buraei & Yang, 2012, ‘the first HCN4 mutation to be unequivocally identified as a direct cause of arrhythmia’ was the heterozygous mutation S672R described in 2006 in a family with asymptomatic sinus bradycardia (Milanesi et al. 2006; Fig. 4). The mutation was expressed in a substantial fraction of the family (15 out of 27 members); importantly, all the individuals with the mutation had low resting heart rates (≤60 beats min−1, mean rate = 52.2 beats min−1), while all the wild-type individuals had normal rates (>60 beats min−1, mean rate = 73.2 beats min−1) (Fig. 4A). These results indicated that expression of the mutation and the bradycardic phenotype were highly correlated; the LOD (logarithm of odds) score of the correlation was indeed very high (5.47).
Figure 4.
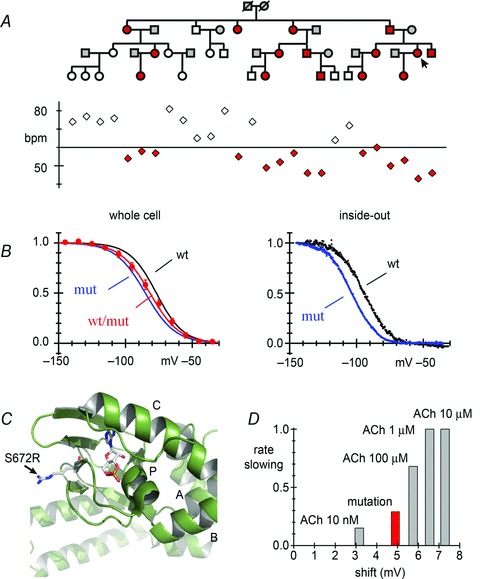
A, top, pedigree of a family with asymptomatic sinus bradycardia (arrow: index patient). Red symbols represent individuals with the heterozygous HCN4 mutation S672R, white symbols individuals with wild-type HCN4. A, bottom, resting heart rates of individuals with (red) or without the mutation (white) plotted in correspondence with A, top. B, mean activation curves of IHCN4 recorded from HEK293 cells in whole-cell (left) and inside-out patch clamp experiments (right) upon expression of wild-type (black), homomeric S672R mutant (blue) and heteromeric wild-type/mutant channels (red, only whole-cell), as indicated. Relative to wild-type channels, the shift in whole-cell experiments was 8.4 mV for homomeric mutant and 4.9 mV for heteromeric wild-type/mutant channels; in inside-out experiments a similar shift was obtained (9.1 mV for homomeric mutant channels). C, model reconstruction of the C-linker–CNBD of HCN4 showing the position of residue S672 (mutated to R); S572R and cAMP molecule drawn in stick mode. α-helices of the CNBD (A, B, C and P) are also indicated. The reconstruction was based on a published HCN4 CNBD crystal structure (PDB entry: 3U11, Lolicato et al. 2011). D, plot comparing the mutation-induced bradycardia with the rate slowing caused by ACh. The graph plots the slowing of rate caused by different ACh concentrations and that caused by the S672R mutation against the shift of the If activation curve induced by the same agents. ACh data from DiFrancesco et al. 1989. Plots in A and B are modified from Milanesi et al. 2006. Copyright © 2006 Massachusetts Medical Society. Reprinted with permission.
S672 is a highly conserved residue of the CNBD of the channel, located near the cAMP-binding pocket (Fig. 4C). This suggested that the mutation could affect cAMP-induced activation (DiFrancesco & Tortora, 1991) of the channel. However, functional studies showed that wild-type and mutant channels had similar responses to cAMP, implying a similar cAMP dependence; instead, mutant channels had a more negative activation voltage range than wild-type channels (Fig. 4B), indicating intrinsic new gating properties of the mutated channel. S672R can be considered, therefore, as a loss-of-function mutation which causes a decreased inward current flow during diastole, explaining the slower than normal heart rate of individuals with the mutation.
Shifting of the voltage dependence of current activation to more negative voltages is an action similar to that induced by ACh and consequent inhibition of adenylate cyclase and reduction of cAMP synthesis (DiFrancesco & Tromba, 1988; DiFrancesco et al. 1989). In fact, it is possible to verify that the S672R-induced negative shift of 4.9 mV is adequate to account for the slowing observed (29%). This can be done by comparing these data with existing data on the effects of ACh on the If activation curve and rate slowing in single cells (DiFrancesco et al. 1989). In Fig. 4D, the fractional rate slowing is plotted against the shift of the If activation curve corresponding to the same ACh concentration (range 10 nm to 10 μm, grey bars). Plotting in the same graph the mutation-induced shift and the corresponding slowing measured in the bradycardic family (red bar) shows that these values fit well the trend of the curve; these values are in fact intermediate between those caused by 10 and 100 nm ACh, a range of concentrations within which slowing is attributable to If inhibition (DiFrancesco et al. 1989). The plot of Fig. 4 supports the view that the mutation-associated shift is fully reponsible for the bradycardic phenotype.
A recent report (Xu et al. 2012) has confirmed the shifting effect of the mutation, in agreement with Milanesi et al. 2006 data, but has proposed that the shift is caused by a reduced cAMP binding to the CNBD and consequent decreased cAMP sensitivity, rather than by constitutive structural modifications. The results from the two studies cannot apparently be reconciled, although several significant differences exist in the experimental settings. A major difference concerns the types of channels investigated, since while Milanesi et al. used full-length human HCN4, Xu et al. used a chimaera obtained by replacing the C-terminus of mouse hcn2 with a portion of the human HCN4 C-terminus; these latter channels therefore did not faithfully reproduce HCN4 channels.
Differences in the results obtained in the two studies indeed support the notion of a different behaviour of the two types of channels. For example, activation curves of wild-type channels have substantially more negative V1/2 values in Xu et al.'s than in Milanesi et al.'s study (by some 10–15 mV in whole-cell and 20–35 mV in inside-out patches). Part of this difference may derive from the different protocols used to measure activation curves. While Milanesi et al. apply steps of variable duration, lasting longer at depolarized than hyperpolarized voltages, so as to reach steady state at all levels, Xu et al. apply, instead, steps of fixed 3 s duration, which may not be sufficient in the more depolarized range. This protocol may lead to underestimation of the degree of activation at depolarized voltages, with a resulting negative shift of the activation curve.
However, this is unlikely to explain other discrepancies. Considering for example the cAMP dependence of the activation curve, K1/2 and the maximal shift from Hill fitting were 0.2 μm and 17.5 mV, respectively, for the chimaeric channel of Xu et al. and 1.53 μm and 9.8 mV, respectively, for HCN4 of Milanesi et al. It is also worth noting that the cAMP dependence of S672R-mutated mouse hcn2-h4 channels, by predicting significant activation only at high concentrations (10–100 μm), would imply a nearly complete lack of sensitivity at physiological cAMP levels. The clinical consequence of this feature would be chronotropic incompetence, a feature not found in the asymptomatic patients of Milanesi et al.'s study.
Taken together, these data point to substantial functional differences between the two types of channels. Clearly more experimental data are required to investigate these differences.
G480R and A485V
The heterozygous mutation G480R was found in a family with asymptomatic sinus bradycardia evident from a young age (Nof et al. 2007); affected members of the family did not have other rhythm or conduction abnormalities and had normal chronotropic competence. The mutation co-segregated with the bradycardic phenotype in most patients of the family.
Unlike the HCN4 mutations reported earlier, this was the first to occur in the extracellular pore (P)-loop of the channel (Fig. 1). More specifically, the mutation involves a glycine residue belonging to the GYG selectivity filter motif typical of K+-selective channels. This might suggest that the G480R mutation affects channel permeabilty and/or conductance. Indeed the mutation was found to cause a reduced current in heterologous expression experiments. However, the current carried by mutant G480R channels had the same reversal potential than wild-type channels, suggesting no significant change in the Na+/K+ permeability ratio; possible changes in the channel conductance were not investigated. Analysis by Western blot and electrophysiology indicated that the current decrease was attributable to a negative shift of the voltage dependence and slowing of current activation, and to a decreased synthesis and trafficking to the plasma membrane of mutated channels. The effect of the loss-of-function G480R mutation therefore confirms the idea that reduced HCN4 contribution to the activity of pacemaker cells slows the heart.
The same team (Laish-Farkash et al. 2010) later identified a second extracellular single-point mutation (A485V), located in the loop connecting P to S6 (Fig. 1), potentially associated with sinus bradycardia in unrelated individuals belonging to the same ethnic group of Moroccan Jews (Fig. 5). Unlike carriers of the G480R mutation, carriers of the A485V mutation were symptomatic with dizziness, presyncopal episodes and cardiac arrest, the latter occurring in one index patient.
Figure 5.
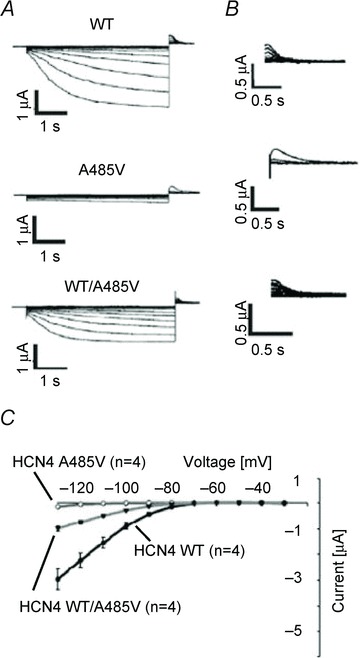
Sample activation (A) and deactivation traces (B) and mean current–voltage relations of IHCN4 currents (C) recorded from Xenopus oocytes expressing wild-type, homomeric A485V mutant and heterozygous wild-type/mutant channels, showing the reduction of current caused by the mutation. From Laish-Farkash et al. 2010. Reproduced with permission.
It is somewhat unexpected that despite the different symptoms, the A485V mutation is reported to affect channel function in ways similar to those found with G480R, i.e. a decrease in the synthesis and membrane expression of channel protein, with no change in ion selectivity, and a negative shift of the voltage range of current activation (Laish-Farkash et al. 2010).
To explain the modifications induced by the mutation on cardiac rhythm, including the fact that mutation carriers had a normal chronotropic and exercise capacity despite functional changes of mutated channels, the authors discussed possible effects of heteromeric assembly of wild-type and A485V mutant HCN4 channels, or even of heteromeric assembly comprising HCN2 subunits. A more precise interpretation of the functional effects of the A485V mutation will require a more in-depth analysis of the detailed biophysical and cellular changes associated with the mutation itself.
K530N
The screening of patients affected by atrial and/or ventricular tachyarrhythmias has recently led to the identification of a new mutation in the HCN4 C-linker (K530N) associated with familial tachycardia–bradycardia syndrome and atrial fibrillation (Duhme et al. 2012). Following identification of a proband with paroxysmal AF, several members of the same family with similar symptoms were found to carry the same heterozygous mutation, although one individual with the mutation had no symptoms.
Functional studies revealed that K530N is a loss-of-function mutation whose effect is to shift the current activation curve to more negative voltages (Fig. 6). An atypical property of this mutation is that a negative shift was observed in heterologous wild-type/mutant channels, which mimics the condition of individuals heterozygous for the mutation, but not in homomeric mutant channels, whose kinetic behaviour was indisinguishable from that of wild-type channels (Fig. 6B and C). The authors interpreted this surprising finding by assuming that homomeric wild-type or mutant channels have a more stable ‘tetrameric’ arrangement than heteromeric channels, where the ‘dimer of dimers’ configuration is favoured instead, leading to increased tonic inhibition of activity. However, no evidence was provided to support this hypothesis.
Figure 6.
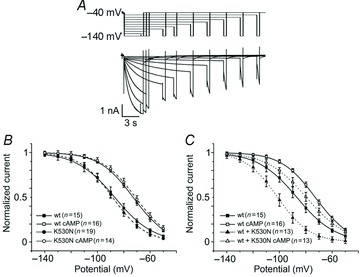
Sample traces (A) and mean current–voltage activation curves of IHCN4 currents measured in whole-cell recordings from HEK293 cells expressing wild-type, homomeric K530N mutant (B) or heterozygous wild-type/mutant channels (C) in the presence and in the absence of 10 mm cAMP in the patch pipette, as indicated. These data show that the activation curve of heteromeric wild-type/mutant, but not of homomeric mutant channels, was shifted to more negative voltages relative to wild-type channels. All types of channels responded to cAMP. From Duhme et al. 2012. Reproduced with permission.
As discussed above for the mutation D553N, it is unclear how a simple modification like a more negative range of current activation can be responsible for a complex phenotype such as the one observed in this family, especially because the same type of action exerted by other mutations (for example S672R) is associated with milder arrhythmic phenotypes. According to the authors, slow heart rate increases susceptibility to ectopic activity, particularly during adrenergic stress, and may eventually trigger AF onset by inducing premature beats or pulmonary vein activity and re-entry, especially under conditions of conduction heterogeneity in diseased/ageing heart. However, while highlighting an interesting, potential functional role of the f-channels in AF, this proposed mechanism remains speculative.
Conclusion
Recent studies investigating the association of HCN4 mutations with inheritable forms of arrhythmias have provided growing evidence for an important role of dysfunctional f-channels in rhythm disorders. These studies, along with evidence from the investigation of knockout animals (Baruscotti et al. 2011) and from the clinical use of the If blocker ivabradine (DiFrancesco & Camm, 2004; DiFrancesco & Borer, 2007; Cappato et al. 2012), have shown that as well as being involved in the generation and control of basal physiological rhythm of the SAN and autonomic rhythm control, proper If function is relevant to the maintenance of normal rhythmic activity in a broader context. Dysfunctional f-channels and/or altered f-channel expression can thus be associated with asymptomatic and symptomatic sinus bradycardia, tachycardia–bradycardia syndrome, atrioventricular node block, atrial fibrillation and contribute to more complex types of arrhythmias. For example, If has a pathophysiological role in hypertrophied ventricles of the failing human heart, where increased transcription of HCN channels predisposes to enhanced ventricular automaticity (Cerbai & Mugelli, 2006). Also, dysfunctional If may also have a role in atrial fibrillation originating from pulmonary veins, since f-channel-expressing pacemaker cells are present in pulmonary veins, where they can contribute to ectopic beat generation and atrial arrhythmogenesis (Chen et al. 2009; Suenari et al. 2012). Interestingly, a recent genome-wide association study of atrial fibrillation conducted on European individuals has identified, among others, a susceptibility locus in the first intron of HCN4 (Ellinor et al. 2012).
It is worth noticing that all HCN4 mutations described so far are loss-of-function mutations. It is likely that more mutations, perhaps including gain-of-function mutations, and/or mutations in HCN channel subunits and ancillary proteins, will be found to contribute to other types of arrhythmias in the near future.
Acknowledgments
This work was supported by the Italian Ministry of Education, University and Research grant PRIN 2010BWY8E9.
Glossary
- AF
atrial fibrillation
- CNBD
cyclic nucleotide-binding domain
- HCN
hyperpolarization-activated, cyclic-nucleotide-gated
- LQT
long QT
- SAN
sinoatrial node
References
- Altomare C, Terragni B, Brioschi C, Milanesi R, Pagliuca C, Viscomi C, Moroni A, Baruscotti M, DiFrancesco D. Heteromeric HCN1-HCN4 channels: a comparison with native pacemaker channels from the rabbit sinoatrial node. J Physiol. 2003;549:347–359. doi: 10.1113/jphysiol.2002.027698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbuti A, Baruscotti M, DiFrancesco D. The pacemaker current: from basics to the clinics. J Cardiovasc Electrophysiol. 2007;18:342–347. doi: 10.1111/j.1540-8167.2006.00736.x. [DOI] [PubMed] [Google Scholar]
- Baruscotti M, Bucchi A, Viscomi C, Mandelli G, Consalez G, Gnecchi-Rusconi T, Montano N, Casali KR, Micheloni S, Barbuti A, DiFrancesco D. Deep bradycardia and heart block caused by inducible cardiac-specific knockout of the pacemaker channel gene Hcn4. Proc Natl Acad Sci U S A. 2011;108:1705–1710. doi: 10.1073/pnas.1010122108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buraei Z, Yang J. Not very funny: how a single mutation causes heritable bradycardia. Structure. 2012;20:1991–1992. doi: 10.1016/j.str.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappato R, Castelvecchio S, Ricci C, Bianco E, Vitali-Serdoz L, Gnecchi-Ruscone T, Pittalis M, De Ambroggi L, Baruscotti M, Gaeta M, Furlanello F, DiFrancesco D, Lupo PP. Clinical efficacy of ivabradine in patients with inappropriate sinus tachycardia: a prospective, randomized, placebo-controlled, double-blind, crossover evaluation. J Am Coll Cardiol. 2012;60:1323–1329. doi: 10.1016/j.jacc.2012.06.031. [DOI] [PubMed] [Google Scholar]
- Cerbai E, Mugelli A. If in non-pacemaker cells: role and pharmacological implications. Pharmacol Res. 2006;53:416–423. doi: 10.1016/j.phrs.2006.03.015. [DOI] [PubMed] [Google Scholar]
- Cerbai E, Sartiani L, DePaoli P, Pino R, Maccherini M, Bizzarri F, DiCiolla F, Davoli G, Sani G, Mugelli A. The properties of the pacemaker current IF in human ventricular myocytes are modulated by cardiac disease. J Mol Cell Cardiol. 2001;33:441–448. doi: 10.1006/jmcc.2000.1316. [DOI] [PubMed] [Google Scholar]
- Chen SA, Hsieh MH, Tai CT, Tsai CF, Prakash VS, Yu WC, Hsu TL, Ding YA, Chang MS. Initiation of atrial fibrillation by ectopic beats originating from the pulmonary veins: electrophysiological characteristics, pharmacological responses, and effects of radiofrequency ablation. Circulation. 1999;100:1879–1886. doi: 10.1161/01.cir.100.18.1879. [DOI] [PubMed] [Google Scholar]
- Chen YC, Pan NH, Cheng CC, Higa S, Chen YJ, Chen SA. Heterogeneous expression of potassium currents and pacemaker currents potentially regulates arrhythmogenesis of pulmonary vein cardiomyocytes. J Cardiovasc Electrophysiol. 2009;20:1039–1045. doi: 10.1111/j.1540-8167.2009.01480.x. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D. The role of the funny current in pacemaker activity. Circ Res. 2010;106:434–446. doi: 10.1161/CIRCRESAHA.109.208041. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D, Borer JS. The funny current: cellular basis for the control of heart rate. Drugs. 2007;67(Suppl. 2):15–24. doi: 10.2165/00003495-200767002-00003. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D, Camm JA. Heart rate lowering by specific and selective If current inhibition with ivabradine: a new therapeutic perspective in cardiovascular disease. Drugs. 2004;64:1757–1765. doi: 10.2165/00003495-200464160-00003. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D, Ducouret P, Robinson RB. Muscarinic modulation of cardiac rate at low acetylcholine concentrations. Science. 1989;243:669–671. doi: 10.1126/science.2916119. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D, Noble D. The funny current has a major pacemaking role in the sinus node. Heart Rhythm. 2012;9:299–301. doi: 10.1016/j.hrthm.2011.09.021. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D, Tortora P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature. 1991;351:145–147. doi: 10.1038/351145a0. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D, Tromba C. Muscarinic control of the hyperpolarization-activated current (if) in rabbit sino-atrial node myocytes. J Physiol. 1988;405:493–510. doi: 10.1113/jphysiol.1988.sp017344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhme N, Schweizer PA, Thomas D, Becker R, Schroter J, Barends TR, Schlichting I, Draguhn A, Bruehl C, Katus HA, Koenen M. Altered HCN4 channel C-linker interaction is associated with familial tachycardia-bradycardia syndrome and atrial fibrillation. Eur Heart J. 2012 doi: 10.1093/eurheartj/ehs391. [DOI] [PubMed] [Google Scholar]
- Ellinor PT, Lunetta KL, Albert CM, Glazer NL, Ritchie MD, Smith AV, Arking DE, Muller-Nurasyid M, Krijthe BP, Lubitz SA, Bis JC, Chung MK, Dorr M, Ozaki K, Roberts JD, Smith JG, Pfeufer A, Sinner MF, Lohman K, Ding J, Smith NL, Smith JD, Rienstra M, Rice KM, Van Wagoner DR, Magnani JW, Wakili R, Clauss S, Rotter JI, Steinbeck G, Launer LJ, Davies RW, Borkovich M, Harris TB, Lin H, Volker U, Volzke H, Milan DJ, Hofman A, Boerwinkle E, Chen LY, Soliman EZ, Voight BF, Li G, Chakravarti A, Kubo M, Tedrow UB, Rose LM, Ridker PM, Conen D, Tsunoda T, Furukawa T, Sotoodehnia N, Xu S, Kamatani N, Levy D, Nakamura Y, Parvez B, Mahida S, Furie KL, Rosand J, Muhammad R, Psaty BM, Meitinger T, Perz S, Wichmann HE, Witteman JC, Kao WH, Kathiresan S, Roden DM, Uitterlinden AG, Rivadeneira F, McKnight B, Sjogren M, Newman AB, Liu Y, Gollob MH, Melander O, Tanaka T, Stricker BH, Felix SB, Alonso A, Darbar D, Barnard J, Chasman DI, Heckbert SR, Benjamin EJ, Gudnason V, Kaab S. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet. 2012;44:670–675. doi: 10.1038/ng.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haissaguerre M, Jais P, Shah DC, Takahashi A, Hocini M, Quiniou G, Garrigue S, Le Mouroux A, Le Metayer P, Clementy J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. 1998;339:659–666. doi: 10.1056/NEJM199809033391003. [DOI] [PubMed] [Google Scholar]
- Laish-Farkash A, Glikson M, Brass D, Marek-Yagel D, Pras E, Dascal N, Antzelevitch C, Nof E, Reznik H, Eldar M, Luria D. A novel mutation in the HCN4 gene causes symptomatic sinus bradycardia in Moroccan Jews. J Cardiovasc Electrophysiol. 2010;21:1365–1372. doi: 10.1111/j.1540-8167.2010.01844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakatta EG, DiFrancesco D. What keeps us ticking: a funny current, a calcium clock, or both. J Mol Cell Cardiol. 2009;47:157–170. doi: 10.1016/j.yjmcc.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolicato M, Nardini M, Gazzarrini S, Moller S, Bertinetti D, Herberg FW, Bolognesi M, Martin H, Fasolini M, Bertrand JA, Arrigoni C, Thiel G, Moroni A. Tetramerization dynamics of C-terminal domain underlies isoform-specific cAMP gating in hyperpolarization-activated cyclic nucleotide-gated channels. J Biol Chem. 2011;286:44811–44820. doi: 10.1074/jbc.M111.297606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanesi R, Baruscotti M, Gnecchi-Ruscone T, DiFrancesco D. Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N Engl J Med. 2006;354:151–157. doi: 10.1056/NEJMoa052475. [DOI] [PubMed] [Google Scholar]
- Nof E, Luria D, Brass D, Marek D, Lahat H, Reznik-Wolf H, Pras E, Dascal N, Eldar M, Glikson M. Point mutation in the HCN4 cardiac ion channel pore affecting synthesis, trafficking, and functional expression is associated with familial asymptomatic sinus bradycardia. Circulation. 2007;116:463–470. doi: 10.1161/CIRCULATIONAHA.107.706887. [DOI] [PubMed] [Google Scholar]
- Schulze-Bahr E, Neu A, Friederich P, Kaupp UB, Breithardt G, Pongs O, Isbrandt D. Pacemaker channel dysfunction in a patient with sinus node disease. J Clin Invest. 2003;111:1537–1545. doi: 10.1172/JCI16387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stillitano F, Lonardo G, Zicha S, Varro A, Cerbai E, Mugelli A, Nattel S. Molecular basis of funny current (If) in normal and failing human heart. J Mol Cell Cardiol. 2008;45:289–299. doi: 10.1016/j.yjmcc.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Suenari K, Cheng CC, Chen YC, Lin YK, Nakano Y, Kihara Y, Chen SA, Chen YJ. Effects of ivabradine on the pulmonary vein electrical activity and modulation of pacemaker currents and calcium homeostasis. J Cardiovasc Electrophysiol. 2012;23:200–206. doi: 10.1111/j.1540-8167.2011.02173.x. [DOI] [PubMed] [Google Scholar]
- Ueda K, Nakamura K, Hayashi T, Inagaki N, Takahashi M, Arimura T, Morita H, Higashiuesato Y, Hirano Y, Yasunami M, Takishita S, Yamashina A, Ohe T, Sunamori M, Hiraoka M, Kimura A. Functional characterization of a trafficking-defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J Biol Chem. 2004;279:27194–27198. doi: 10.1074/jbc.M311953200. [DOI] [PubMed] [Google Scholar]
- Ulens C, Siegelbaum SA. Regulation of hyperpolarization-activated HCN channels by cAMP through a gating switch in binding domain symmetry. Neuron. 2003;40:959–970. doi: 10.1016/s0896-6273(03)00753-0. [DOI] [PubMed] [Google Scholar]
- Xu X, Marni F, Wu S, Su Z, Musayev F, Shrestha S, Xie C, Gao W, Liu Q, Zhou L. Local and global interpretations of a disease-causing mutation near the ligand entry path in hyperpolarization-activated cAMP-gated channel. Structure. 2012;20:2116–2123. doi: 10.1016/j.str.2012.09.017. [DOI] [PubMed] [Google Scholar]