

Abstract
Congenital long QT syndrome (LQTS) is caused by single autosomal-dominant mutations in a gene encoding for a cardiac ion channel or an accessory ion channel subunit. These single mutations can cause life-threatening arrhythmias and sudden death in heterozygous mutation carriers. This recognition has been the basis for world-wide staggering numbers of subjects and families counselled for LQTS and treated based on finding (putative) disease-causing mutations. However, prophylactic treatment of patients is greatly hampered by the growing awareness that simple carriership of a mutation often fails to predict clinical outcome: many carriers never develop clinically relevant disease while others are severely affected at a young age. It is still largely elusive what determines this large variability in disease severity, where even within one pedigree, an identical mutation can cause life-threatening arrhythmias in some carriers while in other carriers no disease becomes clinically manifested. This suggests that additional factors modify the clinical manifestations of a particular disease-causing mutation. In this article, potential demographic, environmental and genetic factors are reviewed, which, in conjunction with a mutation, may modify the phenotype in LQTS, and thereby determine, at least partially, the large variability in disease severity.
 |
Arthur A.M. Wilde is a cardiologist-electrophysiologist and head of the Department of Clinical and Experimental Cardiology at the Academic Medical Centre (University of Amsterdam), Amsterdam, The Netherlands. He is a member of the Dutch Academy of Science. His research interest include the genetic basis of cardiac arrhythmias, ranging from the monogenetic diseases to more broader healthcare problems as sudden cardiac death in the setting of ischemic heart disease. Recent studies centre on genotype-phenotype relationships in these disorders and the role of clinical and genetic modifiers in establishing the phenotype.
Congenital long QT syndrome (LQTS) is a cardiac repolarization disease that is characterized by prolonged heart rate-corrected QT interval (QTc) on the electrocardiogram (ECG) and cardiac events (syncope, out-of-hospital cardiac arrest, and sudden cardiac death (SCD)) due to torsades de pointes ventricular tachycardia (TdP) and ventricular fibrillation (VF). The discovery of rare variants in genes encoding for cardiac ion channels as disease-causing mutations in families with LQTS in 1995 (Curran et al. 1995; Wang et al. 1995), more than 30 years after the first description of the disease (Romano et al. 1963; Ward 1964), created great interest for genetic studies in a few research laboratories across the world. This has resulted in the identification of hundreds of mutations in 14 LQTS-susceptibility genes in single individuals or families with LQTS (Table 1). In the last few years, genetic testing has become available for clinical use (Kapplinger et al. 2009) and this has been the basis for a rapidly increasing number of individuals and families worldwide being counselled for LQTS. However, the use of results from genetic testing to identify individuals at the highest risk for prophylactic treatment is greatly hampered by the growing awareness that carriership of a mutation often fails to predict clinical outcome, since many carriers remain asymptomatic (incomplete penetrance), some carriers only show QTc prolongation without cardiac events, while others experience serious cardiac events at a young age (variable expressivity). It remains largely elusive what determines this large variability in disease severity, where even family members that carry an identical mutation display large differences in phenotype and disease severity (Fig. 1). This suggests that additional factors modify the phenotypic/clinical manifestations of a particular disease-causing mutation. This article aims to review potential genetic and non-genetic factors (demographic and environmental) that, in conjunction with a mutation, may modify the phenotype in LQTS, and thereby determine, at least partially, the large variability in disease severity (Fig. 2).
Table 1.
Genetic basis of the long QT syndrome (LQTS)
| Type | Occurrence (or % of genotyped) | Gene | Protein | Protein function | Affected current | Reference |
|---|---|---|---|---|---|---|
| 1 | 42–54% | KCNQ1 | Kv7.1 | α-subunit IKs channel | IKs decrease | Wang et al. 1996 |
| 2 | 35–45% | KCNH2 | Kv11.1 | α-subunit IKr channel | IKr decrease | Curran et al. 1995; Sanguinetti et al. 1995 |
| 3 | 1.7–8% | SCN5A | Nav1.5 | α-subunit INa channel | INaL increase | Wang et al. 1995 |
| 4 | <1% | ANK2 | Ankyrin-B | Adaptor protein | None | Mohler et al. 2003 |
| 5 | <1% | KCNE1 | minK | β-subunit IKs channel | IKs decrease | Barhanin et al. 1996; Sanguinetti et al. 1996 |
| 6 | <1% | KCNE2 | MiRP1 | β-subunit IKr channel | IKr decrease | Abbott et al. 1999 |
| 7 | Rare | KCNJ2 | Kir2.1 | α-subunit IK1 channel | IK1 decrease | Plaster et al. 2001 |
| 8 | Rare | CACNA1C | Cav1.2 | α-subunit ICa,L channel | ICa,L increase | Splawski et al. 2004 |
| 9 | Rare | CAV3 | Caveolin-3 | Component of caveolae (co-localizes with Nav1.5) | INaL increase | Vatta et al. 2006 |
| 10 | <0.1% | SCN4B | β4 | β-subunit INa channel | INaL increase | Medeiros-Domingo et al. 2007 |
| 11 | Rare | AKAP9 | Yotiao | Mediates Kv7.1 phosphorylation | Inadequate IKs increase during β-adrenergic stimulation | Chen et al. 2007 |
| 12 | Rare | SNTA1 | α1-syntrophin | Regulates INa channel function | INaL increase | Ueda et al. 2008 |
| 13 | Rare | KCNJ5 | Kir3.4 | Subunit KACh channel | IKACh decrease | Yang et al. 2010 |
| 14 | Rare | CALM | Calmodulin | Calmodulin | Defective Ca2+ signalling | Crotti et al. 2013 |
Figure 1. Incomplete penetrance and variable expressivity.
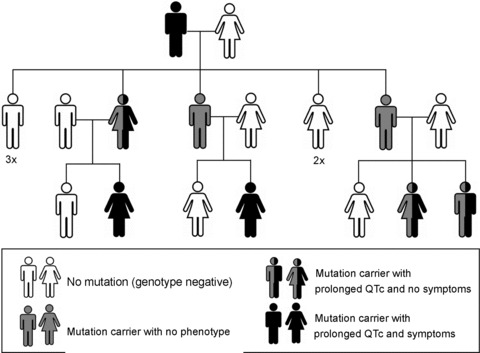
The structure of a representative multigenerational pedigree with type 1 long QT syndrome (LQT1) from the Academic Medical Center (AMC) in Amsterdam. LQT1 is caused by the IVS7+5G>A (c.842+5G>A) mutation in intron 7 of KCNQ1. The mutation displays 75% disease penetrance. Note the differences in disease severity (variable disease expressivity) between the genotype-positive (mutation-carrying) family members.
Figure 2. Modifiers of phenotype in long QT syndrome.

Schematic representation of genetic and non-genetic factors that are known to modify the phenotype in the long QT syndrome, thereby contributing to incomplete penetrance and variable expressivity. Straight line reflects a direct effect (e.g. direct channel block). Dashed line reflects an indirect effect (e.g. altering gene transcription, messenger RNA translation, or channel phosphorylation). 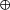 , stimulatory effect;
, stimulatory effect;  , inhibitory effect; 5′UTR, 5′ untranslated region; 3′UTR, 3′ untranslated region; M2-R, M2 muscarinic acetylcholine receptor; β-AR, β-adrenergic receptor; Nav1.5, INa channel protein; Kv11.1, IKr channel protein; Kv7.1, IKs channel protein; TNF, tumour necrosis factor; IL-1, interleukin-1.
, inhibitory effect; 5′UTR, 5′ untranslated region; 3′UTR, 3′ untranslated region; M2-R, M2 muscarinic acetylcholine receptor; β-AR, β-adrenergic receptor; Nav1.5, INa channel protein; Kv11.1, IKr channel protein; Kv7.1, IKs channel protein; TNF, tumour necrosis factor; IL-1, interleukin-1.
The ventricular repolarization
Ventricular repolarization, reflected by the QT interval on the ECG (Fig. 3A), is a complex process which results from a concerted interplay of ion channels or ion channel accessory subunits that are present (expressed) in the sarcolemma or in the cytoplasm. In individual myocytes, repolarization is the net result of inward depolarizing and outward repolarizing currents during phases 1–3 of the action potential (Fig. 3B). The cardiac Na+ channel (Nav1.5), encoded by SCN5A, is responsible for the depolarization during phase 0 of the action potential by permitting an inward current (INa). Phase 1 is accomplished by the repolarizing transient outward K+ current (ITO). Phase 2 reflects a balance between the depolarizing L-type inward Ca2+ current (ICa,L) and the repolarizing rapidly activating delayed outward rectifying currents (IKr) and the slowly activating delayed outward rectifying current (IKs). Phase 3 results from the predominance of the delayed outward rectifying currents after inactivation of the L-type Ca2+ channels. Repolarization ends by K+ efflux through the IK1 channels (Fig. 3C; Amin et al. 2010b).
Figure 3. The cardiac electrical activity and the long QT syndrome.
A, prolongation of the QT interval on the surface electrocardiogram. B, prolongation of the action potential duration. C, schematic representation of major inward and outward currents that contribute to action potential formation in ventricular myocytes in health (straight lines), and ion current dysfunctions linked to different types of long QT syndrome (dashed lines).
Mutations in genes encoding for ion channels or their accessory subunits are linked to different types of LQTS (Table 1). Approximately 90% of LQTS mutation carriers (i.e. genotype-positive patients) carry a mutation in KCNQ1 (type 1 LQTS; LQT1), KCNH2 (type 2 LQTS; LQT2), and SCN5A (type 3 LQTS; LQT3). Loss-of-function mutations in KCNQ1-encoded Kv7.1 channels and KCNH2-encoded Kv11.1 channels lead to a decrease in IKs and IKr, respectively. Normally, due to its fast inactivation (voltage-dependent closing), Nav1.5 does not conduct current (or only minimally) during the repolarization phases of the action potential. However, LQT3-linked mutations in SCN5A impair the fast inactivation of Nav1.5, and this results in a late (sustained or persistent) depolarizing Na+ current (INa,L). Thus, LQTS-linked mutations cause a decrease in the repolarizing currents (LQT1 and LQT2) or an increase in the depolarizing currents (LQT3), thereby delaying the repolarization of the ventricular action potential. This is reflected by QTc durations that are longer than designated as normal by international guidelines (i.e. in men ≥450 ms and in women ≥460 ms; Rautaharju et al. 2009). When the repolarization process is delayed long enough, Cav1.2 channels may be able to recover from inactivation and reactivate during phases 2 or 3 of the action potential, thereby triggering early afterdepolarizations (EADs), the cellular substrate for the initiation of TdP (Amin et al. 2010a).
Non-genetic factors
Age and sex
Age and sex are well-known modifiers of QTc duration and symptomatology in LQTS. Cardiac events in young patients (<20 years) may usually be triggered by adrenergically or vagally mediated stimuli (exercise, arousal, and/or sleep), while in older patients (≥40 years) environmental triggers (e.g. drugs or hypokalaemia) are more often required to express or aggravate the phenotype (Schwartz et al. 2001; Sakaguchi et al. 2008). Moreover, cohort studies have found an age–sex interaction, with male LQTS patients experiencing 90% of the first cardiac events before adolescence, while female patients experience their first events in the post-adolescence period (Locati et al. 1998). The mechanisms underlying the sex-related differences are still poorly understood. It is partially attributed to the effects of sex hormones on the expression of genes encoding cardiac ion channels and their accessory subunits. In female rabbit cardiomyocytes IK1 and IKr are smaller compared to male myocytes (Liu et al. 1998). Similarly, IK1 and IKs are less expressed in female guinea pig cardiomyocytes compared to male myocytes (Zicha et al. 2003; James et al. 2004). In line with animal data, messenger RNA (mRNA) and protein expression levels of channel subunits of IKr (Kv11.1), ITO (Kv1.4), IK1 (Kir2.3) and IKs (minK, the β-subunit) were lower in non-diseased transplant donor hearts of females versus males (Gaborit et al. 2010). In addition, direct (non-genomic) effects of sex hormones on cardiac ion currents are also reported. Testosterone rapidly shortens the action potential duration in guinea pig ventricular myocytes through enhancement of IKs and IK1, and inhibition of ICa,L (Bai et al. 2005). Progesterone shortens the action potential duration in guinea pig ventricular myocytes through enhancement of IKs under basal conditions and inhibition of ICa,L under cAMP-stimulated conditions (mimicking β-adrenergic activity; Nakamura et al. 2007). Conversely, oestrogen prolongs the action potential duration in guinea pig ventricular myocytes by suppression of ICa,L and inhibition of IKr and IKs (Tanabe et al. 1999; Kurokawa et al. 2008). Based on these data, we suggest that the net effect of sex hormones on the expression and function of cardiac ion channels is a lower repolarization reserve in women than in men, rendering women more prone to QTc prolongation and TdP occurrence in the presence of a LQTS-causing mutation.
The risk for cardiac events is reduced in women with LQT2 during pregnancy but significantly increased during the 9 month postpartum period (Khositseth et al. 2004). Risk is also higher in women with LQT2 during the perimenopausal period (Buber et al. 2011). It is not yet known why the postpartum period and the perimenopausal period are associated with increased arrhythmogenicity only in LQT2 (and not for example in LQT1). However, β-blockers reduce the risk for cardiac events during these high-risk periods (Seth et al. 2007; Buber et al. 2011).
The autonomic nervous system
It is well established that the autonomic nervous system influences the phenotype in LQTS. The duration of the ventricular repolarization in healthy hearts is shorter during increased sympathetic activity. This is mainly accomplished by an increase of IKs during β-adrenergic stimulation through Kv7.1 channel phosphorylation (Amin et al. 2010b). Genotype–phenotype correlations have shown differences in the effects of the autonomic nervous system on the phenotype in different types of LQTS. In LQT1, patients are at increased risk for cardiac events in conditions when sympathetic activity is elevated, i.e. during exercise (in particular during swimming; Schwartz et al. 2001). In LQT2, arousal by abrupt auditory triggers, such as the sound of a telephone or an alarm clock, trigger most events. In LQT3, cardiac events occur often during rest or sleep. Furthermore, onset of TdP differs among LQTS genotypes on the ECG level. TdP in LQT1 occurs usually at fast heart rates, while TdP in LQT2 is often preceded by a pause (68% in LQT2 versus 0% in LQT1; Tan et al. 2006), with the R–R interval immediately before TdP (pause interval) being significantly longer in LQT2 than in LQT1 patients. The mechanisms underlying the variable responses to the sympathetic nervous activity in different types of LQTS are not precisely known. Clinical studies indicate that β-adrenergic stimulation with adrenaline (epinephrine) infusion causes a persistent increase of QTc in LQT1, an initial transient (but dramatic) increase of QTc in LQT2 at peak adrenaline levels (with return of QTc to baseline levels at steady state), and a small and transient increase of QTc in LQT3 followed by shortening of QTc to baseline levels (or even below baseline levels) at steady state (Noda et al. 2002; Shimizu et al. 2003). Studies in arterially perfused wedge preparations of canine left ventricle have aimed to define these mechanisms by using β-adrenergic agonists and antagonists for sympathetic modulation and selective blockers of IKs, IKr or enhancers of INa,L for mimicking different types of LQTS (Shimizu & Antzelevitch, 1998, 2000). In the LQT1 model, β-adrenergic stimulation with isoproterenol (isoprenaline) resulted in a persistent prolongation of the QT interval and increase in the transmural dispersion of repolarization (TDR), due to regional (transmural) differences in the action potential duration, which facilitated occurrence of TdP. Of note, in the LQT1 model TDR was not increased under basal conditions, i.e. in the absence of isoproterenol. In contrast, in the LQT2 and LQT3 models, TDR was increased and TdP occurred under basal conditions. In the LQT2 model, isoproterenol caused an initial further prolongation of action potential duration and QT interval and also a further increase in TDP and TdP incidence. However, this was followed by a shortening of the action potential duration and QT interval and a decrease in TDR and TdP, most likely due to an adequate augmentation of IKs in the LQT2 model during continuous β-adrenergic stimulation. Finally, in the LQT3 model isoproterenol caused a decrease in TDP and TdP incidence. Persistent increase of the action potential duration and TDR during β-adrenergic stimulation is in line with the higher risk of cardiac events in LQT1 patients during exercise. However, based on the pause dependence of TdP in LQT2, and the transient dramatic increase of the action potential duration and TDR upon β-adrenergic stimulation, we assume that the occurrence of a ventricular ectopic beat upon sudden arousal initiates TdP in the LQT2 patients. In LQT3, decreased action potential duration and TDR during β-adrenergic stimulation supports the higher incidence of cardiac events in these patients during rest or sleep when the sympathetic activity is low. Finally, the above-mentioned data provide a rationale for the effectiveness of β-blockers to prevent events in LQT1 patients, and to a lesser extent in LQT2 patients (Schwartz et al. 2001).
Furthermore, recent studies suggest that differences in autonomic responses modify disease severity in LQT1. A study in 166 carriers of the A341V mutation in KCNQ1 showed that subjects with lower heart rates are at lower risk of developing symptoms (Brink et al. 2005). In addition, another study in 56 carriers of this mutation showed that the baroreflex sensitivity (measured as the response of blood pressure and heart rate to bolus phenylephrine injections) is lower in asymptomatic than symptomatic mutation carriers, suggesting that a weakened autonomic response is protective in LQT1 (Schwartz et al. 2008). Interestingly, higher baroreflex sensitivity in this cohort was associated with a polymorphism in the α2-adrenergic receptor gene resulting in loss of synaptic autoinhibitory feedback and thereby increased presynaptic release of noradrenaline (norepinephrine), and a polymorphism in the β1-adrenergic receptor gene leading to enhanced β1 activity.
Fever/hyperthermia
In 2008, we first described the repeated occurrence of fever-induced TdP and VF in two related LQT2 patients (father and son) with the A558P mutation in KCNH2 (Amin et al. 2008). ECG analysis showed increased QTc with fever in both patients. Molecular analysis revealed that the A558P mutation disrupts the intracellular trafficking of mutant Kv11.1 proteins, and (when co-expressed with normal Kv11.1 proteins) that the A558P mutant proteins exert dominant-negative effects on the intracellular trafficking of normal Kv11.1 proteins and reduce the temperature-dependent increase of the normal Kv11.1 current. Thus, the mutant Kv11.1 current did not increase to the same extent as the normal Kv11.1 current did at higher temperatures. These findings suggest that the blunted increase of mutant IKr in patients with LQT2 during fever, while the depolarizing ICa,L is significantly enhanced, upsets the balance between the depolarizing and the repolarizing forces in favour of depolarization, and thereby contributes to the development of QTc prolongation and TdP/VF at febrile temperatures (Amin et al. 2008). In line with our data, Burashnikov and colleagues showed that, when IKr is blocked by a Kv11.1 channel blocking agent, hyperthermia can induce EADs and TdP in canine left ventricle wedge preparations (Burashnikov et al. 2008). Together, these data imply that fever can trigger TdP/VF in conditions when the repolarization reserve of myocardium is compromised by defective K+ channels, as in patients with LQT1 and LQT2. However, this notion awaits further clinical confirmation.
Extracellular K+ concentration
Alterations in extracellular K+ concentrations ([K+]o), influence the QTc duration in healthy subjects and in patients with LQTS. The Nernst equation predicts smaller outward K+ currents during hyperkalaemia, i.e. when [K+]o is elevated. However, IK1 (at potentials higher than the resting membrane potential) and IKr display a ‘paradoxical’ increase when [K+]o is elevated. IKr increases as a result of a depolarizing shift in the voltage dependence of inactivation (i.e. towards more positive membrane potentials), whereas the effect of K+ on IK1 is complex and beyond the scope of this review (McAllister & Noble, 1966; Sanguinetti & Jurkiewicz, 1992; Choy et al. 1997). These findings suggest shortening of the action potential duration upon elevation of [K+]o. Indeed, intravenous K+ administration has been shown to decrease the QTc duration in seven LQT2 patients (Compton et al. 1996). However, it must be noted that the patients in the study of Dr Compton and colleagues had markedly prolonged QTc (mean ± SD 627 ± 92 ms), which is much longer than the mean QTc reported in large LQT2 cohorts (∼470 ms to ∼510 ms; Tester et al. 2005; Shimizu et al. 2009). The extreme QTc values in this study may have positively biased the effect of intravenous K+ administration because of the higher chance of spontaneous regression of QTc values towards the mean after the therapy (Morton & Torgerson, 2003). Furthermore, hypokalaemia has been associated with increased arrhythmogenesis in vivo (Sabir et al. 2007), and with further QTc prolongation and cardiac events in a relatively small cohort of LQTS patients (Sakaguchi et al. 2008).
QTc-prolonging drugs
Drugs possess the ability to unmask or aggravate the phenotype in patients with LQTS or to cause acquired LQTS (Sakaguchi et al. 2008; Kannankeril et al. 2010). The underlying mechanism is almost always block of the Kv11.1 (IKr) channels. Because of their molecular structure, Kv11.1 channels are susceptible to direct block by drugs with a wide variety of chemical structures and sizes (Kannankeril et al. 2010). Moreover, drugs can bind to Kv11.1 proteins in the cytoplasm and disrupt their intracellular trafficking to the sarcolemma (Rajamani et al. 2006). In addition, some drugs may prolong repolarization by increasing the inward depolarizing currents ICa,L and INa,L (Kühlkamp et al. 2003; Kuryshev et al. 2006).
Genetic factors
Although the above-mentioned non-genetic (demographic and environmental) factors are important determinants of phenotype in LQTS and can relatively easily be detected by routine medical examination, they explain only a small part of the incomplete disease penetrance and variable expressivity in LQTS. Literature provides several examples of pedigrees with LQTS in which members of the same sex and generation display large differences in disease severity, ranging from a lifelong asymptomatic state to SCD at a young age, with a disease penetrance ranging from 25% to 100% (Vincent et al. 1992; Priori et al. 1999; Viadero et al. 2011). In addition, significant overlap in QTc duration may exist between genotype-positive members and genotype-negative members of a particular LQTS pedigree (Vincent et al. 1992). Moreover, variability in the QTc duration is not limited to the LQTS, since apparently healthy subjects from the general population may display prolonged QTc duration (>460 ms), while patients with a mutation in a LQTS susceptibility gene can have normal QTc duration (Taggart et al. 2007).
In the early nineties, the discovery that the risk for SCD is increased in the first-degree relatives of SCD victims, along with the association of QTc prolongation with an increased risk for SCD in the general population (Algra et al. 1991), led to the hypothesis that QTc variability may have a heritable component. This created the basis for considerable interest and extensive efforts to identify genetic factors that may modify the QTc duration. Ever since, large scale genome-wide association studies (GWAS) and candidate gene studies have searched for genetic variants that impact the QTc duration in the general population (Arking et al. 2006; Newton-Cheh et al. 2009; Pfeufer et al. 2009; Marroni et al. 2009), while relatively small genetic studies have searched for the co-inheritance of (putatively) functional genetic variants that modulate the QTc duration in LQTS cohorts (Crotti et al. 2005, 2009; Nishio et al. 2009). These studies have greatly increased our knowledge about the heritable component of the QTc variability.
The genetic architecture of the QTc duration
Although the genetic architecture of the QTc duration turns out to be pretty complex, it is thought to consist of a spectrum of genetic variants ranging from very rare variants, with <1% minor allele frequency (MAF) but large effect on the QTc duration, to common variants with >5% MAF but small effect on the QTc duration. In between are the ‘common rare’ variants with 1–5% MAF and an intermediate effect on the QTc duration (Fig. 4; Sauer & Newton-Cheh, 2012). Rare variants are expected to be disease-causing mutations in genes encoding ion channels and their accessory proteins that cause abnormally prolonged (or shortened) QTc durations and increase the risk of TdP/VF and SCD. Mutations that are traditionally linked to LQTS in small genetic studies in single pedigrees are examples of such rare variants. These rare variants are not commonly found in the general population. Instead, common variants, identified through GWAS in the general population, are expected to minimally influence (prolong or shorten) the ventricular repolarization (and thus the QTc duration), and confer only small impact on the risk of SCD individually. However, the presence of multiple common variants with the same small effect on the repolarization in one person may theoretically affect the repolarization time to an extent that might lead to manifestation of disease phenotype. For example, co-inheritance of common variants with minor prolonging effect on the repolarization may explain a prolonged QTc duration in a subject with no mutation in one of the LQTS susceptibility genes. Inversely, co-inheritance of common variants with small shortening effect on the repolarization may theoretically weaken the effect of a LQTS-causing mutation, leading to a normal QTc duration in a genotype-positive patient. However, such a concerted effect of common variants on disease severity has not been described in humans yet. Finally, ‘common rare’ variants with 1–5% MAF are expected to exert an intermediate effect on QTc duration. Examples include disease-causing mutations found in LQTS families with low penetrance and variable expressivity (Priori et al. 1999), variants associated with strong modifying effects on the QTc duration in the general population (Arking et al. 2006; Newton-Cheh et al. 2009; Pfeufer et al. 2009; Marroni et al. 2009), and variants associated with disease only in the co-presence of a non-genetic trigger (a ‘second hit’), such as those found in subjects with drug-induced LQTS (Kannankeril et al. 2010).
Figure 4. Distribution of QTc duration in health and in long QT syndrome.
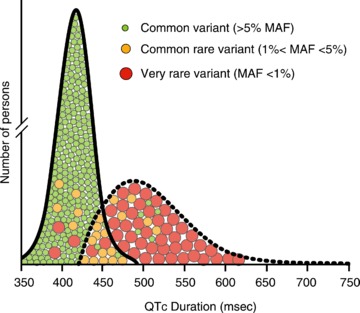
Continuous line represents QTc distribution in health. Dashed line represents QTc distribution in long QT syndrome. The circles display the genetic architecture of the QTc duration consisting of very rare variants with <1% minor allele frequency (MAF) but large effect on the QTc duration, common rare variants with 1–5% MAF with an intermediate effect on QTc duration, and common variants with >5% MAF but small effect on the QTc duration. Rare variants are mutations that are traditionally linked to the long QT syndrome; however, they may be found in persons with normal QTc duration. Common variants are expected to minimally influence (prolong or shorten) the QTc duration. Common rare include variants associated with strong modifying effects on the QTc duration in the general population and variants associated with LQTS only in the co-presence of a non-genetic trigger (a ‘second hit’).
Genetic variants modifying QTc duration in the general population
In general, two types of genetic studies have associated genetic variants with the QTc duration in the general population. Initially, candidate gene studies have searched for associations between the QTc variability in the community and relatively small numbers of variants in ion channel-encoding genes, often non-synonymous (i.e. leading to an amino acid change) single nucleotide polymorphisms (SNPs). More recently, by using maps of hundreds of thousands of common SNPs spread throughout the genome, GWAS have systematically examined whether genomic variations are associated with the QTc variability in the general population. This unbiased approach of GWAS has the advantage of identifying genomic regions that are not previously linked to the QTc duration, but GWAS do not provide information on the underlying causative mechanism.
The most important SNPs that are associated with the QTc duration and/or SCD in the general population by candidate gene approach include K897T in KCNH2-encoded Kv11.1 (Bezzina et al. 2003; Pfeufer et al. 2005; Newton-Cheh et al. 2007; Marjamaa et al. 2009), D85N in KCNE1-encoded minK (Gouas et al. 2005; Marjamaa et al. 2009), and SNPs in the non-coding regions of KCNQ1 and KCNH2 (Newton-Cheh et al. 2009; Pfeufer et al. 2009). GWAS have not only provided further support for the association of K897T, D85N and some of the non-coding SNPs that were found in these small studies, but have also uncovered novel associations between SNPs and the QTc duration in the general population (Table 2). As expected, some of these SNPs are located at genomic regions harbouring ion channel-encoding genes that are implicated in LQTS (KCNQ1, KCNH2, SCN5A and KCNJ2). Moreover, the first GWAS in a community-based cohort from Germany introduced the power of GWAS in discovering unexpected genetic associations by showing a strong association between QTc variability and SNPs in NOS1AP, the gene that encodes the nitric oxide synthase 1 adaptor protein (Arking et al. 2006). NOS1AP had not been linked to cardiac electrical activity before. Importantly, two subsequent meta-analyses of GWAS data of QTc duration from several population-based cohorts of European descent replicated the association between QTc variability and SNPs in NOS1AP (Newton-Cheh et al. 2009; Pfeufer et al. 2009).
Table 2.
Common variants associated with the QTc duration
| Locus | SNP | MAF | Location | Nearest gene | Function | QTc effect |
|---|---|---|---|---|---|---|
| 1q | rs12143842 | 0.16 | Intergenic | NOS1AP | Nitric oxide synthase 1 adaptor protein | ↑ |
| rs2880058 | 0.26 | Intergenic | ↑ | |||
| rs10494366 | 0.33 | Intron | ↓ | |||
| rs12029454 | 0.11 | Intron | ↑ | |||
| rs16857031 | 0.15 | Intron | ↑ | |||
| rs4657178 | 0.18 | Intron | ↑ | |||
| 1q | rs10919071 | 0.11 | Intron | ATP1B1 | β-subunit Na+/K+ ATPase | ↑ |
| 1p | rs846111 | 0.26 | 3′UTR | RNF207 | Ring finger protein | ↑ |
| 3p | rs11129795 | 0.34 | Intergenic | SCN5A | α-subunit INa channel (Nav1.5) | ↓ |
| rs12053903 | 0.29 | Intron | ↓ | |||
| 6q | rs12210810 | 0.08 | Intergenic | C6orf204 | Phosphorylation | ↓ |
| rs11970286 | 0.47 | Intergenic | ↑ | |||
| 7q | rs2968863 | 0.26 | Intergenic | KCNH2 | α-subunit IKr channel (Kv11.1) | ↓ |
| rs4725982 | 0.18 | Intergenic | ↑ | |||
| rs1805123 | 0.24 | Exon (K897T) | ↑↓ | |||
| 11p | rs12296050 | 0.23 | Intron | KCNQ1 | α-subunit IKs channel (Kv7.1) | ↑ |
| rs12576239 | 0.16 | Intron | ↑ | |||
| rs2074238 | 0.08 | Intron | ↓ | |||
| 12q | rs3825214 | 0.22 | Intron | TBX5 | Transcription | ↑ |
| 13q | rs2478333 | 0.35 | Intergenic | SUCLA2 | Mitochondrial enzyme | ↑ |
| 16p | rs8049607 | 0.49 | Intergenic | LITAF | Tumour necrosis factor | ↑ |
| 16q | rs37062 | 0.27 | Intron | CNOT1 | RNA transcription | ↓ |
| 17q | rs17779747 | 0.32 | Intergenic | KCNJ2 | α-subunit IK1 channel (Kir2) | ↓ |
| 17q | rs2074518 | 0.49 | Intron | LIG3 | DNA ligase III | ↓ |
| 21q | rs1805128 | 0.03 | Exon (D85N) | KCNE1 | β-subunit IKs channel (minK) | ↑ |
SNP, single nucleotide polymorphism; MAF, minor allele frequency; ↑, QTc prolongation; ↓, QTc shortening.
Genetic variants modifying QTc duration in LQTS
Genetic studies aiming to unravel QTc- and phenotype-modifying genetic variants in LQTS are mostly based on the theory that co-inheritance of a second genetic variant that influences the ventricular repolarization may affect the final outcome of a disease-causing mutation in an individual carrier, a so-called ‘second hit’ or ‘double hit’ theory.
Double hits may involve mutations in the same gene (compound heterozygosity), mutations in different LQT genes (digenic heterozygosity), or a mutation and a common rare variant in the same gene or in different genes. The prevalence of compound heterozygosity and digenic heterozygosity in LQTS is between 5% and 10%, and is associated with a more severe phenotype (i.e. extensive QTc prolongation and early onset of cardiac events; Schwartz et al. 2003; Westenskow et al. 2004). So far, compound heterozygosity and digenic heterozygosity have only been described for mutations that affect the QTc duration in the same direction by an additive on the ion channel function (i.e. two mutations that exert a prolonging effect on the QTc duration).
Common rare variants that are proposed as genetic modifiers in LQTS include SNPs in KCNH2 (K897T), KCNE1 (D85N) and NOS1AP. SNP rs1805123 (A→C) in KCNH2 has a MAF of ∼24% in populations of European descent in KCNH2 and leads to the substitution of lysine at residue 897 to threonine (K897T) in Kv11.1. Dr Crotti and colleagues showed in a multigenerational pedigree with LQT2 due to the low-penetrant KCNH2 mutation (A1116V) that disease was only manifested in members who co-inherited the minor C allele of SNP rs1805123 on the non-mutant KCNH2 allele (i.e. K897T in the healthy Kv11.1 proteins; Crotti et al. 2005). They also showed that the minor C allele caused a reduction of the Kv11.1 current in heterologous expression systems. However, controversy exists about the effect of K897T on ventricular repolarization, since the minor C allele was associated with a shorter QTc duration in GWAS (Pfeufer et al. 2005), and caused an increase of Kv11.1 current when expressed in different experimental settings (Bezzina et al. 2003).
SNP rs1805128 (G→A) in KCNE1 is present in ∼1% of the general (white) population and leads to the substitution of aspartic acid at residue 85 to asparagine (D85N) in minK, the β-subunit of the IKs channel. In addition, although controversy exists on the role of minK on the regulation of the IKr channel in vivo, minK has been shown to bind to the KCNH2-encoded protein and affect the IKr current density in vitro (McDonald et al. 1997; Ohno et al. 2007). D85N has not only been shown to cause LQTS, but also to contribute to disease penetrance and variable expressivity in patients with LQT1 (Nishio et al. 2009; Lahtinen et al. 2011). Moreover, experimental studies suggest that D85N causes a significant reduction of IKs and IKr (Nishio et al. 2009; Nof et al. 2011), providing a molecular mechanism for the more severe phenotype in LQT1 patients who also carry the minor A allele of SNP rs1805128.
After the discovery of genetic variants in NOS1AP as modifiers of QTc duration in the general population, two studies have established the role of NOS1AP as a genetic modifier of disease severity in LQTS. A study in a South African LQT1 cohort, segregating the A341V mutation in KCNQ1 (including 205 mutation carriers and 295 non-carriers), showed that the minor alleles of two SNPs in the non-coding regions of NOS1AP are associated with longer QTc durations and more symptoms (cardiac arrest and SCD) in this population (Crotti et al. 2009). This study suggested that mutation carriers who also carried the minor allele of one of these two SNPs had a greater risk for experiencing life-threatening cardiac events than the mutation carriers without the minor allele. A second study in 901 LQTS patients, mainly LQT1, LQT2 and LQT3, showed that genetic variation in NOS1AP also modifies disease severity in other types of LQTS (Tomás et al. 2010). Finally, the recent association of SNPs in NOS1AP with drug-induced LQTS further supports the ‘second hit’ theory; in this case disease manifestation through the conjoint effect of a common rare genetic variant with an environmental trigger (i.e. a QT-prolonging drug; Jamshidi et al. 2012). Since SNPs in NOS1AP associated with disease severity in LQTS are located in the non-coding regions of the gene, they are presumed to influence the transcription of the NOS1AP allele in which they are located (cis-acting regulation). Cellular studies suggest that NOS1AP-encoded protein (CAPON) decreases ICa,L density and slightly increases IKr density, leading to a shortening of the action potential (Chang et al. 2008). Minor alleles of SNPs in NOS1AP may reduce CAPON levels by impairing transcription, thereby attenuating the effect of CAPON on the action potential duration.
Genetic variants modifying QTc duration in LQTS in an allele-specific manner
In 2012, we proposed a novel mechanism for genetic variants to modify disease severity in LQTS by describing an association between SNPs in the 3′ untranslated region (3′UTR) of KCNQ1 and the QTc duration and symptomatology in patients with LQT1 (Amin et al. 2012). The 3′UTR is a region of the mRNA that starts with the nucleotide immediately following the stop codon of the coding region. The 3′UTR contains binding sites for microRNAs (miRNAs). By virtue of the repressive effects of the miRNAs on the translation, the 3′UTR determines in a cis-regulatory fashion whether and to what extent mRNA is translated to protein (Chen & Rajewsky, 2006; Huntzinger & Izaurralde, 2011). SNPs in the miRNA binding sites of the 3′UTR have been implicated in the clinical manifestation of various non-cardiac diseases, including asthma, cancer, diabetes mellitus, hypertension and Parkinson's disease (Borel & Antonarakis, 2008). However, most of these SNPs were located outside the gene harbouring the (putative) pathogenic mutation or involved in the pathophysiology of complex diseases. Therefore, clear evidence for allele-specific effects of 3′UTR SNPs, via cis-regulatory actions, on the expression of mutation-carrying genes was lacking. We hypothesized that in an autosomal-dominant disease such as LQTS genetic variation in the 3′UTR of either the normal or rather the mutant allele may influence miRNA binding, and thereby alter the relative amount of proteins stemming from either of these alleles. Our group demonstrated in a combined cohort of 168 LQT1 patients with different KCNQ1 mutations from the United States and the Netherlands that the SNPs rs2519184 (G→A), rs8234 (A→G), and rs10798 (A→G), located in the 3′UTR of KCNQ1, were associated with the QTc duration and symptom occurrence (syncope, TdP, VF, SCD) in an allele-specific manner. Patients with the minor 3′UTR haplotype (A-G-G or G-G-G) on their mutant KCNQ1 allele had shorter QTc and fewer symptoms, while patients with the minor 3′UTR haplotype on their healthy KCNQ1 allele had significantly longer QTc and more symptoms (Fig. 5A). Furthermore, our luciferase reporter assays showed that the expression of KCNQ1's 3′UTR with the minor SNP variants was lower than the expression of the 3′UTR with the major SNP variants (Fig. 5B). Taken together, our clinical and experimental data showed that occurrence of ‘suppressive’ 3′UTR SNPs in cis (on the same allele) to the mutation attenuates disease severity by lowering the abundance of mutant Kv7.1 proteins, while occurrence of ‘suppressive’ 3′UTR SNPs in trans (on the opposite allele) to the mutation aggravates clinical phenotype by reducing the translation of the normal KCNQ1 allele (Fig. 5C). This discovery not only introduced novel genetic modifiers of LQTS, with an effect outweighing by far that of previously identified modifying SNPs, but it also uncovered a mechanism that greatly influences the severity of disease-causing mutation in a Mendelian inherited disease, since in many cases the unaffected parent may alter disease severity by bringing in naturally occurring variation in the 3′UTR. However, it must be noted that the effects of the above-mentioned 3′UTR SNPs are not yet supported by GWAS or genetic studies in other LQTS cohorts. Furthermore, further research is necessary to unravel the precise mechanism by which 3′UTR SNPs influence gene expression and disease severity, e.g. the role of specific miRNAs.
Figure 5. Allele-specific effects of variants in the 3 ′untranslated region (3′UTR) of KCNQ1 on the QTc duration and symptomatology in long QT syndrome type 1 (LQT1).
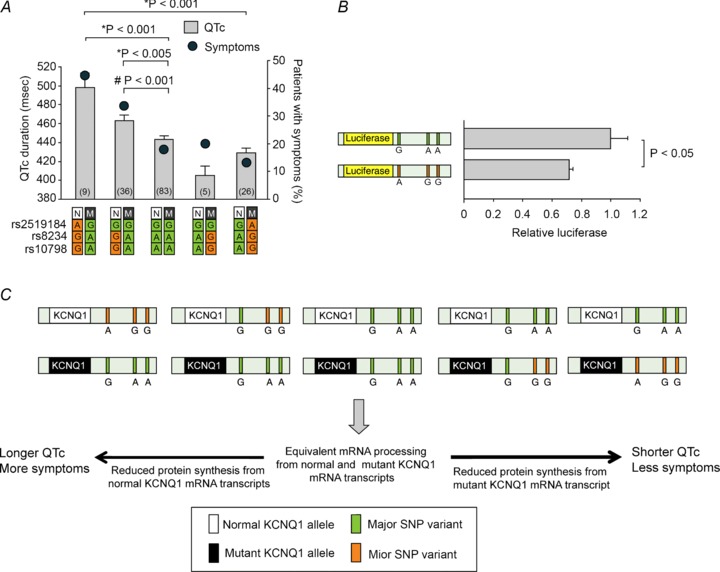
A, allele-specific effects of SNPs rs2519184, rs8234 and rs10798 in the 3′UTR of KCNQ1 on the QTc and symptomatology in patients with LQT1. B, luciferase assays in primary neonatal rat cardiomyocytes transfected with two independent reporter plasmids containing the major or the minor haplotype of SNPs in 3′UTR of KCNQ1. C, the possible mechanism where SNPs in the 3′UTR of KCNQ1 modify phenotype in LQT1. SNPs in the 3′UTR may alter the expression of the normal and the mutant KCNQ1 alleles in an allele-specific manner, and, as a result, the balance between the normal and the mutant Kv7.1 proteins within the tetrameric IKs channels. Numbers in bars denote group sizes. Data are presented as mean; error bars represent SEM. N, normal KCNQ1 allele; M, mutant KCNQ1 allele. Reproduced from Amin et al. (2012) © 2012 with permission from Oxford University Press. * indicates comparisons between QTc durations. # indicates comparison between symptomatology.
Conclusion
LQTS is known as a monogenetic disorder, with several well-recognized non-genetic modifiers (among others, age and sex) as important modifiers. In recent years it has also become increasingly clear that other genetic variants play an equally important modulatory role in establishing the severity of the phenotype. Detailed understanding of these modifying (genetic and non-genetic) factors is mandatory for accurate risk stratification and the subsequent therapeutic choices. Active ‘cascade screening’ in affected families leads to a rapidly increasing number of genotype-positive phenotype-negative individuals, with an ill-defined prognosis. Further appreciation of their actual risk from life-threatening arrhythmias would therefore be more than welcome.
Glossary
- 3′UTR
3′ untranslated region
- 5′UTR
5′ untranslated region
- EAD
early afterdepolarization
- ECG
electrocardiogram
- GWAS
genome-wide association studies
- LQTS
long QT syndrome
- LQT1
long QT syndrome type 1
- LQT2
long QT syndrome type 2
- LQT3
long QT syndrome type 3
- MAF
minor allele frequency
- miRNA
microRNA
- QTc
heart rate-corrected QT interval
- SNP
single nucleotide polymorphism
- TdP
torsades de pointes ventricular tachycardia
- TDR
transmural dispersion of repolarization
- VF
ventricular fibrillation
Additional information
Competing interests
None declared.
Funding
This study was supported by the Center for Translational Molecular Medicine (COHFAR, A.A.M.W; ARENA, Y.M.P.) and the Netherlands Foundation For Cardiovascular Excellence (A.S.A.).
References
- Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SA. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–187. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- Algra A, Tijssen JG, Roelandt JR, Pool J, Lubsen J. QTc prolongation measured by standard 12-lead electrocardiography is an independent risk factor for sudden death due to cardiac arrest. Circulation. 1991;83:1888–1894. doi: 10.1161/01.cir.83.6.1888. [DOI] [PubMed] [Google Scholar]
- Amin AS, Asghari-Roodsari A, Tan HL. Cardiac sodium channelopathies. Pflugers Arch. 2010a;460:223–237. doi: 10.1007/s00424-009-0761-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin AS, Giudicessi JR, Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA, Tanck MW, Kapplinger JD, Hofman N, Sinner MF, Müller M, Wijnen WJ, Tan HL, Bezzina CR, Creemers EE, Wilde AA, Ackerman MJ, Pinto YM. Variants in the 3′ untranslated region of the KCNQ1-encoded Kv7.1 potassium channel modify disease severity in patients with type 1 long QT syndrome in an allele-specific manner. Eur Heart J. 2012;33:714–723. doi: 10.1093/eurheartj/ehr473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin AS, Herfst LJ, Delisle BP, Klemens CA, Rook MB, Bezzina CR, Underkofler HA, Holzem KM, Ruijter JM, Tan HL, January CT, Wilde AA. Fever-induced QTc prolongation and ventricular arrhythmias in individuals with type 2 congenital long QT syndrome. J Clin Invest. 2008;118:2552–2561. doi: 10.1172/JCI35337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin AS, Tan HL, Wilde AA. Cardiac ion channels in health and disease. Heart Rhythm. 2010b;7:117–126. doi: 10.1016/j.hrthm.2009.08.005. [DOI] [PubMed] [Google Scholar]
- Arking DE, Pfeufer A, Post W, Kao WH, Newton-Cheh C, Ikeda M, West K, Kashuk C, Akyol M, Perz S, Jalilzadeh S, Illig T, Gieger C, Guo CY, Larson MG, Wichmann HE, Marbán E, O’Donnell CJ, Hirschhorn JN, Kääb S, Spooner PM, Meitinger T, Chakravarti A. A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet. 2006;38:644–651. doi: 10.1038/ng1790. [DOI] [PubMed] [Google Scholar]
- Bai CX, Kurokawa J, Tamagawa M, Nakaya H, Furukawa T. Nontranscriptional regulation of cardiac repolarization currents by testosterone. Circulation. 2005;112:1701–1710. doi: 10.1161/CIRCULATIONAHA.104.523217. [DOI] [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. KVLQT1 and IsK (minK) proteins associate to form the IKS cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- Bezzina CR, Verkerk AO, Busjahn A, Jeron A, Erdmann J, Koopmann TT, Bhuiyan ZA, Wilders R, Mannens MM, Tan HL, Luft FC, Schunkert H, Wilde AA. A common polymorphism in KCNH2 (HERG) hastens cardiac repolarization. Cardiovasc Res. 2003;59:27–36. doi: 10.1016/s0008-6363(03)00342-0. [DOI] [PubMed] [Google Scholar]
- Borel C, Antonarakis SE. Functional genetic variation of human miRNAs and phenotypic consequences. Mamm Genome. 2008;19:503–509. doi: 10.1007/s00335-008-9137-6. [DOI] [PubMed] [Google Scholar]
- Brink PA, Crotti L, Corfield V, Goosen A, Durrheim G, Hedley P, Heradien M, Geldenhuys G, Vanoli E, Bacchini S, Spazzolini C, Lundquist AL, Roden DM, George AL, Jr, Schwartz PJ. Phenotypic variability and unusual clinical severity of congenital long-QT syndrome in a founder population. Circulation. 2005;112:2602–2610. doi: 10.1161/CIRCULATIONAHA.105.572453. [DOI] [PubMed] [Google Scholar]
- Buber J, Mathew J, Moss AJ, Hall WJ, Barsheshet A, McNitt S, Robinson JL, Zareba W, Ackerman MJ, Kaufman ES, Luria D, Eldar M, Towbin JA, Vincent M, Goldenberg I. Risk of recurrent cardiac events after onset of menopause in women with congenital long-QT syndrome types 1 and 2. Circulation. 2011;123:2784–2791. doi: 10.1161/CIRCULATIONAHA.110.000620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burashnikov A, Shimizu W, Antzelevitch C. Fever accentuates transmural dispersion of repolarization and facilitates development of early afterdepolarizations and torsade de pointes under long-QT conditions. Circ Arrhythm Electrophysiol. 2008;1:202–208. doi: 10.1161/CIRCEP.107.691931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KC, Barth AS, Sasano T, Kizana E, Kashiwakura Y, Zhang Y, Foster DB, Marban E. CAPON modulates cardiac repolarization via neuronal nitric oxide synthase signaling in the heart. Proc Natl Acad Sci U S A. 2008;105:4477–4482. doi: 10.1073/pnas.0709118105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Marquardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kass RS. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc Natl Acad Sci U S A. 2007;104:20990–20995. doi: 10.1073/pnas.0710527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Rajewsky N. Natural selection on human microRNA binding sites inferred from SNP data. Nat Genet. 2006;38:1452–1456. doi: 10.1038/ng1910. [DOI] [PubMed] [Google Scholar]
- Choy AM, Lang CC, Chomsky DM, Rayos GH, Wilson JR, Roden DM. Normalization of acquired QT prolongation in humans by intravenous potassium. Circulation. 1997;96:2149–2154. doi: 10.1161/01.cir.96.7.2149. [DOI] [PubMed] [Google Scholar]
- Compton SJ, Lux RL, Ramsay MR, Strelich KR, Sanguinetti MC, Green LS, Keating MT, Mason JW. Genetically-defined therapy of inherited long-QT syndrome: correction of abnormal repolarization by potassium. Circulation. 1996;94:1018–1022. doi: 10.1161/01.cir.94.5.1018. [DOI] [PubMed] [Google Scholar]
- Crotti L, Johnson CN, Graf E, De Ferrari GM, Cuneo BF, Ovadia M, Papagiannis J, Feldkamp MD, Rathi SG, Kunic JD, Pedrazzini M, Wieland T, Lichtner P, Beckmann BM, Clark T, Shaffer C, Benson DW, Kääb S, Meitinger T, Strom TM, Chazin WJ, Schwartz PJ, George AL., Jr Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation. 2013;127:1009–1017. doi: 10.1161/CIRCULATIONAHA.112.001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, Vicentini A, Yang P, Roden DM, George AL, Jr, Schwartz PJ. KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome. Circulation. 2005;112:1251–1258. doi: 10.1161/CIRCULATIONAHA.105.549071. [DOI] [PubMed] [Google Scholar]
- Crotti L, Monti MC, Insolia R, Peljto A, Goosen A, Brink PA, Greenberg DA, Schwartz PJ, George AL., Jr NOS1AP is a genetic modifier of the long-QT syndrome. Circulation. 2009;120:1657–1663. doi: 10.1161/CIRCULATIONAHA.109.879643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- Gaborit N, Varro A, Le Bouter S, Szuts V, Escande D, Nattel S, Demolombe S. Gender-related differences in ion-channel and transporter subunit expression in non-diseased human hearts. J Mol Cell Cardiol. 2010;49:639–646. doi: 10.1016/j.yjmcc.2010.06.005. [DOI] [PubMed] [Google Scholar]
- Gouas L, Nicaud V, Berthet M, Forhan A, Tiret L, Balkau B, Guicheney P D.E.S.I.R. Study Group. Association of KCNQ1KCNE1KCNH2 and SCN5A polymorphisms with QTc interval length in a healthy population. Eur J Hum Genet. 2005;13:1213–1222. doi: 10.1038/sj.ejhg.5201489. [DOI] [PubMed] [Google Scholar]
- Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- James AF, Arberry LA, Hancox JC. Gender-related differences in ventricular myocyte repolarization in the guinea pig. Basic Res Cardiol. 2004;99:183–192. doi: 10.1007/s00395-003-0451-6. [DOI] [PubMed] [Google Scholar]
- Jamshidi Y, Nolte IM, Dalageorgou C, Zheng D, Johnson T, Bastiaenen R, Ruddy S, Talbott D, Norris KJ, Snieder H, George AL, Marshall V, Shakir S, Kannankeril PJ, Munroe PB, Camm AJ, Jeffery S, Roden DM, Behr ER. Common variation in the NOS1AP gene is associated with drug-induced QT prolongation and ventricular arrhythmia. J Am Coll Cardiol. 2012;60:841–850. doi: 10.1016/j.jacc.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannankeril P, Roden DM, Darbar D. Drug-induced long QT syndrome. Pharmacol Rev. 2010;62:760–781. doi: 10.1124/pr.110.003723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapplinger JD, Tester DJ, Salisbury BA, Carr JL, Harris-Kerr C, Pollevick GD, Wilde AA, Ackerman MJ. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 2009;6:1297–1303. doi: 10.1016/j.hrthm.2009.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khositseth A, Tester DJ, Will ML, Bell CM, Ackerman MJ. Identification of a common genetic substrate underlying postpartum cardiac events in congenital long QT syndrome. Heart Rhythm. 2004;1:60–64. doi: 10.1016/j.hrthm.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Kühlkamp V, Mewis C, Bosch R, Seipel L. Delayed sodium channel inactivation mimics long QT syndrome 3. J Cardiovasc Pharmacol. 2003;42:113–117. doi: 10.1097/00005344-200307000-00017. [DOI] [PubMed] [Google Scholar]
- Kurokawa J, Tamagawa M, Harada N, Honda S, Bai CX, Nakaya H, Furukawa T. Acute effects of oestrogen on the guinea pig and human IKr channels and drug-induced prolongation of cardiac repolarization. J Physiol. 2008;586:2961–2973. doi: 10.1113/jphysiol.2007.150367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryshev YA, Wang L, Wible BA, Wan X, Ficker E. Antimony-based antileishmanial compounds prolong the cardiac action potential by an increase in cardiac calcium currents. Mol Pharmacol. 2006;69:1216–1225. doi: 10.1124/mol.105.019281. [DOI] [PubMed] [Google Scholar]
- Lahtinen AM, Marjamaa A, Swan H, Kontula K. KCNE1 D85N polymorphism – a sex-specific modifier in type 1 long QT syndrome. BMC Med Genet. 2011;12:11. doi: 10.1186/1471-2350-12-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitinen P, Fodstad H, Piippo K, Swan H, Toivonen L, Viitasalo M, Kaprio J, Kontula K. Survey of the coding region of the HERG gene in long QT syndrome reveals six novel mutations and an amino acid polymorphism with possible phenotypic effects. Hum Mutat. 2000;15:580–581. doi: 10.1002/1098-1004(200006)15:6<580::AID-HUMU16>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Liu XK, Katchman A, Drici MD, Ebert SN, Ducic I, Morad M, Woosley RL. Gender difference in the cycle length-dependent QT and potassium currents in rabbits. J Pharmacol Exp Ther. 1998;285:672–679. [PubMed] [Google Scholar]
- Locati EH, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, Towbin JA, Priori SG, Napolitano C, Robinson JL, Andrews M, Timothy K, Hall WJ. Age- and sex-related differences in clinical manifestations in patients with congenital long-QT syndrome: findings from the International LQTS Registry. Circulation. 1998;97:2237–2244. doi: 10.1161/01.cir.97.22.2237. [DOI] [PubMed] [Google Scholar]
- McAllister RE, Noble D. The time and voltage dependence of the slow outward current in cardiac Purkinje fibres. J Physiol. 1966;186:632–662. doi: 10.1113/jphysiol.1966.sp008060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald TV, Yu Z, Ming Z, Palma E, Meyers MB, Wang KW, Goldstein SA, Fishman GI. A minK–HERG complex regulates the cardiac potassium current IKr. Nature. 1997;388:289–292. doi: 10.1038/40882. [DOI] [PubMed] [Google Scholar]
- Marjamaa A, Newton-Cheh C, Porthan K, Reunanen A, Lahermo P, Väänänen H, Jula A, Karanko H, Swan H, Toivonen L, Nieminen MS, Viitasalo M, Peltonen L, Oikarinen L, Palotie A, Kontula K, Salomaa V. Common candidate gene variants are associated with QT interval duration in the general population. J Intern Med. 2009;265:448–458. doi: 10.1111/j.1365-2796.2008.02026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marroni F, Pfeufer A, Aulchenko YS, Franklin CS, Isaacs A, Pichler I, Wild SH, Oostra BA, Wright AF, Campbell H, Witteman JC, Kääb S, Hicks AA, Gyllensten U, Rudan I, Meitinger T, Pattaro C, van Duijn CM, Wilson JF, Pramstaller PP EUROSPAN Consortium. A genome-wide association scan of RR and QT interval duration in 3 European genetically isolated populations: the EUROSPAN project. Circ Cardiovasc Genet. 2009;2:322–328. doi: 10.1161/CIRCGENETICS.108.833806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B, Valdivia C, Ueda K, Canizales-Quinteros S, Tusié-Luna MT, Makielski JC, Ackerman MJ. SCN4B-encoded sodium channel β4 subunit in congenital long-QT syndrome. Circulation. 2007;116:134–142. doi: 10.1161/CIRCULATIONAHA.106.659086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, Song LS, Haurogné K, Kyndt F, Ali ME, Rogers TB, Lederer WJ, Escande D, Le Marec H, Bennett V. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–639. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- Morton V, Torgerson DJ. Effect of regression to the mean on decision making in health care. BMJ. 2003;326:1083–1084. doi: 10.1136/bmj.326.7398.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H, Kurokawa J, Bai CX, Asada K, Xu J, Oren RV, Zhu ZI, Clancy CE, Isobe M, Furukawa T. Progesterone regulates cardiac repolarization through a nongenomic pathway: an in vitro patch-clamp and computational modelling study. Circulation. 2007;116:2913–2922. doi: 10.1161/CIRCULATIONAHA.107.702407. [DOI] [PubMed] [Google Scholar]
- Newton-Cheh C, Eijgelsheim M, Rice KM, de Bakker PI, Yin X, Estrada K, Bis JC, Marciante K, Rivadeneira F, Noseworthy PA, Sotoodehnia N, Smith NL, Rotter JI, Kors JA, Witteman JC, Hofman A, Heckbert SR, O’Donnell CJ, Uitterlinden AG, Psaty BM, Lumley T, Larson MG, Stricker BH. Common variants at ten loci influence QT interval duration in the QTGEN Study. Nat Genet. 2009;41:399–406. doi: 10.1038/ng.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton-Cheh C, Guo CY, Larson MG, Musone SL, Surti A, Camargo AL, Drake JA, Benjamin EJ, Levy D, D’Agostino RB, Sr, Hirschhorn JN, O’Donnell CJ. Common genetic variation in KCNH2 is associated with QT interval duration: the Framingham Heart Study. Circulation. 2007;116:1128–1136. doi: 10.1161/CIRCULATIONAHA.107.710780. [DOI] [PubMed] [Google Scholar]
- Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ, Yamamoto S, Ozawa T, Ding WG, Toyoda F, Kawamura M, Akao M, Matsuura H, Kimura T, Kita T, Horie M. D85N, a KCNE1 polymorphism, is a disease-causing gene variant in long QT syndrome. J Am Coll Cardiol. 2009;54:812–819. doi: 10.1016/j.jacc.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Noda T, Takaki H, Kurita T, Suyama K, Nagaya N, Taguchi A, Aihara N, Kamakura S, Sunagawa K, Nakamura K, Ohe T, Horie M, Napolitano C, Towbin JA, Priori SG, Shimizu W. Gene-specific response of dynamic ventricular repolarization to sympathetic stimulation in LQT1, LQT2 and LQT3 forms of congenital long QT syndrome. Eur Heart J. 2002;23:975–983. doi: 10.1053/euhj.2001.3079. [DOI] [PubMed] [Google Scholar]
- Nof E, Barajas-Martinez H, Eldar M, Urrutia J, Caceres G, Rosenfeld G, Bar-Lev D, Feinberg M, Burashnikov E, Casis O, Hu D, Glikson M, Antzelevitch C. LQT5 masquerading as LQT2: a dominant negative effect of KCNE1-D85N rare polymorphism on KCNH2 current. Europace. 2011;13:1478–1483. doi: 10.1093/europace/eur184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S, Zankov DP, Yoshida H, Tsuji K, Makiyama T, Itoh H, Akao M, Hancox JC, Kita T, Horie M. N- and C-terminal KCNE1 mutations cause distinct phenotypes of long QT syndrome. Heart Rhythm. 2007;4:332–340. doi: 10.1016/j.hrthm.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Pfeufer A, Jalilzadeh S, Perz S, Mueller JC, Hinterseer M, Illig T, Akyol M, Huth C, Schöpfer-Wendels A, Kuch B, Steinbeck G, Holle R, Näbauer M, Wichmann HE, Meitinger T, Kääb S. Common variants in myocardial ion channel genes modify the QT interval in the general population: results from the KORA study. Circ Res. 2005;96:693–701. doi: 10.1161/01.RES.0000161077.53751.e6. [DOI] [PubMed] [Google Scholar]
- Pfeufer A, Sanna S, Arking DE, Müller M, Gateva V, Fuchsberger C, Ehret GB, Orrú M, Pattaro C, Köttgen A, Perz S, Usala G, Barbalic M, Li M, Pütz B, Scuteri A, Prineas RJ, Sinner MF, Gieger C, Najjar SS, Kao WH, Mühleisen TW, Dei M, Happle C, Möhlenkamp S, Crisponi L, Erbel R, Jöckel KH, Naitza S, Steinbeck G, Marroni F, Hicks AA, Lakatta E, Müller-Myhsok B, Pramstaller PP, Wichmann HE, Schlessinger D, Boerwinkle E, Meitinger T, Uda M, Coresh J, Kääb S, Abecasis GR, Chakravarti A. Common variants at ten loci modulate the QT interval duration in the QTSCD Study. Nat Genet. 2009;41:407–414. doi: 10.1038/ng.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaster NM, Tawil R, Tristani-Firouzi M, Canún S, Bendahhou S, Tsunoda A, Donaldson MR, Iannaccone ST, Brunt E, Barohn R, Clark J, Deymeer F, George AL, Jr, Fish FA, Hahn A, Nitu A, Ozdemir C, Serdaroglu P, Subramony SH, Wolfe G, Fu YH, Ptácek LJ. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell. 2001;105:511–519. doi: 10.1016/s0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QT syndrome: clinical impact. Circulation. 1999;99:529–533. doi: 10.1161/01.cir.99.4.529. [DOI] [PubMed] [Google Scholar]
- Rajamani S, Eckhardt LL, Valdivia CR, Klemens CA, Gillman BM, Anderson CL, Holzem KM, Delisle BP, Anson BD, Makielski JC, January CT. Drug-induced long QT syndrome: hERG K+ channel block and disruption of protein trafficking by fluoxetine and norfluoxetine. Br J Pharmacol. 2006;149:481–489. doi: 10.1038/sj.bjp.0706892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautaharju PM, Surawicz B, Gettes LS, Bailey JJ, Childers R, Deal BJ, Gorgels A, Hancock EW, Josephson M, Kligfield P, Kors JA, Macfarlane P, Mason JW, Mirvis DM, Okin P, Pahlm O, van Herpen G, Wagner GS, Wellens H American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST segment, T and U waves, and the QT interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol. 2009;53:982–991. doi: 10.1016/j.jacc.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Romano C, Gemme G, Pongiglione R. Aritmie cardiache rare dell’età pediatrica. Clin Pediatr (Bologna) 1963;45:656–683. [PubMed] [Google Scholar]
- Sabir IN, Killeen MJ, Goddard CA, Thomas G, Gray S, Grace AA, Huang CL. Transient alterations in transmural repolarization gradients and arrhythmogenicity in hypokalaemic Langendorff-perfused murine hearts. J Physiol. 2007;581:277–289. doi: 10.1113/jphysiol.2007.128637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi T, Shimizu W, Itoh H, Noda T, Miyamoto Y, Nagaoka I, Oka Y, Ashihara T, Ito M, Tsuji K, Ohno S, Makiyama T, Kamakura S, Horie M. Age- and genotype-specific triggers for life-threatening arrhythmia in the genotyped long QT syndrome. J Cardiovasc Electrophysiol. 2008;19:794–799. doi: 10.1111/j.1540-8167.2008.01138.x. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of KVLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jurkiewicz NK. Role of external Ca2+ and K+ in gating of cardiac delayed rectifier K+ currents. Pflugers Arch. 1992;420:180–186. doi: 10.1007/BF00374988. [DOI] [PubMed] [Google Scholar]
- Sauer AJ, Newton-Cheh C. Clinical and genetic determinants of torsade de pointes risk. Circulation. 2012;125:1684–1694. doi: 10.1161/CIRCULATIONAHA.111.080887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz PJ, Priori SG, Napolitano C. How really rare are rare diseases?: The intriguing case of independent compound mutations in the long QT syndrome. J Cardiovasc Electrophysiol. 2003;14:1120–1121. doi: 10.1046/j.1540-8167.2003.03339.x. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, Guicheney P, Breithardt G, Keating MT, Towbin JA, Beggs AH, Brink P, Wilde AA, Toivonen L, Zareba W, Robinson JL, Timothy KW, Corfield V, Wattanasirichaigoon D, Corbett C, Haverkamp W, Schulze-Bahr E, Lehmann MH, Schwartz K, Coumel P, Bloise R. Genotype–phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, Vanoli E, Crotti L, Spazzolini C, Ferrandi C, Goosen A, Hedley P, Heradien M, Bacchini S, Turco A, La Rovere MT, Bartoli A, George AL, Jr, Brink PA. Neural control of heart rate is an arrhythmia risk modifier in long QT syndrome. J Am Coll Cardiol. 2008;51:920–929. doi: 10.1016/j.jacc.2007.09.069. [DOI] [PubMed] [Google Scholar]
- Seth R, Moss AJ, McNitt S, Zareba W, Andrews ML, Qi M, Robinson JL, Goldenberg I, Ackerman MJ, Benhorin J, Kaufman ES, Locati EH, Napolitano C, Priori SG, Schwartz PJ, Towbin JA, Vincent GM, Zhang L. Long QT syndrome and pregnancy. J Am Coll Cardiol. 2007;49:1092–1098. doi: 10.1016/j.jacc.2006.09.054. [DOI] [PubMed] [Google Scholar]
- Shimizu W, Antzelevitch C. Cellular basis for the ECG features of the LQT1 form of the long-QT syndrome: effects of β-adrenergic agonists and antagonists and sodium channel blockers on transmural dispersion of repolarization and torsade de pointes. Circulation. 1998;98:2314–2322. doi: 10.1161/01.cir.98.21.2314. [DOI] [PubMed] [Google Scholar]
- Shimizu W, Antzelevitch C. Differential effects of beta-adrenergic agonists and antagonists in LQT1, LQT2 and LQT3 models of the long QT syndrome. J Am Coll Cardiol. 2000;35:778–786. doi: 10.1016/s0735-1097(99)00582-3. [DOI] [PubMed] [Google Scholar]
- Shimizu W, Moss AJ, Wilde AA, Towbin JA, Ackerman MJ, January CT, Tester DJ, Zareba W, Robinson JL, Qi M, Vincent GM, Kaufman ES, Hofman N, Noda T, Kamakura S, Miyamoto Y, Shah S, Amin V, Goldenberg I, Andrews ML, McNitt S. Genotype-phenotype aspects of type 2 long QT syndrome. J Am Coll Cardiol. 2009;54:2052–2062. doi: 10.1016/j.jacc.2009.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu W, Noda T, Takaki H, Kurita T, Nagaya N, Satomi K, Suyama K, Aihara N, Kamakura S, Sunagawa K, Echigo S, Nakamura K, Ohe T, Towbin JA, Napolitano C, Priori SG. Epinephrine unmasks latent mutation carriers with LQT1 form of congenital long QT syndrome. J Am Coll Cardiol. 2003;41:633–642. doi: 10.1016/s0735-1097(02)02850-4. [DOI] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Taggart NW, Haglund CM, Tester DJ, Ackerman MJ. Diagnostic miscues in congenital long-QT syndrome. Circulation. 2007;115:2613–2620. doi: 10.1161/CIRCULATIONAHA.106.661082. [DOI] [PubMed] [Google Scholar]
- Tan HL, Bardai A, Shimizu W, Moss AJ, Schulze-Bahr E, Noda T, Wilde AA. Genotype-specific onset of arrhythmias in congenital long-QT syndrome: possible therapy implications. Circulation. 2006;114:2096–2103. doi: 10.1161/CIRCULATIONAHA.106.642694. [DOI] [PubMed] [Google Scholar]
- Tanabe S, Hata T, Hiraoka M. Effects of estrogen on action potential and membrane currents in guinea pig ventricular myocytes. Am J Physiol Heart Circ Physiol. 1999;277:H826–H833. doi: 10.1152/ajpheart.1999.277.2.H826. [DOI] [PubMed] [Google Scholar]
- Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- Tomás M, Napolitano C, De Giuli L, Bloise R, Subirana I, Malovini A, Bellazzi R, Arking DE, Marban E, Chakravarti A, Spooner PM, Priori SG. Polymorphisms in the NOS1AP gene modulate QT interval duration and risk of arrhythmias in the long QT syndrome. J Am Coll Cardiol. 2010;55:2745–2752. doi: 10.1016/j.jacc.2009.12.065. [DOI] [PubMed] [Google Scholar]
- Ueda K, Valdivia C, Medeiros-Domingo A, Tester DJ, Vatta M, Farrugia G, Ackerman MJ, Makielski JC. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A. 2008;105:9355–9360. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, Tester DJ, Balijepalli RC, Foell JD, Li Z, Kamp TJ, Towbin JA. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114:2104–2112. doi: 10.1161/CIRCULATIONAHA.106.635268. [DOI] [PubMed] [Google Scholar]
- Viadero MT, Rubin E, Amigo T, Gonzalez-Lamuno D. Three generations of hereditary long-QT syndrome with complete penetrance caused by the p.G316E KCNQ1 mutation. Pediatr Cardiol. 2011;32:102–104. doi: 10.1007/s00246-010-9821-7. [DOI] [PubMed] [Google Scholar]
- Vincent GM, Timothy KW, Leppert M, Keating M. The spectrum of symptoms and QT intervals in carriers of the gene for the long-QT syndrome. N Engl J Med. 1992;327:846–852. doi: 10.1056/NEJM199209173271204. [DOI] [PubMed] [Google Scholar]
- Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- Ward OC. A new familial cardiac syndrome in children. J Ir Med Assoc. 1964;54:103–106. [PubMed] [Google Scholar]
- Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Compound mutations: a common cause of severe long-QT syndrome. Circulation. 2004;109:1834–1841. doi: 10.1161/01.CIR.0000125524.34234.13. [DOI] [PubMed] [Google Scholar]
- Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet M, Olesen SP, Rasmussen HB, Ellinor PT, Gao L, Lin X, Li L, Wang L, Xiao J, Liu Y, Liu Y, Zhang S, Liang D, Peng L, Jespersen T, Chen YH. Identification of a Kir3.4 mutation in congenital long QT syndrome. Am J Hum Genet. 2010;86:872–880. doi: 10.1016/j.ajhg.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zicha S, Moss I, Allen B, Varro A, Papp J, Dumaine R, Antzelevich C, Nattel S. Molecular basis of species-specific expression of repolarizing K+ currents in the heart. Am J Physiol Heart Circ Physiol. 2003;285:H1641–H1649. doi: 10.1152/ajpheart.00346.2003. [DOI] [PubMed] [Google Scholar]