

Abstract
In addition to acting on mineralocorticoid receptors, aldosterone has been recently shown to activate the G protein-coupled oestrogen receptor (GPER) in vascular cells. In light of the newly identified role for GPER in vagal cardiac control, we examined whether or not aldosterone activates GPER in rat nucleus ambiguus. Aldosterone produced a dose-dependent increase in cytosolic Ca2+ concentration in retrogradely labelled cardiac vagal neurons of nucleus ambiguus; the response was abolished by pretreatment with the GPER antagonist G-36, but was not affected by the mineralocorticoid receptor antagonists, spironolactone and eplerenone. In Ca2+-free saline, the response to aldosterone was insensitive to blockade of the Ca2+ release from lysosomes, while it was reduced by blocking the Ca2+ release via ryanodine receptors and abolished by blocking the IP3 receptors. Aldosterone induced Ca2+ influx via P/Q-type Ca2+ channels, but not via L-type and N-type Ca2+ channels. Aldosterone induced depolarization of cardiac vagal neurons of nucleus ambiguus that was sensitive to antagonism of GPER but not of mineralocorticoid receptor. in vivo studies, using telemetric measurement of heart rate, indicate that microinjection of aldosterone into the nucleus ambiguus produced a dose-dependent bradycardia in conscious, freely moving rats. Aldosterone-induced bradycardia was blocked by the GPER antagonist, but not by the mineralocorticoid receptor antagonists. In summary, we report for the first time that aldosterone decreases heart rate by activating GPER in cardiac vagal neurons of nucleus ambiguus.
Key points
Faster cellular effects of aldosterone incompatible with the genomic effects mediated by mineralocorticoid receptors have been proposed for 40 years but the receptors remained elusive.
Recently, aldosterone has been shown to activate the G protein-coupled oestrogen receptor (GPER) in the vasculature.
Our results indicate that aldosterone activates the GPER in cardiac vagal neurons of nucleus ambiguus leading to an increase in cytosolic Ca2+ concentration and depolarization; in addition, in vivo studies indicate that microinjection of aldosterone in nucleus ambiguus produces bradycardia in conscious rats.
In summary, our results identified a new role for aldosterone in the modulation of cardiac vagal tone via GPER activation in nucleus ambiguus.
Introduction
Aldosterone, a member of the renin–angiotensin–aldosterone system, is classically involved in the regulation of salt and water homeostasis by acting on mineralocorticoid receptors in the kidney. Activation of mineralocorticoid receptors by aldosterone leads to genomic effects, modulation of gene transcription and protein synthesis, characterized by a delayed onset of action. Faster actions of aldosterone, insensitive to blockade of mineralocorticoid receptors, have been described in vitro and in vivo (Schneider et al. 1997; Wehling et al. 1998; Liu et al. 2003; Schmidt et al. 2003; Lösel et al. 2004). Moreover, rapid non-genomic aldosterone effects were reported in the mineralocorticoid receptor knockout mouse suggesting that they were produced by a receptor distinct from the intracellular mineralocorticoid receptor (Haseroth et al. 1999). Recently, aldosterone has been reported to act rapidly via the G protein-coupled oestrogen receptor (GPER; Gros et al. 2011a,b, 2013; Feldman & Gros, 2011).
In addition to the peripheral effects, aldosterone acts centrally to stimulate sympathetic tone and to increase the blood pressure (Yu et al. 2008; Zhang et al. 2008). The effects of aldosterone on vagal tone are poorly understood: aldosterone has been reported to increase (Heindl et al. 2006), decrease (Yee & Struthers, 1998; Schmidt et al. 2005) or to produce biphasic (Schmidt et al. 1999) effects on the cardiac vagal tone and baroreflex. As we recently identified that activation of GPER in the nucleus ambiguus increases vagal tone and produces bradycardia (Brailoiu et al. 2013), in the current study we examined the in vitro and in vivo effects of aldosterone mediated by GPER in cardiac preganglionic neurons of nucleus ambiguus.
Methods
Ethical approval
Animal protocols were approved by the Institutional Animal Care and Use Committee of Thomas Jefferson University and Temple University.
Chemicals
Aldosterone, 2-aminoethoxydiphenyl borate (2-APB), spironolactone, ω-conotoxin MVIIC and ω-conotoxin GVIA were from Sigma-Aldrich (St Louis, MO, USA); eplerenone was from Tocris Bioscience (R&D Systems, Minneapolis, MN, USA); xestospongin C and ryanodine were from EMD Chemicals Inc. (San Diego, CA, USA); and G-1 and G-36 were synthesized by J. B. Arterburn.
Animals
Neonatal Sprague–Dawley rats (1–2 days old) were used for retrograde tracing, and neuronal culture. Adult male Sprague–Dawley rats (200–250 g) were used for telemetry experiments. At the end of the experiments, anesthetized adult rats were euthanized by CO2 inhalation followed by decapitation.
Neuronal labelling and culture
Cardiac vagal preganglionic neurons of nucleus ambiguus were retrogradely labelled by intrapericardial injection of rhodamine (X-RITC, 40 μl, 0.01%, Invitrogen, Carlsbad, CA, USA), as previously described (Brailoiu et al. 2012, 2013). Medullary neurons were dissociated and cultured, 24 h after rhodamine injection. Briefly, the brains were quickly removed and immersed in ice-cold Hanks’ balanced salt solution (Mediatech, Manassas, VA, USA). The ventral side of the medulla (containing nucleus ambiguus) was dissected, minced, and the cells were dissociated by enzymatic digestion with papain, followed by mechanical trituration. After centrifugation at 500 g, fractions enriched in neurons were collected and resuspended in culture medium containing Neurobasal-A (Invitrogen), which promotes the survival of postnatal neurons, 1% GlutaMax (Invitrogen), 2% penicillin–streptomycin–amphotericin B solution (Mediatech) and 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA, USA). Cells were plated on round 25 mm glass coverslips previously coated with poly-l-lysine (Sigma-Aldrich) in 6-well plates. Cultures were maintained at 37°C in a humidified atmosphere with 5% CO2. The mitotic inhibitor cytosine β-arabino furanoside (1 μm; Sigma-Aldrich) was added to the culture to inhibit glial cell proliferation (Schöniger et al. 2001). Cells were used for calcium imaging after 2–4 days in culture.
Calcium imaging
Measurements of [Ca2+]i were carried out in rhodamine-labelled neurons as previously described (Brailoiu et al. 2012, 2013). Cells were incubated with the acetoxymethyl ester form of fura-2 (fura-2 AM; 4 μm; Invitrogen) in Hanks’ balanced salt solution (HBSS) at room temperature for 45 min, in the dark, washed three times with dye-free HBSS, and then incubated for another 45 min to allow for complete de-esterification of the dye. Coverslips (25 mm diameter) were subsequently mounted in an open bath chamber (RP-40LP, Warner Instruments, Hamden, CT, USA) on the stage of an inverted microscope Nikon Eclipse TiE (Nikon Inc., Melville, NY, USA). The microscope was equipped with a Perfect Focus System and a Photometrics CoolSnap HQ2 CCD camera. During the experiments, the Perfect Focus System was activated. Rhodamine (emission 580 nm)-labelled neurons were identified after excitation at 510 nm. Fura-2 AM fluorescence (emission 510 nm), following alternate excitation at 340 and 380 nm, was acquired at a frequency of 0.25 Hz. Images were acquired and analysed using NIS-Elements AR software (Nikon). The ratio of the fluorescence signals (340 nm/380 nm) was converted to Ca2+ concentrations (Grynkiewicz et al. 1985).
Measurement of membrane potential
The relative changes in membrane potential of single neurons were evaluated using bis-(1,3-dibutylbarbituric acid) trimethine oxonol (DiBAC4(3); Invitrogen), a slow response voltage-sensitive dye, as previously described (Brailoiu et al. 2010, 2013). Upon membrane hyperpolarization, the dye concentrates in the cell membrane, leading to a decrease in fluorescence intensity, while depolarization induces the sequestration of the dye into the cytosol, resulting in an increase of the fluorescence intensity (Brauner et al. 1984). Cultured neurons were incubated for 30 min in HBSS containing 0.5 mm DiBAC4(3) and fluorescence monitored at 0.17 Hz (excitation/emission 480 nm/540 nm). Calibration of DiBAC4(3) fluorescence following background subtraction was performed using the Na+–K+ ionophore gramicidin in Na+-free physiological solution and various concentrations of K+ (to alter membrane potential) and N-methylglucamine (to maintain osmolarity; Brauner et al. 1984). Under these conditions the membrane potential is approximately equal to the K+ equilibrium potential determined by the Nernst equation. The intracellular K+ and Na+ concentration were assumed to be 130 mm and 10 mm, respectively.
Surgical procedures
Male Sprague–Dawley rats (200–250 g) were anaesthetized with an intraperitoneal injection of a mixture of ketamine hydrochloride (100–150 mg kg−1) and acepromazine maleate (0.2 mg kg−1). Animals were placed into a stereotaxic instrument; a guide C315G cannula (PlasticsOne, Roanoke, VA, USA) was bilaterally inserted into the nucleus ambiguus and fixed in position with dental cement. The stereotaxic coordinates for identification of nucleus ambiguus were: 12.24 mm posterior to bregma, 2.1 mm from the midline and 8.2 mm ventral to the dura mater (Paxinos & Watson, 1998). A C315DC cannula dummy (PlasticsOne) of identical length was inserted into the guide cannula to prevent any contamination. For the implantation of transmitters, an incision 2 cm in length was made along the linea alba, and the underlying tissue was dissected and retracted. A calibrated transmitter (E-mitters, series 4000; Mini Mitter, Sunriver, OR, USA) was inserted in the intraperitoneal space, as previously described (Benamar et al. 2010). After the transmitter was passed through the incision, the abdominal musculature and dermis were sutured independently, and the animals were returned to individual cages. The animals were observed daily to ensure health and recovery.
Microinjection into nucleus ambiguus
One week after surgery, either vehicle, or the compound tested was bilaterally microinjected into the nucleus ambiguus, using a C315I internal cannula (33 gauge, PlasticsOne), without handling the rats. At least 2 h were allowed between two injections for recovery. In all in vivo experiments, two types of controls were used: microinjection of saline was used as a negative control and microinjection of l-glutamate (5 mm, 50 nl with a Neuros Hamilton syringe, Model 7000.5 KH SYR) was used for the functional identification of nucleus ambiguus (Chitravanshi et al. 2009; Brailoiu et al. 2013). In some experiments, rats bearing a cannula in nucleus ambiguus and the intraperitoneal transmitter were anaesthetized with ketamine hydrochloride (100–150 mg kg−1) and acepromazine maleate (0.2 mg kg−1), and one of the femoral veins was cannulated for intravenous injection of atropine.
Telemetric heart rate monitoring
The signal generated by transmitters was collected via series 4000 receivers (Mini Mitter) placed underneath the home cage. VitalView software (Mini Mitter) was used for data acquisition. Each data point represents the average heart rate per 1 min.
Statistical analysis
Data were expressed as mean ± standard error of mean (SEM). One-way ANOVA followed by post hoc analysis using Bonferonni and Tukey tests was used to evaluate significant differences between groups; P < 0.05 was considered statistically significant.
Results
Aldosterone increases [Ca2+]i in preganglionic neurons of nucleus ambiguus
In rhodamine-labelled neurons, aldosterone (10−8 m) produced a fast and long-lasting increase in cytosolic Ca2+ concentration ([Ca2+]i; Fig. 1A and B); the increase in [Ca2+]i reached a peak within 2 min after aldosterone treatment and maintained a long-lasting plateau (for about 10 min). G-36 (10−6 m), the GPER antagonist (Dennis et al. 2011), did not elicit a change in [Ca2+]i, but abolished the response to aldosterone (10−8 m; Fig. 1A and B); cells were treated with G-36 for 10 min before aldosterone and for the duration of aldosterone treatment. The mineralocorticoid receptor antagonists spironolactone (10−6 m) and eplerenone (10−6 m) did not significantly affect the Ca2+ response to aldosterone (Fig. 1A–B); the neurons were treated with spironolactone and eplerenone for 30 min before aldosterone and for the duration of aldosterone administration. Aldosterone (10−10, 10−9, 10−8 and 10−7 m) produced a dose-dependent increase in [Ca2+]i by 14 ± 1.6 nm, 98 ± 1.7 nm, 214 ± 3.4 nm and 239 ± 3.1 nm, respectively (n= 6 for each concentration tested). Pretreatment with the GPER antagonist G-36 (10−6 m, 10 min) abolished the response to aldosterone (10−8 m); Δ[Ca2+]i= 17 ± 1.4 nm in the presence of G-36 as compared to 214 ± 3.4 nm in the absence of the antagonist, while spironolactone or eplerenone did not significantly affect the aldosterone-induced Ca2+ response; Δ[Ca2+]i= 209 ± 3.1 nm in the presence of spironolactone, and 211 ± 2.8 nm in the presence of eplerenone (Fig. 1B). Representative examples of changes in 340/380 fluorescence ratio in rhodamine-labelled neurons of nucleus ambiguus in response to aldosterone in the absence and presence of the GPER and mineralocorticoid receptor antagonists are shown in Fig. 1C.
Figure 1. Aldosterone increases [Ca2+]i in cardiac vagal preganglionic neurons of nucleus ambiguus via GPER activation.
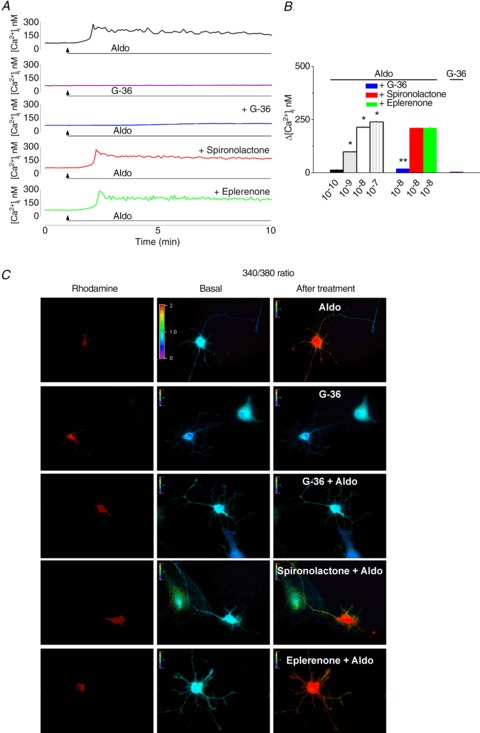
A, representative examples of Ca2+ responses upon administration of aldosterone (10−8 m) in the absence and presence of GPER antagonist G-36 and mineralocorticoid antagonists spironolactone and eplerenone. G-36 did not affect the [Ca2+]i, but abolished the response elicited by aldosterone. B, comparison of the Ca2+ responses elicited by increasing concentrations of aldosterone (Aldo, 10−10, 10−9, 10−8 and 10−7 m) in the presence of G-36 (10−6 m), spironolactone (10−6 m) and eplerenone (10−6 m); *P < 0.05 as compared to basal [Ca2+]i levels; **P < 0.05 as compared with Ca2+ response induced by aldosterone (10−8 m). C, Fura-2 AM fluorescence ratio (340 nm/380 nm) of rhodamine-labelled neurons, before and after treatment with aldosterone (Aldo, 10−8 m) in the absence and presence of G-36, spironolactone and eplerenone; hot colours indicate an increased fluorescence ratio, cold colours indicate low fluorescence ratio.
Aldosterone promotes Ca2+ release via inositol 1,4,5-trisphosphate (IP3) and ryanodine receptors
In Ca2+-free saline, aldosterone (10−8 m) produced a fast and transitory increase in [Ca2+]i (Fig. 2A) with a lower amplitude than that occurring in Ca2+-containing saline (Fig. 1A and B). The response elicited by aldosterone in Ca2+-free saline was not significantly affected by blocking Ca2+ release from endo-lysosomes with Ned-19 (5 μm, 15 min; Naylor et al. 2009), while it was reduced by antagonism of ryanodine receptors with ryanodine (10 μm, 1 h), and abolished by blockade of IP3 receptors with xestospongin C (XeC, 10 μm, 15 min) and 2-APB (100 μm, 15 min; Fig. 2A). Representative examples of aldosterone-induced Ca2+ responses are shown in Fig. 2A and the comparison of the amplitude of Ca2+ increases produced by aldosterone in Ca2+-free saline, in the absence and presence of the antagonists, is shown in Fig. 2B. Aldosterone-induced increase in [Ca2+]i in Ca2+-free saline was 143 ± 2.9 nm; in the presence of Ned-19, Δ[Ca2+]i= 135 ± 3.2 nm (n= 6, P > 0.05); the response decreased to 81 ± 1.6 nm in the presence of ryanodine (n= 6, P < 0.05) and was practically abolished (Δ[Ca2+]i= 12 ± 1.8 nm) in the presence of xestospongin C and 2-APB (Fig. 2B).
Figure 2. Aldosterone promotes Ca2+ release via IP3 and ryanodine receptors.
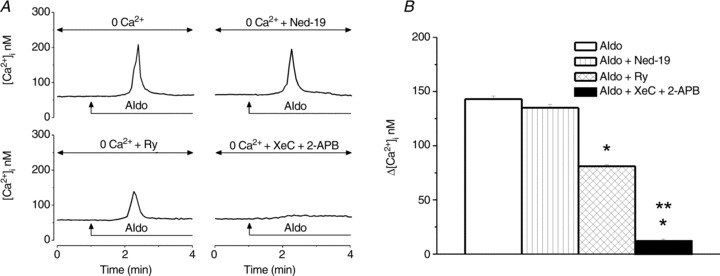
A, representative examples of increases in [Ca2+]i produced by aldosterone (10−8 m) in Ca2+-free saline before (top left) or after treatment with Ned-19 (1 μm, top right), ryanodine (Ry, 10 μm, bottom left) or xestospongin C (XeC, 10 μm) and 2-APB (100 μm, bottom right); the arrows indicate the treatment with aldosterone. B, comparisons of mean amplitude ± SEM of [Ca2+]i increases produced by aldosterone in Ca2+-free saline and the blocker(s) mentioned (*P < 0.05 compared to the response to aldosterone alone; **P < 0.05 as compared to [Ca2+]i increases produced by aldosterone in the presence of ryanodine).
Aldosterone promotes Ca2+ influx via voltage-gated P/Q channels
Since the Ca2+ response to aldosterone was reduced in Ca2+-free saline, this indicates that Ca2+ influx, in addition to Ca2+ release from internal stores, is involved in aldosterone-induced increase in [Ca2+]i. We assessed the contribution of Ca2+ influx via voltage-gated L-, N- and P/Q-type Ca2+ channels in the Ca2+ response elicited by aldosterone. Blockade of L-type Ca2+ channels with nifedipine (1 μm, 10 min) or that of N-type channels with ω-conotoxin GVIA (100 nm, 10 min) did not significantly affect the response to aldosterone, while blockade of voltage-gated P/Q Ca2+ channels with ω-conotoxin MVIIC (100 nm, 10 min) markedly reduced the response; examples of representative recordings are shown in Fig. 3A. We assessed the amplitude of the increase in [Ca2+]i produced by aldosterone at the peak and on the plateau phase of the response; comparison of the changes in amplitude in the presence of the Ca2+ channels blockers are shown in Fig. 3B. The mean amplitude of the aldosterone-induced increase in [Ca2+]i in the absence of the blockers, on the plateau phase was 189 ± 2.8 nm; nifedipine and conotoxin GVIA did not significantly affect the Ca2+ response to aldosterone; on the plateau phase, Δ[Ca2+]i= 173 ± 3.4 nm, in the presence of nifedipine, and 186 ± 2.8 nm in the presence of conotoxin GVIA, as compared to 189 ± 2.8 nm in the absence of the blockers (n= 6 for each condition, P > 0.05), while conotoxin MVIIC (100 nm) abolished the plateau phase of the response to aldosterone (Δ[Ca2+]i= 6 ± 1 nm; n= 6, P < 0.05) and reduced the peak of the response (Fig. 3B).
Figure 3. Aldosterone promotes Ca2+ influx through P/Q-type voltage-gated Ca2+ channels.
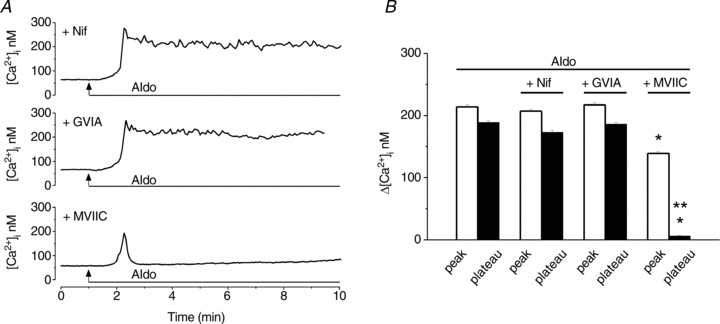
A, representative traces illustrating the Ca2+ responses of nucleus ambiguus neurons to aldosterone (10−8 m) treatment in the presence of nifedipine (L-type Ca2+ channel blocker, 1 μm, top), ω-conotoxin GVIA (GVIA, N-type Ca2+ channel blocker, 100 nm, middle) or ω-conotoxin MVIIC (MVIIC, P/Q-type Ca2+ channel blocker, 100 nm, bottom). B, comparison of the mean amplitude ± SEM of [Ca2+]i increases produced by aldosterone at the peak and on the plateau of the response in the indicated conditions; P < 0.05 as compared to the peak (*) or plateau (**) of the Ca2+ response produced by aldosterone alone.
Aldosterone depolarizes preganglionic vagal neurons of nucleus ambiguus
Aldosterone (10−8 m) produced a long-lasting depolarization of rhodamine-labelled neurons of nucleus ambiguus (Fig. 4A). The GPER antagonist G-36 (10−6 m), while not producing any change in membrane potential, when applied alone abolished the depolarization induced by aldosterone (10−8 m; Fig. 4A). On the other hand, aldosterone-induced depolarization was not affected by spironolactone (10−6 m) or eplerenone (10−6 m); the neurons were treated with the mineralocorticoid antagonists for 30 min before aldosterone and for the duration of aldosterone application (Fig. 4A). The mean amplitude of depolarization produced by aldosterone (10−10, 10−9, 10−8 and 10−7 m) was 0.27 ± 0.16 mV, 1.19 ± 0.37 mV, 5.8 ± 0.81 mV and 6.34 ± 0.63 mV, respectively (n= 6 for each concentration). Pretreatment with G-36 (10−6 m, 10 min) abolished the response to aldosterone (Δvoltage =−0.43 ± 0.09 mV), while pretreatment with spironolactone (10−6 m) or eplerenone (10−6 m) did not significantly affect the depolarization induced by aldosterone (Δvoltage = 5.7 ± 0.72 mV and 5.5 ± 0.84 mV in the presence of spironolactone and eplerenone, respectively, as compared to 5.8 ± 0.81 mV in the absence of the antagonists; n= 6, P > 0.05; Fig. 4B).
Figure 4. Aldosterone elicits depolarization of cardiac preganglionic neurons of nucleus ambiguus.
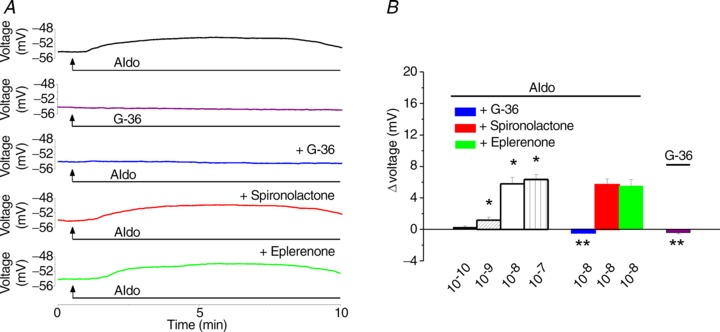
A, representative recordings illustrating changes in membrane potential of cardiac vagal neurons upon application of aldosterone (10−8 m), G-36 (10−6 m) alone, and aldosterone (10−8 m), in the presence of the GPER antagonist G-36 (10−6 m) or of the mineralocorticoid receptor antagonists spironolactone (10−6 m) and eplerenone (10−6 m). B, aldosterone (10−10, 10−9, 10−8 and 10−7 m) produces a concentration-dependent depolarization of nucleus ambiguus neurons. The response to 10−8 m aldosterone is sensitive to antagonism of GPER by G-36, but not to the antagonism of mineralocorticoid receptor by spironolactone or eplerenone; P < 0.05 compared to basal membrane potential (*) or to the depolarization induced by 10−8 m aldosterone (**).
Functional identification of nucleus ambiguus
Microinjection of control saline into nucleus ambiguus did not affect the heart rate, while microinjection of l-glutamate (L-Glu, 5 × 10−3 m, 50 nl) at the same site elicited a bradycardic response in anaesthetized rats (Fig. 5A), as previously reported (Chitravanshi et al. 2009, 2012; Brailoiu et al. 2013), indicating the correct placement of the cannula. The bradycardic effect induced by microinjection of l-glutamate was prevented by i.v. injection of atropine (2 mg kg−1), a potent muscarinic receptor antagonist (Fig. 5A), indicating that the response was cholinergic-mediated. The comparison of the amplitude of the heart rate change produced by control saline, l-glutamate, and l-glutamate after atropine, is illustrated in Fig. 5B. In anaesthetized rats, microinjection of l-glutamate into the nucleus ambiguus decreased the heart rate by 76 ± 2.5 beats min−1 (n= 5).
Figure 5. Control microinjections and functional identification of nucleus ambiguus.
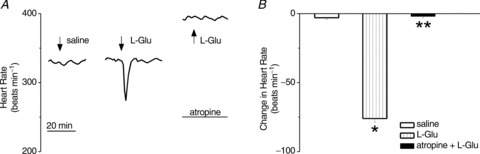
A, examples of changes in heart rate after microinjection of control saline, l-glutamate (L-Glu, 5 × 10−3 m, 50 nl) and L-Glu after atropine (2 mg kg−1) in anaesthetized rats; L-Glu-induced bradycardia was abolished by atropine. B, comparison of the heart rate changes produced by microinjection of saline, L-Glu and L-Glu after atropine; *P < 0.05 and **P > 0.05 as compared to control saline injection.
Microinjection of aldosterone into nucleus ambiguus decreases the heart rate in awake rats
In conscious, freely moving rats bearing a cannula implanted into nucleus ambiguus, microinjection of control saline did not modify the heart rate, while microinjection of l-glutamate (L-Glu, 5 × 10−3 m, 50 nl) elicited a bradycardic response (Fig. 6A) similar to that previously reported in anaesthetized rats (Chitravanshi et al. 2009, 2012; Brailoiu et al. 2013). Microinjection of aldosterone (10−7 m, 50 nl), or G-1 (10−5 m), the synthetic GPER agonist (Bologa et al. 2006), or concomitant microinjection of aldosterone and G-1, at the same site, produced a decrease in heart rate; representative examples are shown in Fig. 6A. A comparison of the amplitude of the heart rate changes produced by microinjection of the compounds is shown in Fig. 6B. In conscious rats, microinjection of l-glutamate (5 × 10−3 m, 50 nl) into the nucleus ambiguus reduced the heart rate by 89 ± 3.5 beats min−1 (n= 5). Microinjection of aldosterone (10−10 m, 10−9 m, 10−8 m, 10−7 m and 10−6 m; 50 nl) produced a dose-dependent decrease in heart rate by 6 ± 2, 18 ± 1.7, 31 ± 2.9, 46 ± 2.8 and 45 ± 3.1 beats min−1, respectively (n= 5). Microinjection of G-1 (10−5 m) that produced a maximal bradycardic response in our previous study in anaesthetized rats (Brailoiu et al. 2013) elicited a decrease in heart rate by 64 ± 3.1 beats min−1 (Fig. 6B). Concomitant microinjection of G-1 (10−5 m) and aldosterone (10−7 m), decreased heart rate by 63 ± 3.4 beats min−1 (Fig. 6B), that was not significantly different from G-1 alone (P > 0.05).
Figure 6. Microinjection of aldosterone into nucleus ambiguus elicits bradycardia in conscious rats.
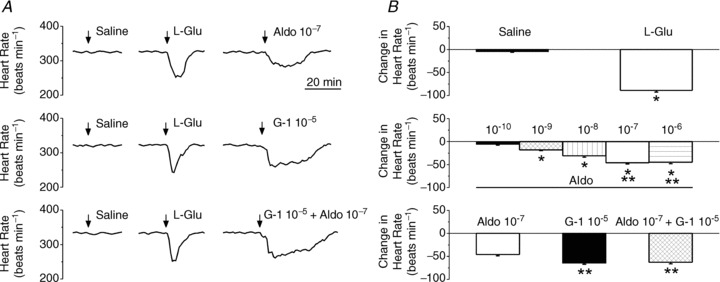
A, example of changes in heart rate after microinjection of control saline, l-glutamate (L-Glu, 5 × 10−3 m, 50 nl), aldosterone (Aldo, 10−7 m, 50 nl), G-1 (10−5 m, 50 nl) and concomitant microinjection of aldosterone (10−7 m) and G-1 (10−5 m). B, comparison of the heart rate changes produced by microinjection in the nucleus ambiguus of saline, L-Glu, aldosterone and G-1, in conscious rats. Aldosterone (10−10 to 10−6 m) produced a dose-dependent decrease in heart rate. Comparison of the amplitude of bradycardia produced by maximal doses of aldosterone (10−7 m) and G-1(10−5 m). *P < 0.05; **P > 0.05.
Aldosterone-induced bradycardia is blocked by the GPER antagonist in awake rats
In the next series of experiments we tested the sensitivity of the bradycardic response elicited by aldosterone to the antagonism of GPER and mineralocorticoid receptors. Microinjection of G-36 (10−6 m), the GPER antagonist, into the nucleus ambiguus, did not produce a significant change in heart rate; concomitant microinjection of G-36 (10−6 m) and aldosterone (10−8 m), abolished the bradycardia elicited by aldosterone (Fig. 7A). Microinjection of spironolactone (10−6 m) or eplerenone (10−6 m) concomitant with aldosterone (10−8 m), did not produce a noticeable effect on aldosterone-induced bradycardia (Fig. 7A). Comparison of the changes in heart rate elicited by the microinjection into nucleus ambiguus of aldosterone in the absence and presence of the GPER and mineralocorticoid antagonists is illustrated in Fig. 7B. The bradycardic response elicited by aldosterone (10−8 m) was abolished by G-36 (10−6 m); in the presence of G-36, aldosterone increased the heart rate by 11 ± 1.8 beats min−1. Microinjection of G-36 (10−6 m) alone produced an insignificant increase in heart rate by 10 ± 1.9 beats min−1 (P > 0.05). Spironolactone (10−6 m) or eplerenone (10−6 m) did not significantly affect the bradycardic response induced by aldosterone; the heart rate decreased by 29 ± 1.6 beats min−1 in the presence of aldosterone and spironolactone and by 32 ± 2.4 beats min−1 in the presence of aldosterone and eplerenone as compared to a decrease by 31 ± 2.9 beats min−1 produced by the same dose (10−8 m) of aldosterone alone (n= 5; Fig. 7B).
Figure 7. Aldosterone-induced bradycardia is mediated by GPER.
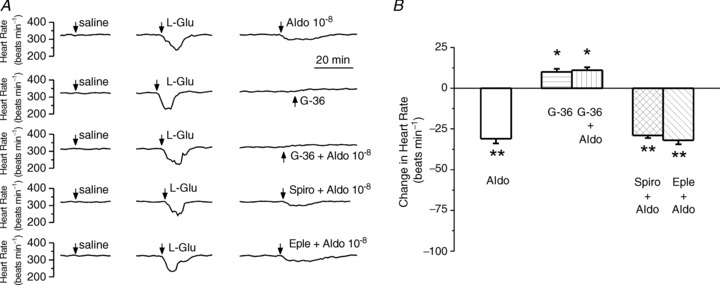
A, example of changes in heart rate after microinjection of control saline, l-glutamate (L-Glu, 5 × 10−3 m, 50 nl), aldosterone (Aldo, 10−8 m, 50 nl), G-36 (10−6 m) and aldosterone (Aldo, 10−8 m, 50 nl) in the presence of G-36 (10−6 m), spironolactone (10−6 m) or eplerenone (10−6 m). The decrease in heart rate produced by aldosterone (10−8 m) was abolished by the GPER antagonist, G-36 (10−6 m), while not affected by the mineralocorticoid receptor antagonists spironolactone (10−6 m) and eplerenone (10−6 m). B, comparison of the change in heart rate induced by microinjection into nucleus ambiguus of aldosterone in the absence and presence of GPER and mineralocorticoid receptor antagonists. *P < 0.05 and **P > 0.05 as compared to bradycardia induced by 10−8 m aldosterone.
Discussion
Increasing evidence supports the non-genomic actions of aldosterone (for review see Funder, 2005). Despite the fact that the receptors mediating faster effects of aldosterone remained unclear, recent evidence indicates that GPER mediates non-genomic effects of aldosterone (Gros et al. 2011a,b, 2013; Feldman & Gros, 2011; Funder, 2011). We have previously reported that GPER is expressed in nucleus ambiguus neurons (Brailoiu et al. 2007) and its activation leads to bradycardia (Brailoiu et al. 2013). In the current study we examined the effect of aldosterone on cardiac vagal neurons of nucleus ambiguus using in vitro and in vivo approaches.
Since one of the non-genomic effects associated with aldosterone action is an increase in cytosolic Ca2+ concentration ([Ca2+]i; Wehling et al. 1995; Schneider et al. 1997; Estrada et al. 2000; Harvey & Higgins, 2000), and GPER activation increases cytosolic Ca2+ in several cellular models (Revankar et al. 2005; Brailoiu et al. 2007; Tica et al. 2011: Deliu et al. 2012; Brailoiu et al. 2013) we first used calcium imaging to examine the effects of aldosterone in cardiac-projecting neurons of nucleus ambiguus. Neurons were retrogradely labelled with rhodamine, a reliable marker for identification of cardiac preganglionic vagal neurons (Bouairi et al. 2006; Brailoiu et al. 2012, 2013).
Aldosterone increased cytosolic Ca2+ concentration in rhodamine-labelled cardiac vagal neurons of nucleus ambiguus in a dose-dependent manner. Similarly, aldosterone has been reported to increase cytosolic Ca2+ in other cell models such as human colon cells (Doolan et al. 1998), rat vascular smooth muscle cells (Wehling et al. 1995), and porcine endothelial cells (Schneider et al. 1997). In addition, in nucleus ambiguus neurons, the Ca2+ response elicited by aldosterone was prevented by G-36, an antagonist of GPER, supporting the involvement of GPER. We have recently reported that another GPER synthetic agonist, G-1 (Bologa et al. 2006) increased cytosolic Ca2+ in cardiac vagal neurons of nucleus ambiguus; similarly, the effect of G-1 on Ca2+ was abolished by G-36 (Brailoiu et al. 2013). With respect to the increase in cytosolic Ca2+ produced, aldosterone was more potent, but less efficacious than G-1. The increase in cytosolic Ca2+ produced by aldosterone was faster, reaching a peak within 2 min after aldosterone treatment, and maintained a long-lasting plateau (for about 10 min). Similarly, aldosterone produced a fast-onset increase in cytosolic Ca2+ followed by a plateau, in skin cells from mineralocorticoid receptor knockout mice (Haseroth et al. 1999). On the other hand, in nucleus ambiguus neurons, the increase in cytosolic Ca2+ occurred 5–6 min after G-1 administration and was also followed by a sustained increase (Brailoiu et al. 2013). The delayed response to G-1 was similar to our previous findings in hypothalamus neurons (Brailoiu et al. 2007), spinal cord neurons (Deliu et al. 2012), or in myometrial cells (Tica et al. 2011). These differences in the Ca2+ response elicited by G-1 and aldosterone may be due to differences in drug–receptor interaction for GPER agonists. While the activation of GPER by oestrogen, an endogenous agonist, and G-1, a synthetic agonist, has been extensively studied (Revankar et al. 2005; Bologa et al. 2006; Prossnitz & Barton, 2011), there is limited information on the pharmacology of aldosterone–GPER interaction.
Another notable observation was that aldosterone-induced increase in cytosolic Ca2+ was not affected by administration of mineralocorticoid antagonists spironolactone and eplerenone, in concentrations that reduced the response to aldosterone in other cellular models (Gros et al. 2011b). The aldosterone-induced Ca2+ response in skeletal muscle cells was also insensitive to spironolactone (Estrada et al. 2000).
With respect to the source of Ca2+, we found that both Ca2+ release from internal stores and Ca2+ influx were involved. In human colon cells (Doolan et al. 1998), vascular smooth muscle cells (Wehling et al. 1995), and porcine endothelial cells (Schneider et al. 1997), the predominant mechanism of aldosterone-induced Ca2+ increase was Ca2+ release from internal stores. In nucleus ambiguus neurons, aldosterone releases Ca2+ via activation of IP3 receptors, and less via ryanodine receptors. IP3 receptors and ryanodine receptors are Ca2+-release channels located predominantly on the endoplasmic reticulum (Berridge, 1998). The endoplasmic reticulum Ca2+ store is sensitive to thapsigargin, an inhibitor of the sarcoplasmic/endoplasmic reticulum ATPase. Similarly, in rat and rabbit aortic smooth muscle cells, aldosterone mobilizes Ca2+ from endoplasmic reticulum thapsigargin-sensitive stores (Wehling et al. 1995). On the other hand, endo-lysosomal Ca2+ stores did not seem to play a role in Ca2+ mobilization by aldosterone in nucleus ambiguus, as blocking of Ca2+ release from endo-lysosomes with Ned-19 (Naylor et al. 2009) failed to affect the Ca2+ response elicited by aldosterone.
In addition to Ca2+ release from endoplasmic reticulum, aldosterone induced Ca2+ influx via P/Q-type Ca2+ channels, but not via L-type and N-type Ca2+ channels in nucleus ambiguus neurons. P/Q channels are the predominant type of voltage-gated Ca2+ channels identified in these neurons (Irnaten et al. 2003). Similar to aldosterone, urocortin 3 also activates P/Q Ca2+ channels in nucleus ambiguus neurons (Chitravanshi et al. 2012, Brailoiu et al. 2012).
Further, aldosterone induced a depolarization of nucleus ambiguus neurons that was sensitive to antagonism of GPER but not of mineralocorticoid receptors. The depolarization had a similar time course to the Ca2+ increase elicited by aldosterone; it is possible that the depolarization induced by aldosterone triggered the Ca2+ influx by P/Q channels. The Ca2+ influx may further increase Ca2+ signals by activating ryanodine receptors via a Ca2+-induced Ca2+ release mechanism. Similar to the Ca2+ signal elicited by aldosterone, with respect to the depolarization induced, aldosterone was more potent but less efficacious than G-1. The depolarization induced by aldosterone also had a faster onset: it started around 1 min after the administration of aldosterone and it was long-lasting, while the depolarization to G-1 was slower, reaching the peak within 5 min (Brailoiu et al. 2013), suggesting an agonist-selective interaction with the receptor.
We tested the significance of our in vitro findings by in vivo telemetric measurement of the heart rate in response to microinjection of aldosterone into the nucleus ambiguus. To prevent any potential interference of anaesthetics with the cardiovascular reflexes (Irnaten et al. 2002a,b) as well as handling of the rats, which may act as a stressor (McDougall et al. 2004), we examined the effect of aldosterone in conscious, freely moving rats. Microinjection of aldosterone into the nucleus ambiguus induces bradycardia, as previously reported by the synthetic GPER agonist, G-1, in anaesthetized rats (Brailoiu et al. 2013). Aldosterone-induced bradycardia was dose-dependent and not affected by the mineralocorticoid antagonists spironolactone and eplerenone, unlike previous findings in vascular smooth muscle and endothelial cells, where both GPER and mineralocorticoid receptors contributed to the effects of aldosterone (Gros et al. 2011a,b). Aldosterone-induced bradycardia was abolished by the GPER antagonist G-36. Similarly, a recent study indicates that the vascular effects of aldosterone or G-1 were blocked by another GPER antagonist, G-15 (Gros et al. 2013).
To further verify whether or not GPER is a common effector for G-1 and aldosterone, we tested the effect of concomitant microinjection of maximal doses of G-1 and aldosterone into the nucleus ambiguus; the two agonists did not produce a higher bradycardic response, as compared to G-1 alone, supporting GPER as a common pathway for the effects of G-1 and aldosterone.
Our results indicate that the activation of nucleus ambiguus neurons by aldosterone is mediated by GPER without an involvement of mineralcorticoid receptors, as previously reported in other cellular models (Gros et al. 2011a,b). In addition, our results do not support a role for eplerenone as a GPER antagonist, as previously suggested (Funder, 2011). Lower concentrations of aldosterone tested in our study mimic those present in physiological settings (Schirpenbach et al. 2006), while higher concentrations are associated with hypertension, hyperaldosteronism and heart failure (Weber, 2001; Losel et al. 2004). Moreover, aldosterone synthesis occurs in the brain and has been involved in cardiovascular regulation (Gomez-Sanchez et al. 2010). For example, the aldosterone concentration in the rat hypothalamus is around 350 pg g−1 (Huang et al. 2009).
The physiological and pathophysiological significance of the increase in cardiac vagal tone by aldosterone remains to be determined; it may be part of a compensatory mechanism. Our results also propose a new mechanism for an earlier reported increase in the cardiac vagal tone by aldosterone in healthy humans (Heindl et al. 2006). Moreover, the current findings indicate that GPER, in addition to mediating the effects of aldosterone on cell growth and vascular reactivity (Gros et al. 2011a,b, 2013), also mediates neuronal effects of aldosterone.
Acknowledgments
None.
Glossary
- 2-APB
2-aminoethoxydiphenyl borate
- [Ca2+]i
cytosolic Ca2+ concentration
- GPER
G protein-coupled oestrogen receptor
- HBSS
Hanks’ balanced salt solution
- IP3
inositol 1,4,5-trisphosphate
Additional information
Competing interests
None.
Author contributions
G.C.B., K.B., J.B.A., E.G., J.E.R. and W.J.K. contributed to the collection, analysis and interpretation of data, and drafting and revising the manuscript. E.B. contributed to the conception and design of the experiments, collection, analysis and interpretation of data, and drafting and revising the manuscript. All authors approved the final version of the manuscript. In vitro and in vivo experiments were carried out at Thomas Jefferson University and Temple University; synthesis of G-1 and G-36 was carried out at New Mexico State University.
Funding
This work was supported by NIH grants HL090804 (to E.B.), HL091096 and HL091799 (to J.E.R.) and CA127731 (to J.B.A.).
References
- Benamar K, Addou S, Yondorf M, Geller EB, Eisenstein TK, Adler MW. Intrahypothalamic injection of the HIV-1 envelope glycoprotein induces fever via interaction with the chemokine system. J Pharmacol Exp Ther. 2010;332:549–553. doi: 10.1124/jpet.109.160309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signalling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, Oprea TI, Prossnitz ER. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. 2006;2:207–212. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- Bouairi E, Kamendi H, Wang X, Gorini C, Mendelowitz D. Multiple types of GABAA receptors mediate inhibition in brain stem parasympathetic cardiac neurons in the nucleus ambiguus. J Neurophysiol. 2006;96:3266–3272. doi: 10.1152/jn.00590.2006. [DOI] [PubMed] [Google Scholar]
- Brailoiu E, Dun SL, Brailoiu GC, Mizuo K, Sklar LA, Oprea TI, Prossnitz ER, Dun NJ. Distribution and characterization of estrogen receptor G protein-coupled receptor 30 in the rat central nervous system. J Endocrinol. 2007;193:311–321. doi: 10.1677/JOE-07-0017. [DOI] [PubMed] [Google Scholar]
- Brailoiu GC, Arterburn JB, Oprea TI, Chitravanshi VC, Brailoiu E. Bradycardic effects mediated by activation of G protein-coupled estrogen receptor in rat nucleus ambiguus. Exp Physiol. 2013;98:679–691. doi: 10.1113/expphysiol.2012.069377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brailoiu GC, Deliu E, Tica AA, Chitravanshi VC, Brailoiu E. Urocortin 3 elevates cytosolic calcium in nucleus ambiguus neurons. J Neurochem. 2012;122:1129–1136. doi: 10.1111/j.1471-4159.2012.07869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brailoiu GC, Gurzu B, Gao X, Parkesh R, Aley PK, Trifa DI, Galione A, Dun NJ, Madesh M, Patel S, Churchill GC, Brailoiu E. Acidic NAADP-sensitive calcium stores in the endothelium: agonist-specific recruitment and role in regulating blood pressure. J Biol Chem. 2010;285:37133–37137. doi: 10.1074/jbc.C110.169763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauner T, Hulser DF, Strasser RJ. Comparative measurements of membrane potentials with microelectrodes and voltage-sensitive dyes. Biochim Biophys Acta. 1984;771:208–216. doi: 10.1016/0005-2736(84)90535-2. [DOI] [PubMed] [Google Scholar]
- Chitravanshi VC, Bhatt S, Sapru HN. Microinjections of α-melanocyte stimulating hormone into the nucleus ambiguus of the rat elicit vagally mediated bradycardia. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1402–R1411. doi: 10.1152/ajpregu.90978.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitravanshi VC, Kawabe K, Sapru HN. Bradycardic effects of microinjections of urocortin 3 into the nucleus ambiguus of the rat. Am J Physiol Regul Integr Comp Physiol. 2012;303:R1023–1030. doi: 10.1152/ajpregu.00224.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deliu E, Brailoiu GC, Arterburn JB, Oprea TI, Benamar K, Dun NJ, Brailoiu E. Mechanisms of G protein-coupled estrogen receptor-mediated spinal nociception. J Pain. 2012;13:742–754. doi: 10.1016/j.jpain.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MK, Field AS, Burai R, Ramesh C, Petrie WK, Bologa CG, Oprea TI, Yamaguchi Y, Hayashi S, Sklar LA, Hathaway HJ, Arterburn JB, Prossnitz ER. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J Steroid Biochem Mol Biol. 2011;127:358–366. doi: 10.1016/j.jsbmb.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolan CM, O'Sullivan GC, Harvey BJ. Rapid effects of corticosteroids on cytosolic protein kinase C and intracellular calcium concentration in human distal colon. Mol Cell Endocrinol. 1998;138:71–79. doi: 10.1016/s0303-7207(98)00020-3. [DOI] [PubMed] [Google Scholar]
- Estrada M, Liberona JL, Miranda M, Jaimovich E. Aldosterone- and testosterone-mediated intracellular calcium response in skeletal muscle cell cultures. Am J Physiol Endocrinol Metab. 2000;279:E132–E139. doi: 10.1152/ajpendo.2000.279.1.E132. [DOI] [PubMed] [Google Scholar]
- Feldman RD, Gros R. Unraveling the mechanisms underlying the rapid vascular effects of steroids: sorting out the receptors and the pathways. Br J Pharmacol. 2011;163:1163–1169. doi: 10.1111/j.1476-5381.2011.01366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funder JW. The nongenomic actions of aldosterone. Endocr Rev. 2005;26:313–321. doi: 10.1210/er.2005-0004. [DOI] [PubMed] [Google Scholar]
- Funder JW. GPR30, mineralocorticoid receptors, and the rapid vascular effects of aldosterone. Hypertension. 2011;57:370–372. doi: 10.1161/HYPERTENSIONAHA.110.165076. [DOI] [PubMed] [Google Scholar]
- Gomez-Sanchez EP, Gomez-Sanchez CM, Plonczynski M, Gomez-Sanchez CE. Aldosterone synthesis in the brain contributes to Dahl salt-sensitive rat hypertension. Exp Physiol. 2010;95:120–130. doi: 10.1113/expphysiol.2009.048900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros R, Ding Q, Davis M, Shaikh R, Liu B, Chorazyczewski J, Pickering JG, Feldman RD. Delineating the receptor mechanisms underlying the rapid vascular contractile effects of aldosterone and estradiol. Can J Physiol Pharmacol. 2011a;89:655–663. doi: 10.1139/y11-062. [DOI] [PubMed] [Google Scholar]
- Gros R, Ding Q, Liu B, Chorazyczewski J, Feldman RD. Aldosterone mediates its rapid effects in vascular endothelial cells through GPER activation. Am J Physiol Cell Physiol. 2013;304:C532–C540. doi: 10.1152/ajpcell.00203.2012. [DOI] [PubMed] [Google Scholar]
- Gros R, Ding Q, Sklar LA, Prossnitz EE, Arterburn JB, Chorazyczewski J, Feldman RD. GPR30 expression is required for the mineralocorticoid receptor-independent rapid vascular effects of aldosterone. Hypertension. 2011b;57:442–451. doi: 10.1161/HYPERTENSIONAHA.110.161653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Harvey BJ, Higgins M. Nongenomic effects of aldosterone on Ca2+ in M-1 cortical collecting duct cells. Kidney Int. 2000;57:1395–1403. doi: 10.1046/j.1523-1755.2000.00981.x. [DOI] [PubMed] [Google Scholar]
- Haseroth K, Gerdes D, Berger S, Feuring M, Gunther A, Herbst C, Christ M, Wehling M. Rapid nongenomic effects of aldosterone in mineralocorticoid-receptor-knockout mice. Biochem Biophys Res Commun. 1999;266:257–261. doi: 10.1006/bbrc.1999.1771. [DOI] [PubMed] [Google Scholar]
- Heindl S, Holzschneider J, Hinz A, Sayk F, Fehm HL, Dodt C. Acute effects of aldosterone on the autonomic nervous system and the baroreflex function in healthy humans. J Neuroendocrinol. 2006;18:115–121. doi: 10.1111/j.1365-2826.2005.01392.x. [DOI] [PubMed] [Google Scholar]
- Huang BS, White RA, Jeng AY, Leenen FH. Role of central nervous system aldosterone synthase and mineralocorticoid receptors in salt-induced hypertension in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol. 2009;296:R994–R1000. doi: 10.1152/ajpregu.90903.2008. [DOI] [PubMed] [Google Scholar]
- Irnaten M, Aicher SA, Wang J, Venkatesan P, Evans C, Baxi S, Mendelowitz D. μ-Opioid receptors are located postsynaptically and endomorphin-1 inhibits voltage-gated calcium currents in premotor cardiac parasympathetic neurons in the rat nucleus ambiguus. Neuroscience. 2003;116:573–582. doi: 10.1016/s0306-4522(02)00657-7. [DOI] [PubMed] [Google Scholar]
- Irnaten M, Wang J, Chang KS, Andresen MC, Mendelowitz D. Ketamine inhibits sodium currents in identified cardiac parasympathetic neurons in nucleus ambiguus. Anesthesiology. 2002a;96:659–666. doi: 10.1097/00000542-200203000-00023. [DOI] [PubMed] [Google Scholar]
- Irnaten M, Wang J, Venkatesan P, Evans CBA, Chang KS, Andresen MC, Mendelowitz D. Ketamine inhibits presynaptic and postsynaptic nicotinic excitation of identified cardiac parasympathetic neurons in nucleus ambiguus. Anesthesiology. 2002b;96:667–674. doi: 10.1097/00000542-200203000-00024. [DOI] [PubMed] [Google Scholar]
- Liu SL, Schmuck S, Chorazcyzewski JZ, Gros R, Feldman RD. Aldosterone regulates vascular reactivity: short-term effects mediated by phosphatidylinositol 3-kinase-dependent nitric oxide synthase activation. Circulation. 2003;108:2400–2406. doi: 10.1161/01.CIR.0000093188.53554.44. [DOI] [PubMed] [Google Scholar]
- Losel R, Schultz A, Boldyreff B, Wehling M. Rapid effects of aldosterone on vascular cells: clinical implications. Steroids. 2004;69:575–578. doi: 10.1016/j.steroids.2004.05.005. [DOI] [PubMed] [Google Scholar]
- McDougall SJ, Lawrence AJ, Widdop RE. Differential cardiovascular responses to stressors in hypertensive and normotensive rats. Exp Physiol. 2005;90:141–150. doi: 10.1113/expphysiol.2004.028308. [DOI] [PubMed] [Google Scholar]
- Naylor E, Arredouani A, Vasudevan SR, Lewis AM, Parkesh R, Mizote A, Rosen D, Thomas JM, Izumi M, Ganesan A, Galione A, Churchill GC. Identification of a chemical probe for NAADP by virtual screening. Nat Chem Biol. 2009;5:220–226. doi: 10.1038/nchembio.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Fourth Edition. San Diego: Academic Press; 1998. [Google Scholar]
- Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol. 2011;16:715–726. doi: 10.1038/nrendo.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signalling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- Schirpenbach C, Seiler L, Maser-Gluth C, Beuschlein F, Reincke M, Bidlingmaier M. Automated chemiluminescence-immunoassay for aldosterone during dynamic testing: comparison to radioimmunoassays with and without extraction steps. Clin Chem. 2006;52:1749–1755. doi: 10.1373/clinchem.2006.068502. [DOI] [PubMed] [Google Scholar]
- Schmidt BM, Horisberger K, Feuring M, Schultz A, Wehling M. Aldosterone blunts human baroreflex sensitivity by a nongenomic mechanism. Exp Clin Endocrinol Diabetes. 2005;113:252–256. doi: 10.1055/s-2005-837650. [DOI] [PubMed] [Google Scholar]
- Schmidt BM, Montealegre A, Janson CP, Martin N, Stein-Kemmesies C, Scherhag A, Feuring M, Christ M, Wehling M. Short term cardiovascular effects of aldosterone in healthy male volunteers. J Clin Endocrinol Metab. 1999;84:3528–3533. doi: 10.1210/jcem.84.10.6020. [DOI] [PubMed] [Google Scholar]
- Schmidt BM, Oehmer S, Delles C, Bratke R, Schneider MP, Klingbeil A, Fleischmann EH, Schmieder RE. Rapid nongenomic effects of aldosterone on human forearm vasculature. Hypertension. 2003;42:156–160. doi: 10.1161/01.HYP.0000083298.23119.16. [DOI] [PubMed] [Google Scholar]
- Schneider M, Ulsenheimer A, Christ M, Wehling M. Nongenomic effects of aldosterone on intracellular calcium in porcine endothelial cells. Am J Physiol Endocrinol Metab. 1997;272:E616–E620. doi: 10.1152/ajpendo.1997.272.4.E616. [DOI] [PubMed] [Google Scholar]
- Schöniger S, Wehming S, Gonzalez C, Schöbitz K, Rodríguez E, Oksche A, Yulis CR, Nürnberger F. The dispersed cell culture as model for functional studies of the subcommissural organ: preparation and characterization of the culture system. J Neurosci Methods. 2001;107:47–61. doi: 10.1016/s0165-0270(01)00351-x. [DOI] [PubMed] [Google Scholar]
- Tica AA, Dun EC, Tica OS, Gao X, Arterburn JB, Brailoiu GC, Oprea TI, Brailoiu E. G protein-coupled estrogen receptor 1-mediated effects in the rat myometrium. Am J Physiol Cell Physiol. 2011;301:C1262–C1269. doi: 10.1152/ajpcell.00501.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber KT. Aldosterone in congestive heart failure. N Engl J Med. 2001;345:1689–1697. doi: 10.1056/NEJMra000050. [DOI] [PubMed] [Google Scholar]
- Wehling M. Specific, nongenomic actions of steroid hormones. Annu Rev Physiol. 1997;59:365–393. doi: 10.1146/annurev.physiol.59.1.365. [DOI] [PubMed] [Google Scholar]
- Wehling M, Neylon CB, Fullerton M, Bobik A, Funder JW. Nongenomic effects of aldosterone on intracellular Ca2+ in vascular smooth muscle cells. Circ Res. 1995;76:973–979. doi: 10.1161/01.res.76.6.973. [DOI] [PubMed] [Google Scholar]
- Wehling M, Spes CH, Win N, Janson CP, Schmidt BM, Theisen K, Christ M. Rapid cardiovascular action of aldosterone in man. J Clin Endocrinol Metab. 1998;83:3517–3522. doi: 10.1210/jcem.83.10.5203. [DOI] [PubMed] [Google Scholar]
- Yee KM, Struthers AD. Aldosterone blunts the baroreflex response in man. Clin Sci (Lond) 1998;95:687–692. doi: 10.1042/cs0950687. [DOI] [PubMed] [Google Scholar]
- Yu Y, Wei SG, Zhang ZH, Gomez-Sanchez E, Weiss RM, Felder RB. Does aldosterone upregulate the brain renin-angiotensin system in rats with heart failure. Hypertension. 2008;51:727–733. doi: 10.1161/HYPERTENSIONAHA.107.099796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZH, Yu Y, Kang YM, Wei SG, Felder RB. Aldosterone acts centrally to increase brain renin-angiotensin system activity and oxidative stress in normal rats. Am J Physiol Heart Circ Physiol. 2008;294:H1067–H1074. doi: 10.1152/ajpheart.01131.2007. [DOI] [PubMed] [Google Scholar]