

Abstract
Pancreatic cancer is a highly fatal disease characterized by a dominant stroma formation. Exploring new biological targets, specifically those overexpressed in stroma cells, holds significant potential for the design of specific nanocarriers to attain homing of therapeutic and imaging agents to the tumor. In clinical specimens of pancreatic cancer, we found increased expression of CD59 in tumor associated endothelial cells as well as infiltrating cells in the stroma as compared to uninvolved pancreas. We explored this dual targeting effect using orthotopic human pancreatic cancer in nude mice. By immunofluorescence analysis, we confirmed the increased expression of Ly6C, mouse homolog of CD59, in tumor associated endothelial cells as well as in macrophages within the stroma. We decorated the surface of porous silicon nanocarriers with Ly6C antibody. Targeted nanocarriers injected intravenously accumulated to tumor associated endothelial cells within 15 minutes. At 4 hours after administration, 9.8±2.3% of injected dose/g tumor of the Ly6C targeting nanocarriers accumulated in the pancreatic tumors as opposed to 0.5±1.8% with non-targeted nanocarriers. These results suggest that Ly6C (or CD59) can serve as a novel dual target to deliver therapeutic agents to the stroma of pancreatic tumors.
Keywords: Ly6C, nanocarriers, porous silicon, endothelial cells, marker
1. Introduction
Pancreatic cancer is one of the most aggressive malignancies in humans. It is the fourth leading cause of cancer death in the United States with more than 43,000 new cases diagnosed and 37,000 deaths annually [1; 2]. The difficulty in detecting pancreatic cancer at an early stage, the aggressive nature of the disease, and the lack of effective therapy are all responsible for the high mortality from this disease, with a 1-year survival rate of 18%, and a 5-year survival rate of less than 3% [3]. Systemic therapy with gemcitabine, most frequently used against pancreatic cancer, has not increased the median survival of patients beyond 6 months and often leads to resistance [4; 5]. The discouraging situation has remained unchanged over the past 20 years [6]. Although, molecular components and signaling pathways involved in pancreatic carcinogenesis have been targeted with therapeutic intent, erlotinib, an EGFR tyrosine kinase inhibitor, is the only agent that has shown a very limited extension of overall survival (2 weeks) in the clinic. Clearly, there is an urgent need to develop novel therapeutic strategies for the devastating disease. Nevertheless, the main body of the research in the field has neglected the fact that transport and retention of anticancer agents in the tumor can be a major roadblock responsible for clinical failure.
Stroma, which occupies 70-90% of the tumor volume, is a typical histological finding in pancreatic cancer and is considered to be one of the main factors preventing efficient therapy [7]. Recent studies suggest that the stroma is blocking drug penetration and contributing to tumor invasion, survival and metastasis [8]. As such, cellular stroma elements, including tumor associated macrophages, endothelial cells, fibroblasts, lymphocytes and neutrophils represent potential targets for novel cancer therapies. Specific markers for the stroma cells can be explored to concentrate therapeutic carriers with nanoscale features (nanocarriers) in the tumor site. Unlike passive accumulation strategies, collectively termed Enhanced Permeation and Retention (EPR) effect [9], which take advantage of the increased permeability of blood vessels in solid malignancies, a promising approach to achieve the selective delivery of nanocarriers, is the active targeting of tumor specific antigens, employing biomolecule recognition. Various forms of active targeting of nanocarriers to tumor cells and microenvironment have been considered in the past [10]. In pancreatic tumors, the proposed strategies focused mainly on targeting tumor cells. As an example, Khan et al. recently published a study about targeting gold nanoparticles to epidermal growth factor receptor expressed on pancreatic cells. In this study, the authors have demonstrated 1.2-2 times higher accumulation of the targeted nanoparticles as compared to untargeted nanoparticles in the tumor model lacking stroma [11]. In another work, Weissleder et al. screened a library comprising 146 nanoparticles decorated with different synthetic small molecules and found that in ectopic pancreatic tumors (grown in the flank of mice) there was an accumulation of the nanoparticles targeted specifically to pancreatic cancer cells possessing low binding to macrophages and endothelial cells [12].
Specific markers on cells in the tumor microenvironment, e.g. tumor associated endothelial cells, are particularly suitable for enhanced delivery of nanocarriers, since they are readily accessible to the systems injected intravenously [13]. However, in the case of pancreatic tumors, targeting only tumor associated endothelial cells may not be a good strategy for efficient active delivery, due to a general hypovascularization of the lesion [4; 5; 6]. Other cell elements that are present in the stroma of the pancreatic tumors are tumor associated macrophages which have a high potential to engulf foreign materials [14]. Finding molecular signatures expressed by several cell populations in the stroma should increase specific delivery of the nanocarreirs.
Here we propose a new targeting strategy to enable accumulation of drug carriers in the tumor stroma. Previous studies have shown increased gene expression of CD59, GPI-linked cell surface glycoprotein, in some inflammatory conditions and malignancies [15; 16; 17]. Our data show that CD59, and its murine analogue Ly6C are overexpressed by tumor associated stroma cells in clinical specimens and in vivo orthotopic models of human pancreatic cancer, respectively. The markers are present on various subpopulations of stroma cells, including tumor associated macrophages and endothelial cells. The nanocarriers employed in this work are comprised of biodegradable and biocompatible nanoporous silicon material[18] and we demonstrate the efficiency of using Ly6C for delivery of nanocarriers to the stroma in the orthotopic pancreatic tumors in mice.
2. Materials and Methods
2.1. Evaluation of CD59 expression in clinical specimens of pancreatic cancer
Human pancreatic cancer specimens (n=6) were obtained by informed consent from patients at the University of Texas M. D. Anderson Cancer Center, Houston, TX. Paraffin embedded sections of the specimen were immunostained using CD59 antibody (Sigma, St. Louis, MO) (Ab) followed by corresponding secondary Ab (Jackson ImmunoResearch, West Grove, PA). Positive reaction was detected by exposure to stable 3,3′-diaminobenzidine (Phoenix Biotechnologies, Huntsville, AL). Immunostaining with CD34-Ab (Biogenex Laboratories, San Ramon, CA) was used for identification of endothelial cells. For double staining of the sections, CD34 was stained in red using Streptavidin AP with chromagen Vulcan fast Red (Biocare Medical, Concord, CA), whereas CD59 was stained in blue using chromagen Ferangi Blue (Biocare Medical).
2.2. Cell lines
Human pancreatic cancer cell lines L3.6pl [19] (originated in Dr. I.J. Fidler’s laboratory) and MPanc96 (kindly provided by Dr Craig Logsdon, MD Anderson Cancer Center, Houston, TX) were validated by short tandem repeat (STR) DNA fingerprinting using the AmpFℓSTR Identifiler kit (Applied Biosystems, Carlsbad, CA) [20]. L3.6pl and MPanc96 were maintained in minimal essential medium supplemented with 10% fetal bovine serum (FBS), sodium pyruvate, nonessential amino acids, L-glutamine, a two-fold vitamin solution (Life Technologies, Inc., Grand Island, NY), and a penicillin-streptomycin mixture (Flow Laboratories, Rockville, MD), as described previously [21].
Murine skin endothelial cells were established from female H-2Kb-tsA58 mice (Charles River Laboratories, Wilmington, MA) as described previously [22].
2.3. Evaluation of Ly6C expression by endothelial cells in vitro
Murine skin endothelial cells were incubated for 12 hours in the culture medium with or without 1 ng/ml of recombinant mouse IFN-γ (R&D Systems, Minneapolis, MN). Following the trypsinization, the cells were stained with fluorescein isothiocyanate (FITC)-labeled Ly6C-Ab or control IgG (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Ly6C expression was analyzed by flow cytometry on a FACScan flow cytometer (Becton Dickinson, Heidelberg, Germany) and the data were analyzed by FlowJo software (Tree Star, Inc., Ashland, OR)[23].
2.4. A model of orthotopic pancreatic tumors in mice
All animal procedures were performed in accordance with the regulations of the M.D. Anderson Cancer center for the Care and Use of Laboratory Animals based on the protocols approved by the IACUC committee. Male athymic nude mice (8-12 weeks, NCI-nu, Bethesda, USA) were maintained in the VAF-barrier facility at M.D. Anderson Cancer center with a regular light-dark cycle. The animals were anesthetized by isoflurane inhalation and, following a small abdominal incision, the pancreas was exposed for tumor implantation. The two orthotopic models investigated in this study were established by injection of either 5×104 L3.6pl or MPanc96 pancreatic tumor cells to produce slow growing tumors as opposed to regular injection of 1×106 cells to establish rapid growing tumors as previously described[20]. This model of slow tumor growth enables formation of increased volume of stroma, mimicking clinical situation. The tumors are formed twelve weeks after the injection.
2.5. Immunofluorescenct analysis of the orthotopic tumors
Twelve weeks after the injection of tumor cells, the mice were anesthetized and euthanized by cervical dislocation. Tumors were harvested and embedded in optimum cutting temperature compound (Miles, Inc., Elkhart, IN), rapidly frozen in liquid nitrogen, and stored at −80°C. Frozen sections were mounted on slides and fixed by cold acetone (5 minutes), acetone/chloroform (1:1 v/v, 5 minutes), and acetone (5 minutes). The sections were treated with protein block solution. Immunofluorescence staining of frozen tissues using phycoerythrin (PE)-labeled CD31 (AbD Serotec, Oxford, UK), FITC-labeled anti-mouse Ly6C antibody (Santa Cruz Biotechnology) and Alexa-647 labeled antibody to CD204 and CD68 (AbD Serotec) was performed. The images were captured by confocal microscope (Carl Zeiss MicroImaging Inc., Thornwood, NY) and analyzed by the associated image analysis software [21].
2.6. Identification of Dividing Cells by BrdU Staining in Vivo
To identify dividing cells in vivo, we injected mice intraperitoneally with 0.2 ml of saline containing 200 μg of BrdU (BD Biosciences, San Jose, CA). The mice were sacrificed 24 hours after the injection, and tumor samples were harvested and frozen as described above. After fixation with acetone, the sections were treated with 1% Triton X-100 in PBS for 8 minutes and incubated for 15 minutes with 2N HCl in PBS at 37°C. Sections were next treated overnight at 4°C with protein blocking solution and primary biotinylated anti-BrdU antibody (Abcam, Inc., Cambridge, MA) and then treated for 1 hour at room temperature with streptavidin-Cy2 (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). Dividing endothelial cells exhibited green nuclei.
2.7. Targeted nanocarriers fabrication and surface modification
Nanocarriers, silicon mesoporous particles (S1MP), were fabricated by semiconductor processing and electrochemical etching in the Microelectronics Research Center at The University of Texas at Austin as previously described [24; 25; 26]. Briefly, hemispherical particles with radius of 0.8μm and 20-50 nm pores were formed by selective electrochemical etch of a SiN masked array of 1μm cylindrical trenches in the silicon in a mixture of hydrofluoric acid (49% HF) and ethanol (3:7 v/v). The hydroxyl groups on the S1MP surface were introduced through oxidation in a piranha solution (1:2 H2O2: concentrated H2SO4 (v/v), Sigma) at 100-110 °C for 2 h. To enable Ly6C-Ab conjugation, S1MP were modified with 3-Aminopropyltriethoxysilane (APTES, Sigma) [25; 26; 27]. APTES modified S1MP were reacted for one hour with 10μM Ly6C-Ab, or control IgG (Santa Cruz Biotechnology) to produce Ly6C-Ab-S1MP or IgG-S1MP, respectively. For in vitro and vivo imaging, NHS-ester Dylight 488 or Dylight 649 (Thermo Fisher Scientific Inc., Rockford, IL) was further conjugated either to the APTES modified surface of S1MP or to Ly6C-Ab or control IgG prior to the chemical conjugation. The particles were then washed (by centrifugation at 4200rpm × 15min) in deionized water 4-6 times to remove any unreacted molecules.
2.8. Targeted nanocarriers characterization
Volumetric particle size, size distribution and count were obtained using a Z2 Coulter® Particle Counter and Size Analyzer (Beckman Coulter, Fullerton, CA) as previously described [25; 26]. The zeta potential of the silicon particles was analyzed using a Zetasizer nano ZS (Malvern Instruments Ltd., Southborough, MA). For the analysis, 2 μL particle suspension containing at least 2×105 particles to give a stable zeta potential values were injected into a sample cell countering filed with phosphate buffer (PB, 1.4 mL, pH 7.3). The analysis was conducted at room temperature (23°C) in triplicates.
Scanning Electron Microscopy (SEM) was applied to examine the structure and morphology of S1MP. Samples were sputter-coated with gold for 2 min at 10nm using a CrC-150 Sputtering System (Torr International, New Windsor, NY) and observed under a FEI Quanta 400 field emission scanning electron microscope (FEI Company, Hillsboro, OR) at an accelerating voltage of 20 kV, chamber pressure of 0.45 Torr and spot size 5.0.
2.9. Evaluation of in vitro binding of the nanocarriers to murine endothelial cells
The endothelial cells cultured in 96 well plates or chamber slides with or without IFN-γ were incubated for 30 minutes with Dylight 649 fluorescently labeled nanocarriers at a concentration of 2 S1MP: 1 cell. The following systems were examined: Ly6C-Ab-S1MP, IgG-S1MP and S1MP (n=3/each). At the end of the incubation, the supernatant was removed and the cells were washed twice with PBS and fixed. The number of the nanocarreirs attached to the endothelial cells was visualized by confocal microscope (Carl Zeiss MicroImaging) excitation/emission=652/670nm, through Cy 5channel and quantified by measuring fluorescent intensities using microplate reader (BioTek, Winooski, VT).
2.10. Evaluation of biodistribution of the nanocarriers
Fluorescently labeled Ly6C-Ab-S1MP, IgG-S1MP or S1MP, were injected to normal mice or L3.6pl tumor bearing mice. The injection volume was 0.1 mL, the particles (5E7) were suspended in sterile PBS and injected via tail-vein of the mice. Fifteen minutes or four hours after the injection, the mice (n=3/each) were euthanized and tumor as well as major organs, such as, brain, lung, heart, liver, spleen, kidneys, thigh bone and pancreas were harvested. Fluorescent images of the organs were captured by IVIS200 imaging system (Caliper Life Sciences, Hopkinton, MA) [27; 28]. For quantification of the accumulation of injected nanocarriers, parts of the organs were weighted and further processed for the elemental silicon analysis by Inductive Couples Plasma Atomic Emission Spectroscopy (ICP-AES) as previously described[27]. Briefly, the organs were homogenized in 20% EtOH in 1N NaOH, left for 48 hours at room temperature for extraction of Si, centrifuged (4200rpm × 10min), diluted and analyzed for Si contents. The Si contents is then recalculated to the total silicon in each sample (100%) which is analyzed in the original nanocarriers suspensions, and also normalized to percentage of total silicon resulting from the number of particles injected, or to the individual organ weight. Other parts of the organs were frozen for immunofluorescence analysis as described above.
2.11. Statistical Analysis
Statistical analysis of the expression of Ly6C by endothelial cells in vitro, the fluorescence intensities of the particles in 96 well plates, the amount of silicon per gram tissue in mouse was performed by the Mann-Whitney U test with a cutoff value of 0.05.
3. Results
3.1. CD59 expression in clinical specimens of pancreatic cancer
Exploring new biological targets that are overexpressed in pancreatic cancer holds an important therapeutic potential. Based on the involvement of CD59 in inflammatory conditions and colon cancer, reported in the literature [15; 16; 17], we examined the expression of this marker in clinical specimens of pancreatic cancer. Hematoxilin and eosin (H&E) staining confirmed the presence of dense stroma surrounding cancer epithelial cells (Figure 1A). Immunohistochemical analysis of the specimens using antibodies to CD34, revealed endothelial cells in the capillaries of both uninvolved pancreas and the pancreatic tumors. In contrast, CD59 staining showed an overexpression of the marker only in the tumor. A double staining of the tissue using two antibodies (CD34/CD59, Figure 1B) showed that CD59 (blue) is expressed by CD34 positive endothelial cells (red) in capillaries (black arrows) as well as in infiltrating stroma cells (red arrows). Cancer cells also expressed high level of CD59 (Fig 1S, Supportive information).
Figure 1.

Hematoxylin and eosin (H&E), CD34 (endothelial cells) and CD59 staining of clinical specimens from pancreatic cancer patients. (A) Cancer epithelial cells were surrounded by high abundance of stroma. Although CD34 positive endothelial cells were found in both uninvolved pancreas and tumor, expression of CD59 was markedly increased only in tumor as compared to those in uninvolved pancreas. (B) In tumor, CD59 (Blue) was expressed by CD34 positive (Red) endothelial cells in capillaries (black arrows) as well as by infiltrating stroma cells (red arrows). Bar=50μm
3.2. Expression of Ly6C, murine homolog of CD59, in stroma of orthotopic human pancreatic cancer in mice
In order to enable in vivo CD59-based targeting of the pancreatic tumors in nude mice, we further evaluated the expression of Ly6C, the murine homolog of CD59, in the stroma of two orthotopic human pancreatic cancer models in mice. Based on our previous data [20], slow growing pancreatic tumor model was used to enable a formation of the extensive stroma, which is a characteristic finding in patients with pancreatic cancer. Histological analysis of orthotopic L3.6pl and MPanc96 tumor showed intensive stroma formation surrounding cancer cells, similarly to clinical specimens (Figure 2, H&E). Immunofluorescent analysis of the tumors and normal pancreas pointed towards increased expression of Ly6C on CD31 positive tumor associated endothelial cells (Ly6C/CD31 superimposition emits yellow fluorescence) in capillaries as compared to those in normal pancreas. Ly6C can be expressed by angiogenic vessels. Supportive information 2S shows an example of a large vessel and a sprouting capillary (arrows) both expressing CD31. Endothelial cells in a sprouting capillary are known to divide actively. Thus, double immunofluorescent staining with Ly6C antibody revealed that only the endothelial cells in the sprouting capillary strongly expressed Ly6C. Infiltrated CD68 positive tumor associated macrophages also expressed Ly6C, indicating infiltration of activated monocytes/macrophages into tumor stroma (Figure 2). These numerous number of Ly6C positive cells also expressed CD204, the surface marker of so-called M2 macrophage, in the stroma of L3.6pl tumors (Fig 3S, Supportive information). In contrast, the number of CD204 and/or Ly6C positive macrophages was obviously less in normal pancreas. The proliferation of the CD204 positive cells in the stroma of the L3.6pl tumor was rare, evaluated by immunefluoresent staining of BrdU positive cells.
Figure 2.
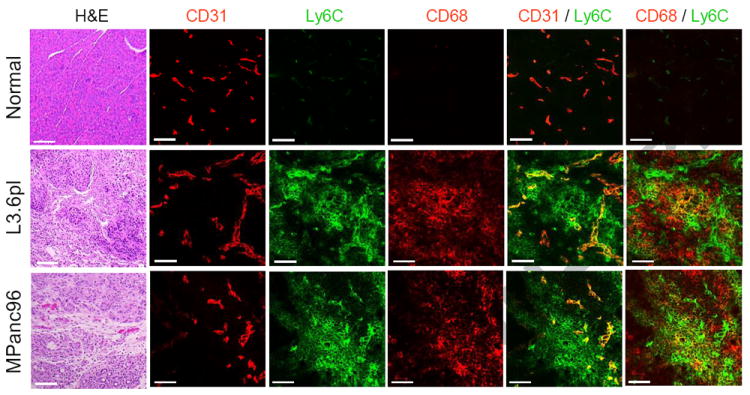
Histological (H&E) and immunofluorescent analysis of orthotopic L3.6pl and MPanc96 human pancreatic cancer in the pancreas of nude mice and normal pancreas. Tumor cells were surrounded by stroma similar to clinical specimens as shown in Figure 1A. Increased expression of Ly6C (green) on CD31 (red) positive endothelial cells or infiltrating CD68 (red) positive tumor associated macrophages as shown by yellow (superimposed) color was found in the tumors as compared to normal pancreas. Bar=50μm
3.3. Conjugation of Ly6C to S1MP surface and Ly6C-Ab-S1MP characterization
Hemispherical S1MP were microfabricated following our previously described protocols [24; 26] (Figure 3). Analysis of hundreds of S1MP visualized by SEM confirmed the reproducibility of the process. The analysis showed 1.6μm diameter (Figure 3A) and 0.6 μm thickness quasi-hemispherical S1MP originated from 1 μm diameter trenches in the Si [24]. Analysis of physical stability of the particles stored in distilled water or isopropyl alcohol performed by ICP-AES has shown no degradation of the particles in one-year storage period (Fig. 4S-A, Supportive information). Microfabricated S1MP originally have a hydrophobic surface. A multi-step process is used to conjugate an antibody to the particle surface, starting from an oxidation of the nanocarriers in strong acidic. The ζ-potential is the electro-kinetic potential directly related to the net surface electrical charge of each particle formulation. Hydroxyl groups on the surface of S1MP impart negative ζ-potential of S1MP (mV, Figure 3B) and can serve as a background moieties for silane-chemistry based reactions. Positively charged APTES molecules are coupled to the surface reversing the net surface charge of the particles to +5.7mV. The amine groups of APTES served as a linker for Ab conjugation. The conjugation of the Ab on the particle neutralized the APTES positive charges resulting in a slightly negative zeta potential (Figure 3B). In addition, to validate the zeta potential, the FTIR spectra showed that upon modification with APTES, new peaks appeared in the range of 1500 – 1700. These peaks can be attributed to the bending bands of NH2 and of protonated amines (-NH3+) [29]. The elemental analysis has shown a significant increase in the normalized values of carbon and nitrogen following the conjugation of isotype and Ly6C antibodies to the S1MP surface. When microfabricated, silicon particles do not include carbon and nitrogen in their structure, thus the increase in these values points toward a successful conjugation of the antibody as also can be evidenced from the confocal microscopy images, showing a stable fluorescent signal from all particles used in this study (Figure 3C-D). When S1MP conjugated to any antibody are used, the antibody was first conjugated to the NHS fluorescent dye and then to the surface of the nanocarrier. In this study we optimized the system based on the binding of fluorescently labeled antibody (FITC-IgG) to the particles surface (Fig 4S-B – Supportive information). Concentration of FITC-IgG in the range of 1.88-45 μM were tested for conjugation to the surface of APTES modified S1MP. The data shows that at 10 μM of FITC-IgG there is a saturation of the amount of the antibody on the surface and further increase in the FITC-IgG concentration in the reaction medium is not reflected in additional molecules bound to the surface. Based on these results, we further used 10 μM for our study.
Figure 3.
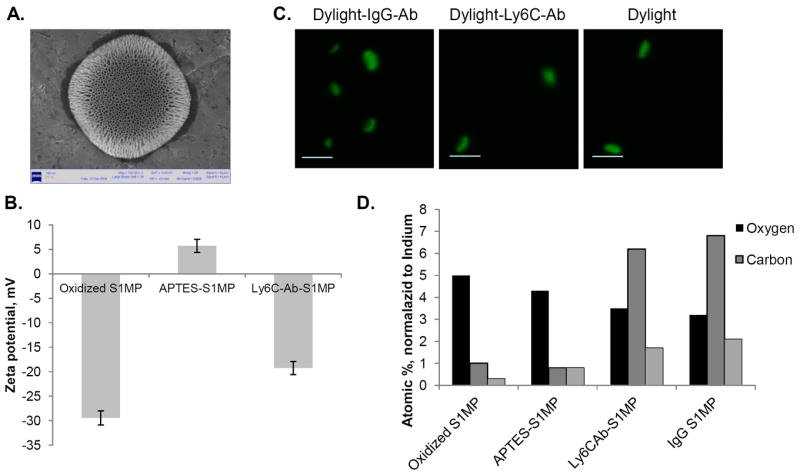
Characterization of conjugation of Ly6C-Ab to the surface of S1MP. (A) Scanning Electron Microscopy image of a single S1MP carriers; (B) Changes in ζ-potential of S1MP as a function of various modification steps (n=6); (C) Confocal microscopic images of S1MP conjugated to either fluorescent probe Dylight488 (Dylight) or to fluorescently labeled isotype Ab (Dylight-IgG-Ab) or Ly6C antibody (Dylight-Ly6C-Ab), Bar=2 μm; (D) Elemental analysis of S1MP as a function of various surface conjugations.
3.4. In vitro targeting of the nanocarriers to endothelial cells
Next, we tested the in vitro association of the nanocarriers with the endothelial cells. The basal expression of Ly6C on murine skin endothelial cells was low, but, as for other reported cell populations [15], incubation of cells with IFN-γ increased the expression of the glycoprotein (Figure 4A). Further, the binding of the nanocarriers with different surface modification to the endothelial cells either stimulated with IFN-γ or unstimulated was evaluated. The tested systems included fluorescently labeled Ly6C-Ab-S1MP, IgG-S1MP and S1MP nanocarriers. Quantified data showed that among the tested systems, Ly6C-Ab-S1MP bound most efficiently to the endothelial cells and the binding efficiency corresponded well to the levels of Ly6C expression. The expression of Ly6C did not affect binding of both S1MP and IgG-S1MP to the endothelial cells, which showed 3-6 times lower fluorescent intensity values in comparison to cells incubated with Ly6C-Ab-S1MP (Figure 4B). Confocal microscopy data confirmed the binding of Ly6C-Ab-S1MP to the stimulated endothelial cells (Figure 4C).
Figure 4.
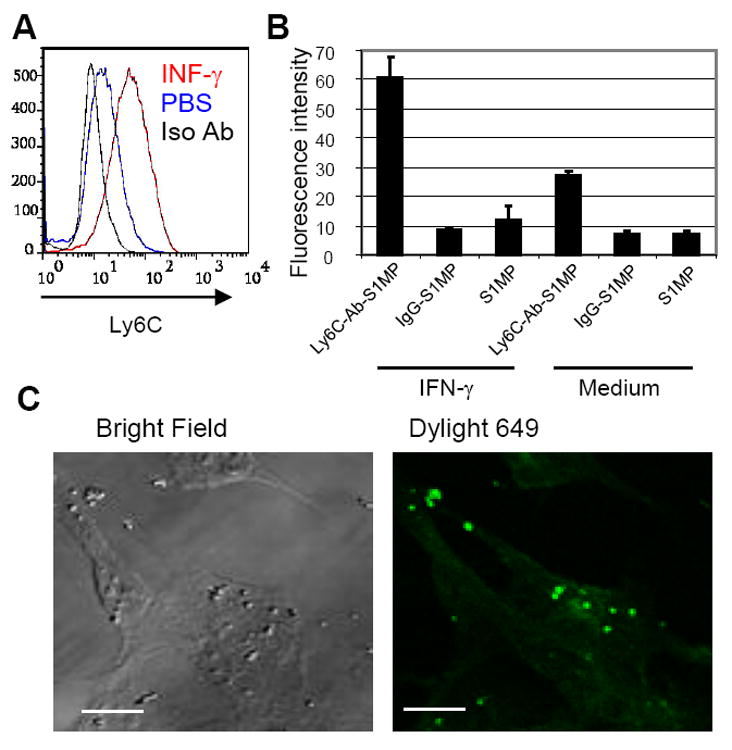
Expression of Ly6C in cultured skin endothelial cells in vitro and affinity of the S1MP carriers to the cells. (A): Flow cytometric analysis of Ly6C expression by skin endothelial cells incubated with or without IFN-γ. Shown in the plots are the cells incubated with IFN-γ in medium and stained with Ly6C antibody (red: INF-γ), stained with iso-type control antibody (black: Iso Ab), or incubated with PBS in medium and stained with Ly6C antibody (blue: PBS). (B): Affinity of the nanocarriers with different surface modifications to the endothelial cells incubated with or without IFN-γ in vitro. (C): Confocal microscopic image of the nanocarriers conjugated with Ly6C Ab (Dylight 649 : pseudo color in green) and endothelial cells (Bright field) stimulated with IFN-γ. These results are representative of two independent experiments. Bar=5μm.
3.5. Biodistribution of the nanocarriers in normal mice and mice with orthotopic L3.6pl human pancreatic cancer
Following the establishment of the pancreatic tumors with significant stroma formation, fluorescent Ly6C-Ab-S1MP and control nanocarriers, IgG-S1MP and S1MP were administered into the tail vein. Healthy (normal) mice receiving similar dose of the nanocarriers served as control. The biodistribution of the systems was examined by a number of techniques, including ex vivo fluorescent imaging of the excised whole organs (IVIS), immunofluorescent staining of tumor sections and quantitative analysis of elemental Si by ICP-AES.
After only 15 minutes, a specific accumulation of the Ly6C-Ab-S1MP nanocarriers in the pancreatic tumors as compared to other organs of the tumor-bearing mice was observed by ex vivo fluorescent imaging (Figure 5A). The highest fluorescent intensity associated with the untargeted control nanocarriers, S1MP and IgG-S1MP, in L3.6pl mice and Ly6C-Ab-S1MP in healthy mice was seen in the lungs of the animals. Low accumulation of the untargeted control IgG-S1MP in the pancreatic tumors and no accumulation of the Ly6C-Ab-S1MP into the healthy pancreas was detected.
Figure 5.

Biodistribution and immunofluorescent analysis of the nanocarriers injected i.v. into the L3.6pl human tumor bearing or normal mice. (A): Ex vivo fluorescent imaging (IVIS) of the organs of normal mouse and the tumor bearing mice injected with the nanocarriers conjugated with Ly6C Ab (Ly6C-Ab-S1MP), control IgG (IgG-S1MP) or unconjugated nanocarriers (S1MP). (B and C): Immunofluorescent analysis of pancreatic tumors in the mouse injected with the nanocarriers conjugated with Ly6C Ab (labeled with Dylight 649 and detected through Cy5 channel). The nanocarriers attached to endothelial cells (CD31) in capillaries which also expressed Ly6C (emitted yellow fluorescence) 15 minutes after i.v. injection (indicated by white arrows). The nanocarriers were further engulfed by CD68 and Ly6C positive tumor associated macrophages as identified 4 hours after the injection. These results are representative of two independent experiments. Bar=5μm; (D) ICP-AES quantitative analysis of targeted (Ly6Cab-S1MP, white) and untargeted (SMP, black) nanocarriers biodistribution in various organs (n=4-5); (E) ICP-AES quantitative analysis of nanocarriers biodistribution revealed that 9.8±2.3% of injected dose/g tumor of the Ly6C-Ab-S1MP accumulated in the pancreatic tumors as opposed to 0.5±1.8% with IgG-S1MP (n=4-5). In normal pancreas, accumulation of S1MP was low, regardless of the surface modification.
Immunofluorescence analysis of resected L3.6pl tumor in the mouse injected with the targeted to Ly6C systems (Ly6C-Ab-S1MP) revealed that the nanocarriers successfully attached to endothelial cells in capillaries (CD31 positive) which also expressed Ly6C (as evidenced by yellow fluorescence), 15 minutes after i.v. injection (Figure 5B). At the later timepoint (4 hours after the injection), the carriers were engulfed by tumor associated macrophages, which also expressed both CD68 and Ly6C (Figure 5C). Lower magnification of the images showed wide distribution of the nanocarriers in the tumor stroma (Fig 5S, Supportive information).
Quantification of the S1MP in the tissues by ICP-AES revealed high levels of the nanocarriers in L3.6pl tumor (Figure 5D). At 4 hours after administration, 9.8±2.3% of injected dose/g tumor of the Ly6C-Ab-S1MP accumulated in the pancreatic tumors as opposed to 0.5±1.8% with IgG-S1MP. In normal pancreas, accumulation of S1MP was low, regardless of the surface modification, which can be explained by low expression of Ly6C in normal pancreas. There is also a significant fraction of the particles which accumulates to liver. We have previously shown that S1MP concentrated in the liver do not produce inflammatory responses and changes in the organ biochemistry in healthy animals [30]. As regarding to clinical consequences, since the main site of metastasis of pancreatic tumors is the liver, we anticipate that the concentration of the particles not only to the primary site (pancreas) but also into the liver could be beneficial for the patients.
4. Discussion
The inability to deliver therapeutically relevant levels of drugs to cancer cells in the tumor microenvironment partially accounts for the lack of efficient therapies for pancreatic cancer [31; 32; 33]. Published reports clearly show that drugs reach their desired lesion targets only in one part per 10,000-100,000 molecules [34; 35]. This unfavorable biodistribution generally decreases the effect of therapeutics at the tumor sites and induces side effects at the other organs. Stroma, occupying up to 90% of the tumor mass, is a typical histological finding in adenocarcinoma of the pancreas. It consists of various cellular elements, including tumor associated endothelial cells, macrophages, fibroblasts, lymphocytes, neutrophils and components of extracellular matrix [36; 37; 38]. The identification of specific markers for stroma cells in pancreatic cancer may allow superior delivery of therapeutic and/or imaging molecules by means of targeted nanocarriers injected intravenously. Moreover, since stroma builds tumor microenviroment favoring cancer cells proliferation, invasion and metastasis, it was shown in the clinic that anti-stroma therapy of various tumors can provide a significant survival benefit to cancer patients [14].
Endothelium comprises the first cell layer to which a substance administered to systemic circulation is exposed, therefore, using this layer as a target to therapeutic nanovectors through molecular signatures overexpressed on tumor-associated vascular cells is an appealing strategy to improve delivery of drugs to tumor tissue [13]. While the vast majority of tumors are hypervascularized, pancreatic tumors are known to be hypovascularized as compared to the surrounding tissue, making targeting only endothelial cells a less efficient strategy for achieve higher concentrations in the disease loci and other targets in the tumor microenvironment should be considered. CD59 is a GPI-anchored membrane protein and originally identified to protect cells against complement attack [15]. CD59 is expressed by peripheral blood lymphocytes, monocytes as well as tissue macrophages and endothelial cells in inflammatory lesion such as atherosclerotic vessels [16]. In malignant disease, increased gene expression of CD59 in colon cancer specimen as compared to corresponding normal tissue has been reported [17]. Overexpression of CD59 on tumors can protect them from direct complement lysis. Moreover, CD59 has been shown to promote tumor growth and decrease survival in vivo [39]. In this study, we identified that CD59 expression is increased in tumor associated endothelial cells as well as infiltrating cells into the tumor stroma in the patients with pancreatic cancer as compared to uninvolved pancreas. Since stroma cells belong to the host and we were aiming to initially conduct a preclinical work, we further evaluated the expression of CD59 murine analogue, Ly6C in orthotopic human pancreatic tumors in mice. Indeed, similarly to its clinical counterpart, increased expression of Ly6C is identified in both tumor associated endothelial cells as well as macrophages. Endothelial cells and macrophages both belong to reticuloendothelial system and, therefore, would share phenotype including protein expression [40; 41; 42].
Here we investigated targeting of silicon mesoporous particles (S1MP) to pancreatic tumors, utilizing their conjugation to Ly6C (or CD59) as a dual targeting of tumor associated endothelial cells and macrophages. Nanoporous silicon is biodegradable and biocompatible as we and others previously reported [26; 43]. The specific non-spherical shape of S1MP is enabled by precise microfabrication through photolithography and electrochemical etching [24; 25]. In general, it allows for better margination in the blood stream and receptor specific adhesion with consequent targeting of tumor vasculature [44; 45; 46]. S1MP constitute the first stage of the multistage nanocarriers developed for delivery of therapeutics to tumors [18; 25]. S1MP carriers can further be functionalized on their surface with targeting ligands [47] and load nanoparticles in their porous structure producing Multi-Stage Nanonovector system [18; 25]. While S1MP is rationally designed to enable targeting tumor microenvironment, the smaller particles which are hosted inside the pores of S1MP and so called second stage nanoparticles (S2NP) are further released to specifically affect cancer cells. Our previous studies have shown that a variety of S2NP or nanoparticle ‘cocktails’ can be loaded into the pores of mesoporous S1MP and efficiently delivered to the disease site enabling simultaneous functions for multiple applications[18; 25; 48]. From the therapeutic point of view, we have shown that administration of a single dose of MSV loaded with neutral nanoliposomes containing small interfering RNA (siRNA) targeted against the EphA2 oncoprotein, resulted in an extremely prolonged gene silencing for at least 21 days associated with significantly reduced tumor burden in orthotopic models of ovarian cancer [48]. Another study has demonstrated that upon loading gold-nanoshells into S1MP and following intratumoral injection to breast cancer xenografts, the efficiency of thermal ablation of the tumor by near-infrared ablation is significantly higher than when compared to the injection of nanoshells alone at the same dose[49].
Ly6C positive ‘inflammatory’ monocytes are recruited from bone marrow to the inflammatory lesions including tumor. Most of these monocytes are thought to differentiate into tumor associated macrophages [50]. Various signals from tumor microenvironment selectively polarize macrophages into specific phenotype which facilitate tumor progression by inducing angiogenesis, remodeling extracellular matrix and promoting tumor cell motility [51]. Therefore these macrophages can be a novel target for cancer therapy, especially for pancreatic cancer due to of high abundance of stroma cells/macrophages in tumors. These macrophages are so-called M2 macrophages and characterized by high expression of both the macrophage scavenger receptor (CD204) and the mannose receptor [52]. In our study, numerous number of Ly6C positive cell also expressed CD204 in the stroma of L3.6pl tumor. On the other hand, the number of CD204 and Ly6C positive macrophages was minimal in normal pancreas. Proliferation of CD204 positive macrophages in the tumor stroma was rare evaluated by BrdU staining and these data indicate the CD204/Ly6C positive tumor associated macrophages are originated by recruitment of Ly6C positive inflammatory monocytes rather than originated by proliferation of resident macrophages in normal pancreas. In the present work, S1MPs were conjugated to antibody against Ly6C or control IgG. The conjugation of the antibody was confirmed by several techniques, including elemental analysis, changes in the net ζ-potential and confocal microscopy. Silicon nanocarriers conjugated with Ly6C antibody but not control IgG or unconjugated nanocarriers showed high affinity to the cells expressing Ly6C in vitro. Further, the nanocarriers injected intravenously to mice with orthotopic human pancreatic cancer showed a high degree of accumulation within a short timeframe (15 minutes-4 hours) into the tumor tissue, as was obvious from macroscopic evaluation of fluorescent intensity in pancreas as compared to various organs using IVIS apparatus and elemental analysis of silicon (which constitutes S1MP) in the tissues. Untargeted control nanocarriers in the tumor bearing mice and Ly6C targeted S1MP in normal mice were observed in the lung tissue within 15 minutes, indicating that these systems are still highly present in the circulation. After targeted delivery of Ly6C-Ab-S1MP to the tumor associated endothelial cells which express Ly6C, MSV extravasated through endothelial cell layer and were engulfed by tumor associated macrophages which expressed Ly6C, confirmed by immunofluorescent stainings. Tumor vessels are well known to be leaky and have large pore sizes, which for some tumor types can be as large as a few micrometers in size [53; 54; 55]. Whether pore size of tumor associated vessels in the pancreatic tumors in patients are also large or not is needed to be determined.
EPR effect on nanoformaultions, discussed earlier in this manuscript, was generally tested using spherical particles. In our recent study, we observed a five-time higher accumulation of non-spherical (discoidal) particulates into the tumor tissue [56]. The particles were associated with endothelial cells, macrophages and other cells in the tumor stroma. Further, in our intravital microscopy studies (data not shown) we have not observed significant uptake of S1MP by blood monocytes. While our in vitro work shows a possibility for S1MP to be internalized by endothelial cells, the transcytosis was not observed with S1MP, but with nanoparticles encapsulated in the porous structure of S1MP [57]
We are further considering to explore this efficient targeting of Ly6C-Ab-S1MP to the tumor tissue pursuing three possible therapeutic options through loading into the pores of S1MP with (1) gemcitabine and exploring the effect of ~20 times (from 0.5% to 9.8 %, based on our in vivo experiments) increased concentration of the drug in the tumor tissue on the therapeutic effect; (2) a combination of nanocarriers loaded with gemcitabine and siRNAs against genes reported to be involved in the resistance, such as NFκB [58; 59]; (3) gold nanoparticles enabling radio-frequency [60]/near-infra-red [61] induced ablation of the tumors. The last option is particularly appealing, since successful therapy will induce apoptosis and necrosis of tumor cells, thus increasing inflammation and recruiting additional monocytes/macrophages into the tumor as a target for delivering Ly6C-Ab-S1MP [40; 62]. Ly6C is expressed by inflammatory monocytes/macrophages and these cells continuously migrate into inflamed area including tumor[50; 63]. Tumors cause constant inflammation like a wound and, as stated by Dvorak in mid 80s’, produces “wound that does not heal”[64]. Therefore increase in the treatment efficacy by sequential therapies can be anticipated and resistant mechanism will not be developed to this innate immune reaction.
In summary, we report that CD59/Ly6C is a novel marker expressed by both tumor associated endothelial cells and macrophages in the stroma of pancreatic cancer. Targeting this marker for active delivery of the nanocarriers conjugated with Ly6C antibody produced a successful accumulation of the nanocarriers into the stroma of orthotopic human pancreatic cancer in nude mice and, therefore, can serve as a basis for improved therapy.
Supplementary Material
Figure 1S: CD59 staining (blue) of clinical specimens from pancreatic cancer patients. In tumor, CD59 was highly expressed by cancer cells (indicated by arrows) as well as stroma cells including endothelial cells. Bar=100μm
Figure 2S: Immunofluorescent analysis of a large vessel with a sprouting capillary (arrows) in L3.6pl pancreatic tumor. The tissue was stained using antibodies to CD31 and Ly6C. Although endothelial cells in both the large vessel and sprouting capillary were CD31 positive (Red), endothelial cells were stained with Ly6C and CD31 (emits yellow) only in the sprouting capillary. Immune cells surrounding the vessels expressed only Ly6C. Scale bar indicates 20 μm.
Figure 3S: (A) Immunofluorescent analysis of normal pancreas and L3.6pl tumors in the pancreas using antibody to CD204 (M2 macrophage marker) and Ly6C. Bar=100 μm.
(B) Immunofluorescent staining of L3.6pl tumor using antibody to CD204 and BrdU (proliferation marker). The tumor bearing mice were sacrificed 24 hours after intra-peritoneal injected with BrdU and tumor sections were stained with antibodies against BrdU (pseudo color in green) and CD204 (red). Bar=50 μm
Figure 4S: (A) Physical stability of the particles as measured by ICP-AES analysis for up to 12 months. The samples were stored in distilled water or isopropyl alcohol. (B) Optimization of antibody conjugation to the surface of S1MP. Concentration of FITC-IgG in the range of 1.88-45 μM were tested for conjugation to the surface of APTES modified S1MP. Following the incubation, S1MP were extensively washed and fluorescent intensity of the pellet was detected. The data shows that at 10 μM there is a saturation of the amount of the antibody on the surface and further increase in the FITC-IgG concentration in the reaction medium is not reflected in additional molecules bound to the surface. Based on these results, we further used 10 μM for our study.
Figure 5S: Lower magnification of the images in Figure 5B (A) and 5C (B) as indicated by white squares, respectively. Arrows in (A) indicate attached nanocarriers to the endothelial cells. Bar=10μm
Acknowledgments
The authors acknowledge a financial support from the following sources: Cancer Center Support Core Grant CA16672, U54CA143837, U54CA151668, DODW81XWH-07-2-0101 and the Ernest Cockrell Jr. Distinguished Endowed Chair. STR DNA fingerprinting was done by the Cell Line Core Facility, MD Anderson Cancer Center, funded in part by Cancer Center Support Grant.
Footnotes
Conflicts of Interest
M. Ferrari is the founding scientist and a member of the Board of Directors of Leonardo Biosystems, a member of Board of Directors of ArrowHead Research Corporation, and hereby discloses potential financial interests in the companies. Other authors disclosed no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Pliarchopoulou K, Pectasides D. Pancreatic cancer: current and future treatment strategies. Cancer Treat Rev. 2009;35:431–436. doi: 10.1016/j.ctrv.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 3.NCI, Pancreatic cancer. [May 2, 2012];2012 http://www.cancer.gov/cancertopics/types/pancreatic.
- 4.Parsons CM, Sutcliffe JL, Bold RJ. Preoperative evaluation of pancreatic adenocarcinoma. J Hepatobiliary Pancreat Surg. 2008;15:429–435. doi: 10.1007/s00534-007-1240-7. [DOI] [PubMed] [Google Scholar]
- 5.Kim MP, Gallick GE. Gemcitabine Resistance in Pancreatic Cancer: Picking the Key Players. Clinical Cancer Research. 2008;14:1284–1285. doi: 10.1158/1078-0432.CCR-07-2247. [DOI] [PubMed] [Google Scholar]
- 6.Vaccaro V, Melisi D, Bria E, Cuppone F, Ciuffreda L, Pino MS, Gelibter A, Tortora G, Cognetti F, Milella M. Emerging pathways and future targets for the molecular therapy of pancreatic cancer. Expert Opin Ther Targets. 2011;15:1183–1196. doi: 10.1517/14728222.2011.607438. [DOI] [PubMed] [Google Scholar]
- 7.Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2007;6:1186–1197. doi: 10.1158/1535-7163.MCT-06-0686. [DOI] [PubMed] [Google Scholar]
- 8.Mueller MM, Fusenig NE. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4:839–849. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- 9.Maeda H, Matsumura Y. EPR effect based drug design and clinical outlook for enhanced cancer chemotherapy. Adv Drug Deliv Rev. 2011;63:129–130. doi: 10.1016/j.addr.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Torchilin VP. Passive and active drug targeting: drug delivery to tumors as an example. Handb Exp Pharmacol. 2010:3–53. doi: 10.1007/978-3-642-00477-3_1. [DOI] [PubMed] [Google Scholar]
- 11.Khan JA, Kudgus RA, Szabolcs A, Dutta S, Wang E, Cao S, Curran GL, Shah V, Curley S, Mukhopadhyay D, Robertson JD, Bhattacharya R, Mukherjee P. Designing Nanoconjugates to Effectively Target Pancreatic Cancer Cells In Vitro and In Vivo. PLoS ONE. 2011;6:e20347. doi: 10.1371/journal.pone.0020347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weissleder R, Kelly K, Sun EY, Shtatland T, Josephson L. Cell-specific targeting of nanoparticles by multivalent attachment of small molecules. Nat Biotech. 2005;23:1418–1423. doi: 10.1038/nbt1159. [DOI] [PubMed] [Google Scholar]
- 13.Ruoslahti E, Bhatia SN, Sailor MJ. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010;188:759–768. doi: 10.1083/jcb.200910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmed F, Steele JC, Herbert JM, Steven NM, Bicknell R. Tumor stroma as a target in cancer. Curr Cancer Drug Targets. 2008;8:447–453. doi: 10.2174/156800908785699360. [DOI] [PubMed] [Google Scholar]
- 15.Philbrick WM, Palfree RG, Maher SE, Bridgett MM, Sirlin S, Bothwell AL. The CD59 antigen is a structural homologue of murine Ly-6 antigens but lacks interferon inducibility. Eur J Immunol. 1990;20:87–92. doi: 10.1002/eji.1830200113. [DOI] [PubMed] [Google Scholar]
- 16.Seifert PS, Roth I, Schmiedt W, Oelert H, Okada N, Okada H, Bhakdi S. CD59 (homologous restriction factor 20), a plasma membrane protein that protects against complement C5b-9 attack, in human atherosclerotic lesions. Atherosclerosis. 1992;96:135–145. doi: 10.1016/0021-9150(92)90060-t. [DOI] [PubMed] [Google Scholar]
- 17.van Beijnum JR, Dings RP, van der Linden E, Zwaans BM, Ramaekers FC, Mayo KH, Griffioen AW. Gene expression of tumor angiogenesis dissected: specific targeting of colon cancer angiogenic vasculature. Blood. 2006;108:2339–2348. doi: 10.1182/blood-2006-02-004291. [DOI] [PubMed] [Google Scholar]
- 18.Godin B, Tasciotti E, Liu X, Serda RE, Ferrari M. Multistage Nanovectors: From Concept to Novel Imaging Contrast Agents and Therapeutics. Acc Chem Res. 2011 doi: 10.1021/ar200077p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruns CJ, Harbison MT, Kuniyasu H, Eue I, Fidler IJ. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia. 1999;1:50–62. doi: 10.1038/sj.neo.7900005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yokoi K, Hawke D, Oborn CJ, Jang JY, Nishioka Y, Fan D, Kim SW, Kim SJ, Fidler IJ. Identification and validation of SRC and phospho-SRC family proteins in circulating mononuclear cells as novel biomarkers for pancreatic cancer. Transl Oncol. 2011;4:83–91. doi: 10.1593/tlo.10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yokoi K, Sasaki T, Bucana CD, Fan D, Baker CH, Kitadai Y, Kuwai T, Abbruzzese JL, Fidler IJ. Simultaneous inhibition of EGFR, VEGFR, and platelet-derived growth factor receptor signaling combined with gemcitabine produces therapy of human pancreatic carcinoma and prolongs survival in an orthotopic nude mouse model. Cancer Res. 2005;65:10371–10380. doi: 10.1158/0008-5472.CAN-05-1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langley RR, Ramirez KM, Tsan RZ, Van Arsdall M, Nilsson MB, Fidler IJ. Tissue-specific microvascular endothelial cell lines from H-2K(b)-tsA58 mice for studies of angiogenesis and metastasis. Cancer Res. 2003;63:2971–2976. [PubMed] [Google Scholar]
- 23.Nagendra S, Schlueter AJ. Absence of cross-reactivity between murine Ly-6C and Ly-6G. Cytometry A. 2004;58:195–200. doi: 10.1002/cyto.a.20007. [DOI] [PubMed] [Google Scholar]
- 24.Chiappini C, Tasciotti E, Fakhoury JR, Fine D, Pullan L, Wang YC, Fu L, Liu X, Ferrari M. Tailored Porous Silicon Microparticles: Fabrication and Properties. Chemphyschem. 2010;11:1029–1035. doi: 10.1002/cphc.200900914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tasciotti E, Liu X, Bhavane R, Plant K, Leonard AD, Price BK, Cheng MM, Decuzzi P, Tour JM, Robertson F, Ferrari M. Mesoporous Silicon Particles as a Multistage Delivery System for Imaging and Therapeutic Applications. Nat Nanotechnol. 2008;3:151–157. doi: 10.1038/nnano.2008.34. [DOI] [PubMed] [Google Scholar]
- 26.Godin B, Gu J, Serda RE, Bhavane R, Tasciotti E, Chiappini C, Liu X, Tanaka T, Decuzzi P, Ferrari M. Tailoring the Degradation Kinetics of Mesoporous Silicon Structures through PEGylation. J Biomed Mater Res A. 2010;94:1236–1243. doi: 10.1002/jbm.a.32807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tasciotti E, Godin B, Martinez JO, Chiappini C, Bhavane R, Liu X, Ferrari M. Near-Infrared Imaging Method for the In Vivo Assessment of the Biodistribution of Nanoporous Silicon Particles. Mol Imaging. 2011;10:56–68. [PMC free article] [PubMed] [Google Scholar]
- 28.Liang M, Liu X, Cheng D, Liu G, Dou S, Wang Y, Rusckowski M, Hnatowich DJ. Multimodality nuclear and fluorescence tumor imaging in mice using a streptavidin nanoparticle. Bioconjug Chem. 2010;21:1385–1388. doi: 10.1021/bc100081h. [DOI] [PubMed] [Google Scholar]
- 29.Xia B, Xiao S-J, Guo D-J, Wang J, Chao J, Liu H-B, Pei J, Chen Y-Q, Tang Y-C, Liu J-N. Biofunctionalisation of porous silicon (PS) surfaces by using homobifunctional cross-linkers. Journal of Materials Chemistry. 2006;16:570–578. [Google Scholar]
- 30.Tanaka T, Godin B, Bhavane R, Nieves-Alicea R, Gu J, Liu X, Chiappini C, Fakhoury JR, Amra S, Ewing A, Li Q, Fidler IJ, Ferrari M. In vivo evaluation of safety of nanoporous silicon carriers following single and multiple dose intravenous administrations in mice. Int J Pharm. 2010;402:190–197. doi: 10.1016/j.ijpharm.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garber K. Stromal Depletion Goes on Trial in Pancreatic Cancer. Journal of the National Cancer Institute. 2010;102:448–450. doi: 10.1093/jnci/djq113. [DOI] [PubMed] [Google Scholar]
- 32.Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, Lolkema MP, Buchholz M, Olive KP, Gress TM, Tuveson DA. Stromal biology and therapy in pancreatic cancer. Gut. 2011;60:861–868. doi: 10.1136/gut.2010.226092. [DOI] [PubMed] [Google Scholar]
- 33.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, DeNicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Rückert F, Grützmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science (New York, N Y) 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jain RK. Barriers to Drug Delivery in Solid Tumors. Sci Am. 1994;271:58–65. doi: 10.1038/scientificamerican0794-58. [DOI] [PubMed] [Google Scholar]
- 35.Jain RK. Transport of Molecules, Particles, and Cells in Solid Tumors. Annu Rev Biomed Eng. 1999;1:241–263. doi: 10.1146/annurev.bioeng.1.1.241. [DOI] [PubMed] [Google Scholar]
- 36.Langley RR, Fidler IJ. The seed and soil hypothesis revisited--the role of tumor-stroma interactions in metastasis to different organs. Int J Cancer. 2011;128:2527–2535. doi: 10.1002/ijc.26031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Erkan M, Reiser-Erkan C, Michalski CW, Kleeff J. Tumor microenvironment and progression of pancreatic cancer. Exp Oncol. 2010;32:128–131. [PubMed] [Google Scholar]
- 38.Chu GC, Kimmelman AC, Hezel AF, DePinho RA. Stromal biology of pancreatic cancer. J Cell Biochem. 2007;101:887–907. doi: 10.1002/jcb.21209. [DOI] [PubMed] [Google Scholar]
- 39.Kolev M, Towner L, Donev R. Complement in cancer and cancer immunotherapy. Arch Immunol Ther Exp (Warsz) 2011;59:407–419. doi: 10.1007/s00005-011-0146-x. [DOI] [PubMed] [Google Scholar]
- 40.Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618–631. doi: 10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- 41.Kim SJ, Kim JS, Papadopoulos J, Wook Kim S, Maya M, Zhang F, He J, Fan D, Langley R, Fidler IJ. Circulating monocytes expressing CD31: implications for acute and chronic angiogenesis. Am J Pathol. 2009;174:1972–1980. doi: 10.2353/ajpath.2009.080819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuwana M, Okazaki Y, Kodama H, Satoh T, Kawakami Y, Ikeda Y. Endothelial differentiation potential of human monocyte-derived multipotential cells. Stem Cells. 2006;24:2733–2743. doi: 10.1634/stemcells.2006-0026. [DOI] [PubMed] [Google Scholar]
- 43.Canham LT INSPEC (Information service) Properties of porous silicon. INSPEC; London: 1987. [Google Scholar]
- 44.Decuzzi P, Ferrari M. The receptor-mediated endocytosis of nonspherical particles. Biophys J. 2008;94:3790–3797. doi: 10.1529/biophysj.107.120238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Decuzzi P, Lee S, Bhushan B, Ferrari M. A theoretical model for the margination of particles within blood vessels. Ann Biomed Eng. 2005;33:179–190. doi: 10.1007/s10439-005-8976-5. [DOI] [PubMed] [Google Scholar]
- 46.Godin B, Driessen WH, Proneth B, Lee SY, Srinivasan S, Rumbaut R, Arap W, Pasqualini R, Ferrari M, Decuzzi P. An Integrated Approach for the Rational Design of Nanovectors for Biomedical Imaging and Therapy. Adv Genet. 2010;69:31–64. doi: 10.1016/S0065-2660(10)69009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mann AP, Tanaka T, Somasunderam A, Liu X, Gorenstein DG, Ferrari M. E-Selectin-Targeted Porous Silicon Particle for Nanoparticle Delivery to the Bone Marrow. Adv Mater. 2011;23:H278–H282. doi: 10.1002/adma.201101541. [DOI] [PubMed] [Google Scholar]
- 48.Tanaka T, Mangala LS, Vivas-Mejia PE, Nieves-Alicea R, Mann AP, Mora E, Han HD, Shahzad MM, Liu X, Bhavane R, et al. Sustained Small Interfering RNA Delivery by Mesoporous Silicon Particles. Cancer Res. 2010;70:3687–3696. doi: 10.1158/0008-5472.CAN-09-3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen H, You J, Zhang G, Ziemys A, Li Q, Bai L, Deng X, Erm DR, Liu X, Li C, Ferrari M. Cooperative, Nanoparticle-Enabled Thermal Therapy of Breast Cancer. Advanced Healthcare Materials. 2012;1:84–89. doi: 10.1002/adhm.201100005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 51.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 52.Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, Rimoldi M, Biswas SK, Allavena P, Mantovani A. Macrophage polarization in tumour progression. Semin Cancer Biol. 2008;18:349–355. doi: 10.1016/j.semcancer.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 53.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Danhier F, Feron O, Preat V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148:135–146. doi: 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 55.Torchilin VP. Targeted pharmaceutical nanocarriers for cancer therapy and imaging. AAPS J. 2007;9:E128–147. doi: 10.1208/aapsj0902015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Godin B, Chiappini C, Srinivasan S, Alexander JF, Yokoi K, Ferrari M, Decuzzi P, Liu X. Discoidal Porous Silicon Particles: Fabrication and Biodistribution in Breast Cancer Bearing Mice. Advanced Functional Materials. 2012:n/a–n/a. doi: 10.1002/adfm.201200869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Serda RE, Mack A, van de Ven AL, Ferrati S, Dunner K, Jr, Godin B, Chiappini C, Landry M, Brousseau L, Liu X, et al. Logic-Embedded Vectors for Intracellular Partitioning, Endosomal Escape, and Exocytosis of Nanoparticles. Small. 2010;6:2691–2700. doi: 10.1002/smll.201000727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kong R, Sun B, Jiang H, Pan S, Chen H, Wang S, Krissansen GW, Sun X. Downregulation of nuclear factor-kappaB p65 subunit by small interfering RNA synergizes with gemcitabine to inhibit the growth of pancreatic cancer. Cancer Lett. 2010;291:90–98. doi: 10.1016/j.canlet.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 59.Yokoi K, Fidler IJ. Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin Cancer Res. 2004;10:2299–2306. doi: 10.1158/1078-0432.ccr-03-0488. [DOI] [PubMed] [Google Scholar]
- 60.Glazer ES, Zhu C, Massey KL, Thompson CS, Kaluarachchi WD, Hamir AN, Curley SA. Noninvasive radiofrequency field destruction of pancreatic adenocarcinoma xenografts treated with targeted gold nanoparticles. Clin Cancer Res. 2010;16:5712–5721. doi: 10.1158/1078-0432.CCR-10-2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bardhan R, Lal S, Joshi A, Halas NJ. Theranostic nanoshells: from probe design to imaging and treatment of cancer. Acc Chem Res. 2011;44:936–946. doi: 10.1021/ar200023x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 63.Movahedi K, Laoui D, Gysemans C, Baeten M, Stange G, Van den Bossche J, Mack M, Pipeleers D, In’t Veld P, De Baetselier P, Van Ginderachter JA. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010;70:5728–5739. doi: 10.1158/0008-5472.CAN-09-4672. [DOI] [PubMed] [Google Scholar]
- 64.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1S: CD59 staining (blue) of clinical specimens from pancreatic cancer patients. In tumor, CD59 was highly expressed by cancer cells (indicated by arrows) as well as stroma cells including endothelial cells. Bar=100μm
Figure 2S: Immunofluorescent analysis of a large vessel with a sprouting capillary (arrows) in L3.6pl pancreatic tumor. The tissue was stained using antibodies to CD31 and Ly6C. Although endothelial cells in both the large vessel and sprouting capillary were CD31 positive (Red), endothelial cells were stained with Ly6C and CD31 (emits yellow) only in the sprouting capillary. Immune cells surrounding the vessels expressed only Ly6C. Scale bar indicates 20 μm.
Figure 3S: (A) Immunofluorescent analysis of normal pancreas and L3.6pl tumors in the pancreas using antibody to CD204 (M2 macrophage marker) and Ly6C. Bar=100 μm.
(B) Immunofluorescent staining of L3.6pl tumor using antibody to CD204 and BrdU (proliferation marker). The tumor bearing mice were sacrificed 24 hours after intra-peritoneal injected with BrdU and tumor sections were stained with antibodies against BrdU (pseudo color in green) and CD204 (red). Bar=50 μm
Figure 4S: (A) Physical stability of the particles as measured by ICP-AES analysis for up to 12 months. The samples were stored in distilled water or isopropyl alcohol. (B) Optimization of antibody conjugation to the surface of S1MP. Concentration of FITC-IgG in the range of 1.88-45 μM were tested for conjugation to the surface of APTES modified S1MP. Following the incubation, S1MP were extensively washed and fluorescent intensity of the pellet was detected. The data shows that at 10 μM there is a saturation of the amount of the antibody on the surface and further increase in the FITC-IgG concentration in the reaction medium is not reflected in additional molecules bound to the surface. Based on these results, we further used 10 μM for our study.
Figure 5S: Lower magnification of the images in Figure 5B (A) and 5C (B) as indicated by white squares, respectively. Arrows in (A) indicate attached nanocarriers to the endothelial cells. Bar=10μm