

SUMMARY
Although protein quality control (PQC) is generally perceived as important for the development of the nervous system, the specific mechanisms of neuronal PQC have remained poorly understood. Here, we report that C. elegans EBAX-1 (Elongin BC-Binding AXon regulator), a conserved BC-box protein, regulates axon guidance through PQC of the SAX-3/Robo receptor. EBAX-1 buffers guidance errors against temperature variations. As a substrate-recognition subunit in the Elongin BC-containing Cullin-RING ubiquitin ligase (CRL), EBAX-1 also binds to DAF-21, a cytosolic Hsp90 chaperone. The EBAX-type CRL and DAF-21 collaboratively regulate SAX-3-mediated axon pathfinding. Biochemical and imaging assays indicate that EBAX-1 specifically recognizes misfolded SAX-3 and promotes its degradation in vitro and in vivo. Importantly, vertebrate EBAX also shows substrate preference towards aberrant Robo3 implicated in horizontal gaze palsy with progressive scoliosis (HGPPS). Together, our findings demonstrate a triage PQC mechanism mediated by the EBAX-type CRL and DAF-21/Hsp90 that maintains the accuracy of neuronal wiring.
Keywords: Cullin, Elongin BC-containing ubiquitin ligase, EBAX-1, ZSWIM8, Hsp90, DAF-21, SAX-3, Robo, protein quality control, axon guidance
INTRODUCTION
Protein homeostasis is a cellular network integrating protein synthesis, folding, trafficking and degradation pathways, which acts to maintain appropriate levels of proteins and counteracts negative effects of aberrant proteins (Tyedmers et al., 2010). Under physiological conditions, a significant fraction of newly translated proteins are defective and must be immediately destroyed by proteasomes (Schubert et al., 2000). Environmental stress can increase the level of unfolded and misfolded protein products. Cells have developed sophisticated compartment-specific protein quality control (PQC) strategies to restrict aberrant proteins to harmless levels through molecular chaperone-facilitated folding/refolding and protein degradation (Tyedmers et al., 2010). By suppressing background noise caused by stochastic environmental variations and translational errors, PQC is essential to ensure the robustness of genetically designed developmental programs (Jarosz et al., 2010).
Processes that require extensive protein turnover impose intense pressure on the biosynthesis and PQC pathways. The development of the nervous system involves many steps occurring at a rapid pace, including progenitor cell migration and differentiation, neuronal wiring, and synapse formation and pruning. In addition to high levels of constitutive protein synthesis demanded by developing neurons, the expression of many proteins such as guidance signaling molecules is also spatially and temporally regulated (Dickson and Gilestro, 2006). For example, surface expression of the Robo receptor in Drosophila commissural axons is transiently downregulated by an endosomal protein Commissureless (Comm) during midline crossing, and this suppression is relieved afterwards to prevent re-crossing of commissural axons (Georgiou and Tear, 2002; Keleman et al., 2002). In vertebrates, the ubiquitin-specific protease 33 (USP33)-mediated deubiquitination and recycling of Robo1 is important for the midline crossing of commissural axons and their responsiveness to Slit (Yuasa-Kawada et al., 2009). Despite being challenged by demands of protein synthesis and adverse intrinsic and extrinsic factors, the development of the nervous system shows striking precision, implying the engagement of powerful PQC mechanisms for suppressing background noise and maintaining developmental stability.
Previous PQC studies in eukaryotic cells have demonstrated the essential roles of protein folding and degradation pathways in PQC. In the endoplasmic reticulum (ER), newly synthesized polypeptides are shaped into native forms with the assistance of molecular chaperones such as Hsp90 and Hsp70 (Buchberger et al., 2010; Taipale et al., 2010). Unfolded proteins are degraded by the ER-associated degradation pathway (ERAD) employing E3 ubiquitin ligases such as HRD1, Gp78, and an F-box type Cullin-RING E3 ligase complex SCFFbx2 (Claessen et al., 2012). In the cytosol, E3 ligases, such as CHIP (C-terminus of Hsc70 interacting protein) in vertebrates and Ubr1 in yeast, promote degradation of numerous misfolded proteins in a chaperone-dependent manner (Buchberger et al., 2010; Heck et al., 2010).
In humans, many neurodegenerative diseases are associated with abnormal aggregation of misfolded proteins and malfunctioning PQC in neurons, including Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and the distal hereditary motor neuropathies (Evgrafov et al., 2004; Irobi et al., 2004; Skovronsky et al., 2006). However, in contrast to the well-perceived importance of PQC in various non-neuronal cell types and aging neurons, PQC mechanisms in developing neurons remain largely unexplored. We report here that the novel conserved BC-box protein, EBAX-1, collaborates with DAF-21/Hsp90 to maintain the accuracy of axon guidance in C. elegans. EBAX-1 is a substrate recognition subunit in the BC-box Cullin-RING E3 ligase (CRL) and binds to DAF-21/Hsp90. EBAX-1 is highly enriched in the developing nervous system and contributes to thermotolerance of axon guidance. In AVM ventral axon growth, the EBAX-1-containing CRL and DAF-21/Hsp90 function cell-autonomously to control protein quality of the SAX-3/Robo receptor. EBAX-1 preferentially binds to a metastable mutant SAX-3 protein that is prone to misfolding, and promotes its degradation. Moreover, the mouse homolog of EBAX-1 (ZSWIM8) shows similar substrate preference towards a human Robo3 mutant receptor associated with horizontal gaze palsy with progressive scoliosis (HGPPS). Our studies uncover a conserved protein degradation complex that regulates the accuracy of guidance signaling during development, and identify in vivo roles for functionally coupled molecular chaperone and protein degradation machinery in neuronal protein quality control.
RESULTS
EBAX-1 is a Conserved Substrate Recognition Subunit in the Elongin BC-Containing Cullin-RING E3 Ligase
C. elegans EBAX-1 (Elongin BC-Binding AXon regulator-1, sequence R09E10.7, previously PQN-55) belongs to a novel BC-box protein family conserved from invertebrates to humans (Figure 1A). This family of proteins contains two N-terminal motifs – the BC-box and the Cul2-box (Mahrour et al., 2008), followed by a SWIM domain (named after SWI2/SNF2 transcription factor and MuDR transposase) (Makarova et al., 2002) and several conserved regions without obvious similarity to known domains (Figure S1A). Animals homozygous for ebax-1 null mutations – ju699 and tm2321 (Figure 2A) – are viable and grossly normal in morphology. However, these mutants show sluggish locomotion, defective egg-laying and impaired male mating. To unveil the molecular function of this family of proteins, we first performed mass spectrometry analysis of proteins associated with the mouse homolog of EBAX-1 (zinc finger SWIM domain-containing protein 8, ZSWIM8) stably expressed in HEK293T cells. Two distinct proteins between 10 and 20 kDa were identified as Elongin B and Elongin C (Figure 1B). An independent study reported that endogenous human ZSWIM8 (clone KIAA0913) in HEK293T cells are also associated with Elongin B and C (Mahrour et al., 2008).
Figure 1. EBAX-1 is a Conserved BC-box Protein Serving as a Substrate Recognition Subunit in a Cullin-RING E3 Ligase Complex (CRL).

(A) Schematic diagrams of Drosophila, C. elegans, mouse and human homologs of EBAX-1. The EBAX family of proteins contain a BC-box (purple), a Cul2-box (green) and a zinc-finger SWIM domain (light blue) at the N-terminus followed by several conserved regions, the largest of which is marked as domain A. Numbers indicate percentages of identity and similarity, respectively.
(B) Endogenous human Elongin B and Elongin C interact with the mouse homolog of EBAX-1 (ZSWIM8). HEK293T cells stably expressing Flag-tagged ZSWIM8 WT, ZSWIM8 ΔBox mutant or an empty vector control were subjected to immunoprecipitation with mouse anti-Flag antibodies. The immunoprecipitants were separated by SDS-PAGE and revealed by silver staining.
(C) Schematic illustration of a BC box-type Cullin-RING E3 ubiquitin ligase (CRL).
(D) Alignment of the BC-box and Cul2-box of EBAX-1 homologs and mutants. Amino acids that are conserved in more than 2 sequences are highlighted: orange (aliphatic residues), green (Thr or Pro), purple (Ala or Ser), and blue (Pro). The consensus BC-box and Cul2-box sequences are indicated above the alignment (Mahrour et al., 2008). Essential amino acids for Elongin B/C and CUL2 interactions (marked in red and underlined) were mutated in EBAX-1 M1 (L111S, I114S) and M2 (I151A, P152A) mutants.
(E) Interactions between C. elegans ELC-1 and EBAX-1 full-length protein or mutants in yeast two-hybrid (Y2H) assays. ++ and +++, intermediate and strong interaction; ±, weak interaction; −, no interaction.
(F) EBAX-1 is expressed in the embryonic nervous system. Representative Pebax-1::EBAX-1::GFP fluorescence and DIC images from the comma stage, the 2-fold stage and the 3-fold stage are shown. The anterior (A) and posterior (P) ends of embryos are marked on images. The bracket in the right panel indicates developing anterior neurons. Scale, 10 μm.
Figure 2. EBAX-1 Regulates the Ventral Axon Guidance of AVM and PVM Neurons.

(A) Illustration of the genomic structure of ebax-1. In the ebax-1(ju699) allele, a region of 1548 bp covering the SWIM domain and the domain A is deleted. In the ebax-1(tm2321) allele, a region of 551 bp in exon 5 is deleted.
(B) ebax-1 mutants show mild egg-laying (Egl) defects. The Egl phenotype can be fully rescued by ebax-1 genomic DNA drive by a pan-neuronal promoter (Prgef-1::EBAX-1), suggesting EBAX-1 functions in neurons that control the egg-laying behavior. Data are shown as means (SD); statistics, t-test. **p<0.01 and ##p<0.01 relative to WT.
(C) Morphological defects of HSN axons labeled by Punc-86::myr::GFP(kyIs262) in WT and ebax-1(ju999) animals. Starting from the L2 stage, the HSN axon migrates ventrally, and then extends along the ventral nerve cord. At the L4 stage, the axon forms synapses with the VC4 motor neuron and vulva muscles (c1). A significant proportion of ebax-1(ju699) animals showed various HSN guidance defects, including parallel or directionless axon growth (c3-c4), or axons reaching the ventral cord but missing the vulva midline (marked by asterisk in c2). Scale, 5 μm.
(D) Genetic interactions between ebax-1 and the unc-6/unc-40 and slt-1/sax-3 pathways in HSN guidance. The ebax-1 mutation enhanced the guidance defects in both unc-6(ev400) and sax-3(ky123) mutant backgrounds. Note that in the unc-6 mutants, HSN neurons already showed highly penetrant guidance defects; loss of ebax-1 increased the penetrance of defects to 100%. Data represent means ± SEM. *p<0.05 and ***p<0.001 relative to indicated controls; t-test.
(E) The effects of EBAX-1 on HSN guidance defects at 25°C. The guidance errors increased in wild-type animals at 25°C. The guidance defects in WT was further increased by the ebax-1(ju699) mutation but suppressed by overexpression of EBAX-1. *p<0.05 and **p<0.01 relative to WT.
(F) Schematic illustration of guidance signals for the ventral growth of AVM, PVM and HSN axons. The repellent guidance cue SLT-1/Slit is secreted by dorsal muscles, and the attractive guidance cue UNC-6/Netrin is secreted by the ventral nerve cord. Growth cones expressing SAX-3/Robo and UNC-40/DCC receptors are guided to grow ventrally.
(G) Representative images of AVM neurons in WT, ebax-1(ju699), unc-6(ev400), and ebax-1(ju699); unc-6(ev400) mutants. ALMR is another touch neuron adjacent to AVM. Scale, 20 μm.
(H-I) Quantitative analysis of AVM (H) and PVM (I) guidance defects in various genetic backgrounds. EVA-1 is a co-receptor of SAX-3 (Fujisawa et al., 2007). Data represent means ± SEM. *p<0.05, **p<0.01 and ***p<0.001 relative to indicated controls; t-test.
(J) Rescue and mosaic analysis of Pebax-1::EBAX-1 in unc-6; ebax-1 and unc-40; ebax-1 mutants. The numbers of scored animals are shown in the bar graph. Data represent means ± SEM. ##p<0.01 and ***p<0.001 relative to indicated controls; one-way ANOVA.
Elongin B and C are components of the BC-box type Cullin-RING E3 ligase (CRL). CRLs are the largest class of E3 ubiquitin ligases and are involved in many physiological and pathological processes (Hua and Vierstra, 2011). The subtypes of CRLs are defined by the cullin scaffold and adaptor proteins. In the BC-box CRL, cullin 2 (CUL2) is responsible for assembling Elongin B, Elongin C, the RING-Box protein Rbx1, and the BC-box protein as a complex. BC-box proteins serve as the substrate recognition subunit to recruit specific substrates for ubiquitination (Figure 1C). The BC-box and the Cul2-box mediate the interaction of BC-box proteins with Elongin B/C and CUL2 respectively (Mahrour et al., 2008). We found that deleting the BC-box and Cul2-box in ZSWIM8 (ZSWIM8 ΔBox) completely abolished the interaction between ZSWIM8 and Elongin B/C in co-immunoprecipitation assays (Figure 1B).
The interaction between EBAX-1 and ELC-1, the C. elegans ortholog of Elongin C, was confirmed by yeast two-hybrid assays (Figure 1E, Figure S1B). To verify the importance of the BC-box for EBAX-1 protein interaction, we designed several deletion mutants and found that an N-terminal fragment of EBAX-1 only including the BC-box, Cul2-box and SWIM domain (N2 fragment), showed strong interaction with ELC-1 (Figure 1E, Figure S1B). While the C-terminal half of EBAX-1 did not interact with ELC-1, removal of the C-terminus (EBAX-1 N1 fragment) or the conserved domain A (EBAX-1 ΔA) from EBAX-1 reduced its binding to ELC-1. These results imply that the C-terminus and the domain A may be involved in EBAX-1 protein stability or conformation in yeast. We further generated mutations of two functionally conserved residues in the BC-box consensus sequence (L111S and I114S, M1 mutant) and found that they markedly reduced the binding between the EBAX-1 N2 fragment and ELC-1. In contrast, point mutations in the Cul2-box (I151A and P152A, M2 mutant) had no effect on the interaction between EBAX-1 and ELC-1 (Figure 1D-E, Figure S1B). The interaction between EBAX homologs and Elongin B/C supports the conclusion that EBAX proteins are conserved substrate recognition subunits in the Cullin2-RING E3 ligase (Figure 1C).
EBAX-1 is Enriched in the Developing Nervous System and Buffers Axon Guidance Errors against Mild Thermal Stress and Genetic Mutations
In C. elegans, ebax-1 transcriptional and translational reporters showed that EBAX-1 is enriched in the developing nervous system. A functional C-terminal GFP-fusion of EBAX-1 (EBAX-1::GFP) driven by the endogenous 2.7 kb promoter showed dynamic expression throughout embryonic and larval stages. Fluorescence was detected from mid-embryogenesis, with a higher level in the anterior half of the embryo (Figure 1F, left panel). The expression at the far posterior end increased at the 2-fold stage (Figure 1F, middle panel). The overall intensity of EBAX-1::GFP peaked at the 3-fold stage, especially around the nerve ring region (Figure 1F, right panel), and dropped after hatching. In the fourth larval stage (L4), EBAX-1::GFP was detected in the nerve ring, the ventral nerve cord, the HSN motor neuron, and some neurons in the tail (Figure S2A). Mos1 transposase-mediated Single Copy Insertion (MosSCI) of Pebax-1::EBAX-1::GFP showed similar expression dynamics, albeit at a lower expression level (data not shown). EBAX-1::GFP showed a punctate pattern in the cytosol of individual neurons (Figure S2A). A similar punctate pattern was also observed in the soma and axons when EBAX-1::GFP was specifically expressed in mechanosensory neurons and GABAergic motor neurons (data not shown). Likewise, GFP-tagged mouse ZSWIM8 displayed a cytosolic expression pattern in cultured heterologous cells (Figure S2B).
To decipher the roles of ebax-1 in the developing nervous system, we first examined the morphology of HSN motor neurons that control the egg-laying behavior, since ebax-1 mutants exhibit modest egg-laying defects that can be rescued by neuronal expression of EBAX-1 (Figure 2B). We found that thirty percent of the ebax-1(ju699) mutants showed HSN axon guidance defects at 20°C (Figure 2C-D). A moderate temperature rise to 25°C increased the guidance errors in wild-type and mutant animals, whereas overexpression of EBAX-1 in the wild-type background significantly improved HSN guidance accuracy (Figure 2E). These observations suggest that the accuracy of HSN axon guidance is temperature dependent and sensitive to the level of EBAX-1. A similar temperature dependency of axon guidance defects was also observed in AVM neurons of ebax-1 mutants (Figure S2E). Through extensive analyses of genetic interactions, we identified a specific role for ebax-1 in the ventral axon guidance of both HSN motor neurons and AVM and PVM mechanosensory neurons (the latter two also called touch neurons). Ventral guidance of HSN and AVM/PVM axons is in response to a combination of attractive Netrin/DCC (UNC-6/UNC-40) and repellent Slit/Robo (SLT-1/SAX-3) signals (Figure 2F) (Desai et al., 1988; Hao et al., 2001; Zallen et al., 1998). Mutations disrupting either pathway partially disrupt ventral guidance, while simultaneous loss of both pathways causes fully penetrant ventral guidance defects (Figure 2D, 2H-I, Figure S2C-D). In AVM neurons, ebax-1 mutants alone did not show any guidance defects at 20°C, but significantly enhanced guidance defects in unc-6(ev400) or unc-40(e1430) mutants (Figure 2H, Figure S2C). In contrast, ebax-1 mutations did not enhance AVM axon guidance defects in the slt-1 or sax-3 mutant background (Figure 2H, Figure S2C). In PVM and HSN, ebax-1 showed synergistic effects with both slt-1/sax-3 and unc-6/unc-40 pathways (Figure 2D and 2I, Figure S2D). These observations suggest that ebax-1 affects both guidance pathways in HSN and PVM, while in AVM it exclusively regulates slt-1/sax-3 signaling.
Next, we determined whether ebax-1 functions in neurons that express guidance receptors or surrounding tissues that secrete guidance cues. We performed mosaic analysis in unc-6; ebax-1 and unc-40; ebax-1 double mutants co-expressing a rescuing transgene Pebax-1::EBAX-1 and a co-injection marker Psur-5::SUR-5::mCherry that labels nuclei of cells carrying the transgenes (Yochem et al., 1998). For simplicity of quantification, we focused on AVM as ebax-1 is exclusively involved in the slt-1/sax-3 pathway during AVM axon guidance. ebax-1; unc-6 or ebax-1; unc-40 double mutant animals universally expressing the transgenes showed full rescue of AVM axon guidance defects (Figure 2J). We then identified animals specifically losing the transgenes either in the AB lineage-derived cells (mainly neurons) or in the P1 lineage-derived cells (mainly non-neuronal cells) and scored them for AVM guidance defects. AB-loss animals showed the same severity of axon guidance defects as ebax-1; unc-6 or ebax-1; unc-40 double mutant animals. In contrast, the guidance defects in P1-loss animals were rescued by neuron-restricted expression of EBAX-1 (Figure 2J). Therefore, we conclude that the cell-autonomous expression of EBAX-1 in neurons is important for regulating AVM axon guidance.
The EBAX-1-Containing Cullin-RING E3 Ligase is Important for AVM Axon Guidance
Next we addressed whether the interactions of EBAX-1 with Elongins and cullin 2 are important for its function in AVM axon guidance. We expressed EBAX-1 mutants (ΔBox, M1 and M2; Figure 3A) deficient in interactions with ELC-1 and/or CUL-2 in the ebax-1(ju699); unc-6(ev400) and unc-40(e1430); ebax-1(ju699) backgrounds, and examined their activity on rescuing AVM guidance defects. All mutants showed similar expression as wild-type proteins (Figure S3 and data not shown). Strikingly, these EBAX-1 mutant proteins completely lost rescuing activity (Figure 3B-C), indicating that the in vivo function of EBAX-1 requires its interactions with ELC-1 and CUL-2.
Figure 3. The EBAX-1-Containing CRL is Important for AVM Guidance.
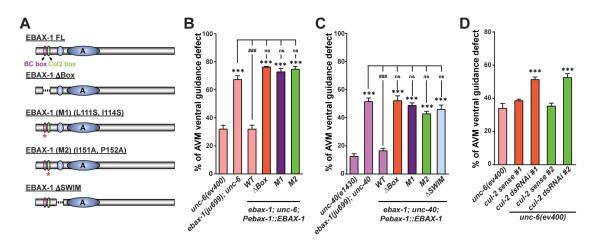
(A) Illustration of EBAX-1 wild-type and mutant constructs. Dashed lines indicate deleted regions in EBAX-1 ΔBox and ΔSWIM mutants. Asterisks mark the BC-box and Cul2-Box with amino acid substitutions in EBAX-1 M1 and M2 mutants, respectively.
(B-C) Quantitative analysis of AVM guidance defects rescued by the EBAX-1 WT, ΔBox, M1, M2, or ΔSWIM mutant driven by the ebax-1 promoter in unc-6 (B) or unc-40 (C) mutant animals. Data combined from 2-4 transgenic lines of each EBAX-1 construct are shown. Data represent means ± SEM. ***p<0.001 and ###p<0.001 relative to indicated controls; ns, not significant; one-way ANOVA.
(D) The effect of touch neuron-specific cul-2 RNAi on AVM guidance in the unc-6(ev400) mutants. cul-2 dsRNAi animals expressed both Pmec-7-driven cul-2 sense and antisense RNA strands. cul-2 sense control animals only expressed the sense RNA strand. Two independent RNAi sequences (dsRNAi #1 and #2) were used. Data represent means ± SEM. ***p<0.001; one-way ANOVA.
We then asked whether other components of the BC box-type Cullin-RING ligase are directly involved in axon guidance. Because Elongin B and Elongin C are also components of cullin 5-containing CRLs (Hua and Vierstra, 2011), we decided to address this question by examining the necessity of CUL-2/cullin 2 in AVM axon guidance. cul-2 was specifically knocked down in touch neurons of unc-6 mutants by co-expressing Pmec-7-driven cul-2 sense and antisense RNAs (Najarro et al., 2012). Touch neuron-specific cul-2 RNAi resulted in a significant enhancement of AVM guidance defects. In contrast, expression of sense RNA alone had no effect (Figure 3D). Taken together, these results demonstrate that the EBAX-1-containing CRL is critical for AVM axon guidance in vivo.
DAF-21/Hsp90 Interacts with EBAX-1 and Regulates AVM Axon Guidance
In our functional rescue experiments, we found that the SWIM domain was also important for the function of EBAX-1 in AVM guidance (Figure 3C). Since the SWIM domain has previously been shown to mediate protein-protein interactions (Makarova et al., 2002), we predicted that unidentified EBAX-1 interactor(s) are involved in the regulation of AVM axon guidance. To expand our knowledge on how the EBAX-1-containing CRL functions in neurons, we performed a yeast two-hybrid screen for other EBAX-1 interacting proteins and identified DAF-21, a cytosolic heat shock protein 90 (Hsp90) homolog in C. elegans (details in Experimental Procedures). Importantly, the interaction with DAF-21 was dependent on the SWIM domain of EBAX-1 (Figures 4A and S4).
Figure 4. DAF-21/Hsp90 Interacts with EBAX-1 and Regulates AVM Guidance.
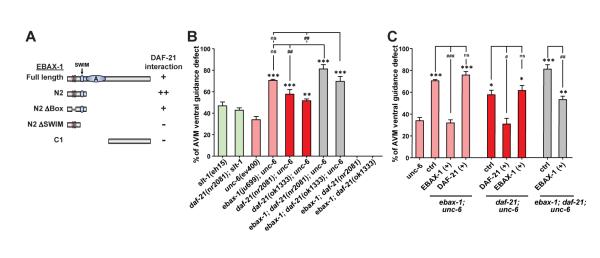
(A) Interactions between DAF-21 and the EBAX-1 WT or truncated mutants in Y2H assays. + and ++, intermediate and strong interaction; –, no interaction.
(B) Genetic effects of daf-21 and ebax-1 mutants on AVM guidance.
(C) Rescue analyses of daf-21 and ebax-1 transgenes in double and triple mutants. EBAX-1 (+) and DAF-21 (+), overexpression of Pebax-1::EBAX-1 WT and Pdaf-21::DAF-21 WT in indicated mutant backgrounds. Data in (B) and (C) represent means ± SEM. *p<0.05, **p<0.01, and ***p<0.001 relative to the unc-6 single mutant; #p<0.05, ##p<0.01, and ###p<0.001 relative to indicated controls; one-way ANOVA.
Hsp90 chaperones are at the center of protein homeostasis and regulate the folding and refolding of many client proteins (Taipale et al., 2010). Compromising the function of Hsp90 leads to decreased developmental stability in Drosophila and zebrafish (Jarosz et al., 2010). In C. elegans, the Hsp90 chaperone DAF-21 buffers stochastic developmental failure caused by genetic variation (Burga et al., 2011). Mice lacking Hsp90β, a member of the cytosolic Hsp90 family, die around embryonic day 9.0/9.5 (Voss et al., 2000). To our knowledge, Hsp90 proteins have not been previously linked to axon guidance. Given the physical interaction between DAF-21/Hsp90 and EBAX-1, we investigated the genetic effect of daf-21 on AVM axon guidance. daf-21 null mutants (nr2081 and ok1333) are arrested at larval stages and can survive until the late L1 to early L2 stage (Birnby et al., 2000). The morphology of AVM neurons is normal in these animals as well as in ebax-1; daf-21 double mutants at 20°C. Interestingly, similar to ebax-1 mutants, daf-21 mutants showed synergistic enhancement of AVM guidance defects in the unc-6 mutant background, while daf-21(nr2081); slt-1 double mutants resembled slt-1 single mutants (Figure 4B). The enhanced defects in unc-6; daf-21 mutants were rescued by expression of DAF-21 in touch neurons, but not in muscles (Figure S4B), indicating a cell-autonomous role of DAF-21.
To address the functional interaction between daf-21 and ebax-1, we analyzed daf-21; ebax-1; unc-6 triple mutants and observed that the severity of guidance defects in the triple mutants was higher than daf-21; unc-6 double mutants, but was not significantly different from ebax-1; unc-6 double mutants (Figure 4B). We further dissected the genetic hierarchy of daf-21 and ebax-1 by assessing the rescuing activity of their transgenes in the double and triple mutants. In these mutant backgrounds, transgenes of wild-type ebax-1 and daf-21 rescued the guidance defects caused by their respective mutations. However, overexpression of ebax-1 did not rescue the guidance defect caused by the daf-21 mutation or vice versa (Figure 4C). These data together suggest that DAF-21/Hsp90 collaborates with EBAX-1 to regulate the slt-1/sax-3 signaling and that EBAX-1 has both DAF-21-dependent and DAF-21-independent functions in vivo.
EBAX-1 is Preferentially Associated with a Temperature-Sensitive and Misfolding-Prone Mutant Form of SAX-3
The coupling of protein degradation and folding machinery is an emerging theme in protein quality control under physiological and stress conditions (Buchberger et al., 2010). Thus, we hypothesized that DAF-21/Hsp90 and EBAX-1 may be involved in suppressing the level of endogenous aberrant misfolded proteins during axon guidance. Numerous human diseases have been associated with amino acid mutations resulting in metastable proteins with temperature-sensitive (ts) misfolding defects (Gelsthorpe et al., 2008; Kjaer and Ibanez, 2003; Pedersen et al., 2003; Singh et al., 1997; Vollrath and Liu, 2006). One well-studied example is the ΔF508 mutant of CFTR (cystic fibrosis transmembrane conductance regulator) identified in cystic fibrosis patients (Lukacs and Verkman, 2012). The advance in therapeutic treatment of cystic fibrosis has heavily relied on PQC studies of the CFTR ΔF508 mutant. Being susceptible targets of the PQC system, such metastable mutant proteins can serve as sensitized probes to examine the function of PQC regulators.
To identify in vivo targets of EBAX-1 and DAF-21, we searched for temperature sensitive mutants with protein misfolding defects in the slt-1/sax-3 pathway. We found that a previously reported ts mutation of sax-3 (ky200) caused striking temperature-dependent misfolding and mislocalization of the SAX-3 receptor in touch neurons. sax-3(ky200) contains a missense mutation at a conserved proline residue (P37S) in the first immunoglobin-like domain (Ig1) (Figure 5A) (Zallen et al., 1998). In the vertebrate Robo1, this amino acid is close to the contact regions between Slit2 and Robo1, but does not directly mediate their interaction (Morlot et al., 2007). sax-3(ky200) mutant animals showed marginal penetrance of AVM guidance defects at the permissive temperature (20°C), suggesting that most mutant SAX-3 is functional under this condition. At the restrictive temperature (22.5°C), the level of defects significantly increased in sax-3(ky200) mutants (Figure 5B). As a control, the guidance defects in sax-3(ky123) null mutants did not show temperature-dependence.
Figure 5. EBAX-1 Preferentially Interacts with a Temperature-Sensitive and Misfolding-Prone Mutant of SAX-3.

(A) Sequence alignment of the Ig1 domain of C. elegans SAX-3, Drosophila Robo and human Robo1, 2 and 3. In the sax-3(ky200) mutant, a conserved Pro37 (marked in red and underlined) is mutated to Ser37. In human Robo3, an I66L mutation (marked in blue and underlined) was found in patients with HGPPS (Jen et al., 2004).
(B) Quantification of AVM guidance defects in the sax-3(ky200) temperature sensitive (ts) and sax-3(ky123) null mutants at permissive (20°C) and restrictive (22.5°C) temperatures. ***p<0.001 relative to the indicated control; t-test.
(C) Subcellular localization of Pmec-7::SAX-3(WT)::GFP and Pmec-7::SAX-3(P37S)::GFP at 20°C and 22.5°C. L1-L2 animals expressing Pmec-7::SAX-3::GFP were imaged for AVM and ALM soma. Yellow arrows indicate cytosolic aggregates of SAX-3(P37S)::GFP. Scale, 2 μm.
(D) FRAP analysis of Pmec-7::SAX-3(WT)::GFP (d1-d2) and Pmec-7::SAX-3(P37S)::GFP (d3-d6) in ALM neurons at the late L1 stage. The fluorescence in regions with strong SAX-3(P37S) aggregates barely recovered after photobleaching (d5-d6), while regions with less aggregation showed a faster recovery rate (d3-d4). The fluorescence intensity is shown as a pseudo-color scale. Scale, 2 μm.
(E) Comparison of the fluorescence recovery rates of SAX-3(WT)::GFP and SAX-3(P37S)::GFP after photobleaching at 20 and 22.5°C. The yellow area indicates the range of SAX-3(WT) samples; the blue area covers mutant samples with slower recovery rates than WT samples. Data shown as scatterplot with means ± SEM. *p<0.05, and **p<0.01; t-test.
(F) Co-immunoprecipitation of Flag-EBAX-1(WT or ΔBox) and SAX-3(WT or P37S)-V5 from HEK293T cell lysates. Immunoprecipitants were subjected to SDS-PAGE separation and immunoblotting (IB) with mouse anti-Flag and mouse anti-V5 antibodies.
(G) Co-immunoprecipitation of Flag-mouse ZSWIM8 (WT) and human Robo3 (WT or I66L)-V5 from HEK293T cell lysates.
To test whether the temperature sensitivity of sax-3(ky200) mutants is caused by protein misfolding, we examined the expression patterns of GFP-tagged SAX-3(WT) and SAX-3(P37S) in touch neurons. SAX-3(WT)::GFP and SAX-3(P37S)::GFP were expressed in the wild-type and sax-3(ky200) mutant backgrounds, respectively, to ensure the homogeneity of endogenous and exogenous proteins. In touch neurons at late L1 to early L2 stages, SAX-3(WT)::GFP was predominantly localized on the cell surface at both 20°C and 22.5°C (Figure 5C). In contrast, SAX-3(P37S)::GFP showed a mixture of cell surface and cytosolic localization with a mild degree of cytosolic aggregation at 20°C (Figure 5C). The cytosolic mislocalization and aggregation of SAX-3(P37S)::GFP were exacerbated at 22.5°C and accompanied with a reduction of surface signals, which was correlated with the aggravated AVM guidance defects in sax-3(ky200) mutants (Figure 5B-C). Moreover, SAX-3(P37S)::GFP showed a significantly slower diffusion rate than SAX-3(WT)::GFP, revealed by fluorescence recovery after photobleaching (FRAP) in live touch neurons (Figure 5D-E). Neurons with strong cytosolic aggregation of SAX-3(P37S)::GFP barely showed any fluorescence recovery 10 minutes after photobleaching (Figure 5D, panels d5-d6). These observations strongly suggest that wild-type SAX-3 is predominantly in its native form and properly delivered to the cell surface, while the sax-3(ky200) mutation results in metastable proteins that are prone to misfolding and tend to form diffusion-limited cytosolic aggregates.
Previous studies have reported that ubiquitin ligases involved in PQC often favor unfolded and misfolded proteins as substrates (Buchberger et al., 2010). In AVM neurons, mCherry-tagged EBAX-1 showed a cytosolic punctate pattern, and more colocalization was observed with SAX-3(P37S)::GFP than with SAX-3(WT)::GFP (Figure S5), suggesting that misfolded SAX-3 may be the substrate of EBAX-1. To test this possibility, we performed co-immunoprecipitation assays using HEK293T cells exogenously expressing EBAX-1 and SAX-3. We found that EBAX-1 showed stronger interaction with SAX-3(P37S) than SAX-3(WT) in the co-IP assays (Figure 5F).
Moreover, we found that the mouse homolog of EBAX-1 (ZSWIM8) also showed preferential binding to a human Robo3 mutant protein identified in horizontal gaze palsy with progressive scoliosis (HGPPS). HGPPS is a complex syndrome that involves severe developmental axon guidance defects, and is associated with missense mutations in the human robo3 gene (Jen et al., 2004). In some patients, a conserved isoleucine residue of Robo3 is mutated to leucine (I66L); this residue is close to the equivalent of the Pro37 residue mutated in sax-3(ky200) (Figure 5A). We found that ZSWIM8 showed a stronger interaction with human Robo3(I66L) compared to human Robo3(WT) in co-immunoprecipitation assays (Figure 5G), suggesting a potential conserved role of EBAX proteins in Robo quality control.
EBAX-1/ZSWIM8 Promotes the Degradation of Metastable SAX-3/Robo
To determine whether EBAX-1 regulates the degradation of aberrant SAX-3, we conducted multiple experiments to assess the degradation rate of SAX-3 under different conditions. First, SAX-3 (WT or P37S) was co-expressed with EBAX-1 (WT or ΔBox) in HEK293T Cells, and the levels of SAX-3 and EBAX-1 were monitored after protein synthesis is blocked. We observed that the degradation of SAX-3(P37S) was significantly delayed in the presence of the EBAX-1 ΔBox mutant, indicating that the interactions of EBAX-1 with other CRL components are important for the degradation of SAX-3(P37S) (Figure 6A). In contrast, the degradation of SAX-3(WT) was less dependent on the BC-box and Cul2-box of EBAX-1 (Figure 6A). Importantly, similar dependence of human Robo3(I66L) degradation on ZSWIM8 was also observed in HEK293T cells (Figure S6A-D), further supporting a conserved role of the EBAX family in PQC.
Figure 6. EBAX-1 Promotes the Degradation of Misfolded SAX-3.

(A) Pulse chase of SAX-3(WT) and SAX-3(P37S) in the presence of EBAX-1 WT or ΔBox. HEK293T cells expressing indicated constructs were treated with 1 μM cycloheximide (CHX) for 0, 2, 4, or 6 hours (h) before collection. An equal amount of protein from each sample was loaded to SDS-PAGE and immunoblotted with mouse anti-Flag, V5 and α-tubulin antibodies.
(B) Representative images of Dendra-tagged SAX-3(WT) and SAX-3(P37S) in AVM neurons at 0 and 7 h after photoconversion. SAX-3(P37S) showed a faster degradation rate (b3-b4) than SAX-3(WT) (b1-b2). The fluorescence intensity is indicated as a pseudocolor scale. Scale, 2 μm.
(C) Comparison of SAX-3(WT)::Dendra and SAX-3(P37S)::Dendra fluorescence intensity 7 h after photoconversion at 20°C and 22.5°C. The yellow area indicates the range of SAX-3(WT) samples and the blue area covers mutant samples with faster degradation rates than WT samples.
(D) The effects of ebax-1(ju699) on the degradation of SAX-3(WT)::Dendra and SAX-3(P37S)::Dendra in AVM neurons. Data represent means ± SEM. *p<0.05, **p<0.01 and ***p<0.001 relative to indicated controls; t-test.
To assess the degradation of SAX-3 in vivo, we generated transgenic animals expressing SAX-3(WT) and SAX-3(P37S) tagged with the green-to-red photo-switchable fluorescent protein Dendra2 (Gurskaya et al., 2006). Without photoconversion, Dendra2 emits green fluorescence. UV illumination converts the pre-existing green fluorescent proteins to red so that they can be distinguished from newly synthesized proteins. We chased the degradation of SAX-3 by measuring the intensity change of red fluorescence at different time points post photoconversion. At 7 hours after photoconversion, an average 35% of photo-converted SAX-3(WT)::Dendra was degraded in AVM at L1-L2 stages, and temperature rise had little effect on their degradation rate (Figure 6B-C). In contrast, over 50% of SAX-3(P37S)::Dendra was degraded at 20°C, and the level of degradation was further increased by temperature shift to 22.5°C (Figure 6C). In touch neurons, a fraction of SAX-3(P37S) was misfolded and either diffused or form aggregates in the cytosol, while most SAX-3(WT) was in a native form and predominantly located on the cell surface (Figure 5C). Therefore, the difference of degradation rates between SAX-3(P37S) and SAX-3(WT) suggests that misfolded SAX-3 is overall more vulnerable to degradation than native SAX-3.
Further, we found that the ebax-1(ju699) null mutation significantly reduced the degradation of SAX-3(P37S) (Figure 6D). Interestingly, a similar reduction was also observed in the degradation rate of SAX-3(WT) in ebax-1 mutants, suggesting that in vivo a pool of wild-type proteins, possibly those in nonnative forms, rely on EBAX-1 for degradation. Supporting this finding, around 44% of AVM neurons in ebax-1 mutants showed aggregation of SAX-3(WT)-GFP, a three-fold increase over neurons in wild-type animals (Figure S6E). Additionally, we found that after enriching misfolded proteins by proteasome inhibition in live worms, a fraction of SAX-3(WT) was detected in the EBAX-1 immunoprecipitant (Figure S6F). In HEK293T cells expressing EBAX-1, ubiquitinated SAX-3(WT) was accumulated after proteasome activity was blocked for 4 hours (Figure S6G). When the function of Hsp90 was further inhibited, the level of SAX-3 ubiquitination was increased (Figure S6G), and SAX-3(WT) was recognized by EBAX-1 (Figure S6H). These data together indicate that EBAX-1 can target misfolded wild-type SAX-3 as well as metastable mutant SAX-3.
EBAX-1 and DAF-21/Hsp90 Collaborate in SAX-3 Protein Quality Control
Consistent with the dependence of misfolded SAX-3 on EBAX-1 for degradation, we found that the AVM guidance defect in the sax-3(ky200) mutant showed strong sensitivity to the protein level of EBAX-1. Loss of endogenous EBAX-1 worsened the guidance defect caused by sax-3(ky200) at 20°C, but did not further enhance the defect at 22.5°C. Overexpression of EBAX-1 significantly suppressed the guidance defect at 22.5°C (Figure 7A). We also found that in the absence of ebax-1, misfolded SAX-3(P37S) caused dominant negative effects on endogenous wild-type SAX-3 and interfered with axon guidance (Figure S7). These results suggest that an endogenous level of EBAX-1 is sufficient and necessary to suppress guidance errors caused by mild misfolding of SAX-3, while elevated levels of EBAX-1 can overcome severe misfolding of SAX-3 caused by thermal stress.
Figure 7. EBAX-1 and DAF-21/Hsp90 Facilitate the Folding and Degradation of Misfolded SAX-3.
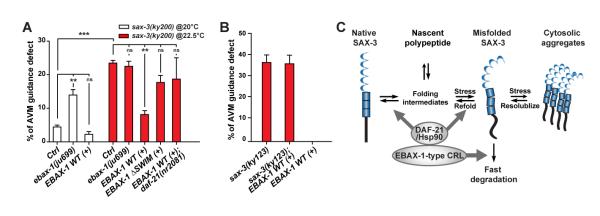
(A) The effects of ebax-1 and daf-21 on AVM guidance in the sax-3(ky200) mutant background. Control (ctrl), the sax-3(ky200) mutant alone. EBAX-1 WT (+) and EBAX-1 ΔSWIM (+), overexpression of Pebax-1::EBAX-1 WT and Pebax-1::EBAX-1 ΔSWIM in the sax-3 mutants. Data represent means ± SEM. **p<0.01 and ***p<0.001 relative to indicated controls; one-way ANOVA.
(B) EBAX-1 overexpression has no effect in sax-3(ky123) null mutants.
(C) Model of the EBAX-1-mediated triage mechanism for SAX-3 quality control during axon guidance. The amount of non-native SAX-3 caused by translational errors, environmental stress or genetic mutations is restricted by collaborative efforts between EBAX-1 and DAF-21/Hsp90. On one hand, EBAX-1 recruits DAF-21/Hsp-90 to promote the folding and refolding of non-native SAX-3. On the other hand, EBAX-1 degrades irreparable proteins through its connection to the CRL complex.
Additionally, we examined whether DAF-21/Hsp90 is essential for EBAX-1-dependent suppression of temperature-sensitive phenotypes of sax-3(ky200). We found that eliminating the daf-21 gene in the sax-3(ky200) mutant abolished the ability of overexpressed EBAX-1 to suppress the guidance defect at 22.5°C (Figure 7A). Overexpression of a mutant of EBAX-1 lacking the SWIM domain (EBAX-1 ΔSWIM) failed to show significant suppression effects (Figure 7A), indicating that the interaction with DAF-21 is important for the function of EBAX-1. As a negative control, overexpression of EBAX-1 had no effect on AVM guidance defects in sax-3(ky123) null mutants (Figure 7B), further supporting the conclusion that EBAX-1 and DAF-21/Hsp90 target the SAX-3 receptor itself.
DISCUSSION
Our findings here identify a neuronal PQC mechanism that coordinates molecular chaperones and protein degradation machinery to ensure the accuracy of axon guidance. We hypothesize that the EBAX-1-containing CRL and the DAF-21/Hsp90 chaperone control the protein quality of SAX-3 receptor via a “triage” mechanism. As a substrate recognition subunit specifically for aberrant proteins, EBAX-1 recruits DAF-21/Hsp90 to facilitate the folding and refolding of SAX-3, while permanently damaged SAX-3 proteins are removed by protein degradation mediated by the EBAX-1-containing CRL (Figure 7C).
A Quality Guard for Axon Guidance Signaling
The protein homeostatic environment in cells is constantly challenged by damaged proteins generated by biosynthetic errors, environmental stress and genetic mutations. Without immediate clearance, lingering defective protein products will impair the proper function of cells by competing with native proteins in a dominant negative fashion and/or forming cytotoxic aggregates. Besides the constitutive PQC system, cells have also evolved the unfolded protein response (UPR) to cope with ER stress caused by unusual concentration changes of misfolded proteins in cells, oxidative stress, disturbed redox balance or calcium homeostasis in the ER lumen (Tabas and Ron, 2011). As one of the downstream targets of UPR, the efficiency of the ER folding and ERAD system is upregulated in order to reduce the workload in the ER and restore protein homeostasis. ER stress can also induce the upregulation of ubiquilins, an evolutionarily conserved protein family involved in the ERAD and autophagy degradation pathways and linked to human neurodegenerative diseases (Deng et al., 2011; Lee and Brown, 2012).
PQC studies in various model organisms and in vitro culture systems have greatly advanced our understanding of protein homeostasis regulation (Gidalevitz et al., 2011; Skovronsky et al., 2006). However, it remains largely unknown how protein quality is controlled in developing neurons to ensure the vitality of neurons and the precision of neuronal development. In addition to stochastic translational errors and genetic variations, environmental stress such as heat exposure can cause protein damage and jeopardize embryonic development. In vertebrates, the developing central nervous system is very sensitive to maternal body temperatures, and hyperthermia-induced severe neurodevelopmental defects have been observed (Edwards et al., 2003; Sharma and Hoopes, 2003). In C. elegans, mild temperature rise hampers the faithfulness of neuronal migration, polarization and axon pathfinding (Fleming et al., 2010). We have also observed that the axon guidance of wild-type HSN motor neurons showed temperature sensitivity (Figure S2E). These studies underscore the sensitivity of the developing nervous system to environmental perturbations and the need for buffering mechanisms.
In the present study, we have found that a conserved EBAX-1-type CRL is functionally coupled with DAF-21/Hsp90 and guards the accuracy of axon guidance. During the ventral guidance of AVM and PVM neurons at the L1 stage, the importance of EBAX-1 was revealed when the strength of guidance signals was weakened in sensitized mutant backgrounds. HSN neurons, whose axon growth occurs in later larval stages, require EBAX-1 for axon guidance in both wild-type and sensitized mutant backgrounds (Figure 2D). Interestingly, ebax-1 functions in both slt-1/sax-3 and unc-6/unc-40 pathways in PVM and HSN neurons (Figures 2D, 2I and S2D), suggesting that EBAX-1 may also target the UNC-40 receptor or its downstream effectors in addition to misfolded SAX-3. Moreover, EBAX-1 facilitates the thermotolerance of axon guidance in HSN and AVM neurons (Figures 2E and S2E). These results indicate that EBAX-1 is essential for sustaining axon pathfinding when the extracellular and intracellular environment of developing neurons is suboptimal. Additionally, the differential dependence of AVM/PVM and HSN neurons on EBAX-1 suggests that the PQC requirement is fine-tuned in individual cells, which may be due to distinct developmental stages and varied reliance on thermosensitive guidance signals.
PQC Mechanisms for SAX-3/Robo in Axon Guidance
Our results have demonstrated that EBAX-1 serves as a substrate recognition subunit to recruit misfolded SAX-3 for quality control. We propose that non-native SAX-3 undergoes a triage decision in the EBAX-type CRL/Hsp90 complex, being either folded into a stable native form by DAF-21/Hsp90 or degraded by the CRL when the damage is irreversible. Previous studies have identified various mechanisms for misfolded protein recognition and fate decision in PQC. Characteristics of misfolded proteins such as abnormally exposed hydrophobic residues can be recognized by chaperones and ubiquitin ligases (Buchberger et al., 2010). A systematic and quantitative analysis for the interaction between HSP90 and its clients reveals that Hsp90 recognizes the thermally unstable status of wild-type kinase clients instead of any particular sequence motifs (Taipale et al., 2012). The yeast nuclear protein quality control E3 ligase San1 uses a “disorder target misorder” mechanism to recognize different misfolded substrates. San1 contains small segments of conserved sequence that serve as substrate-recognition sites, which are interspersed by intrinsically disordered domains. The flexible disordered regions endow San1 with structural plasticity allowing it to bind differently shaped misfolded substrates (Gardner et al., 2005; Rosenbaum et al., 2011). We found that EBAX-1 also contains more than one binding site for SAX-3 (Figure S6H), implying that EBAX-1 might use a similar substrate recognition mechanism as San1 to target thermally unstable or disordered SAX-3.
In yeast and mammalian ER, an N-glycosylation-mediated timer paradigm for PQC of glycosylated proteins has been reported (Buchberger et al., 2010; Roth et al., 2010). In this model, successfully N-glycosylated proteins are rapidly folded by chaperones and sorted into the secretory pathway. If unfolded proteins overly dwell in the ER, the ER mannosidase I and EDEM (ER degradation enhancing alpha-mannosidase-like protein) will trim part of the N-linked glycans off these proteins, thus marking them for the ERAD pathway. Posttranslational modifications can also be used as a strategy to determine the fate of some chaperone/E3 ligase substrates. For example, CHIP degrades the SUMO2/3 protease SENP3 independent of Hsp90 under physiological conditions. Oxidative stress induces thiol modification at SENP3 cysteine residues that are specifically recognized by Hsp90. This resulting ternary SENP3/CHIP/Hsp90 complex promotes the stabilization of SENP3 instead of degradation (Yan et al., 2010). A number of co-chaperones can also regulate the catalytic activity of Hsp70, Hsp90 or CHIP and thus shift the balance between refolding and degradation (Buchberger et al., 2010). Thus, it will be interesting to investigate whether the EBAX-1-type CRL and DAF-21/Hsp90 utilize similar mechanisms to determine the fate of non-native SAX-3 in vivo.
Roles of the EBAX Family in the Nervous System
EBAX-1 and its homologs constitute a conserved family of substrate-recognition subunits of CRLs. In Drosophila, the EBAX-1 homolog (CG34401) regulates R7 photoreceptor axon targeting (M. Morey, A. Nern, and S.L. Zipursky, personal communication). Mouse and human ZSWIM8 are also widely expressed in the brain (Allen Brain Atlas Resources) (Lein et al., 2007). Our data show that mouse ZSWIM8 promotes the degradation of a human Robo3(I66L) mutant protein associated with HGPPS. The human homolog ZSWIM8 has been reported to interact with Ataxin 1 and Atrophin 1, two spinocerebellar ataxia-causing proteins (Lim et al., 2006). It will be of interest to explore the role of EBAX family members in the vertebrate nervous system both during development and in disease. In multiple species, Hsp90 is essential for silencing phenotypic manifestation of genetic variations under physiological conditions and potentiating the creation of new inheritable variants against environmental stress (Burga et al., 2011; Jarosz et al., 2010). However, the underlying molecular mechanisms are still open questions. Our studies raise an intriguing question whether EBAX-1 and its homologs participates in Hsp90-mediated genetic capacitance against genetic and environmental perturbations in neurons. Identifying PQC regulators guarding the accuracy of neuronal development and neuronal wiring will be a fertile area for future investigations.
EXPERIMENTAL PROCEDURES
Strains and Constructs
N2 and mutant C. elegans strains were maintained on NGM plates using standard methods (Brenner, 1974). Animals were grown at 20°C, 22.5°C or 25°C as noted. Constructs are listed in Tables S1-S3. Strains and alleles are listed in Table S4.
Fluorescence Microscopy
P0 animals were grown at 20°C, 22.5°C or 25°C as indicated. F1 animals were immobilized in 1 mM levamisole solution and scored using a Zeiss Axioplan 2 microscope equipped with Chroma HQ filters. GFP and mCherry images were taken using 488 nm and 594 nm lasers and band-pass filters on a Zeiss LSM510 scanning confocal microscope.
Fluorescence Recovery after Photobleaching (FRAP) Analysis
L1 animals expressing GFP-tagged SAX-3(WT) or SAX-3(P37S) were loaded to 4% agar pads and immobilized by 1 mM levamisole solution. A single focal plane image of ALM neuron was taken using a 63x objective lens on a Zeiss LSM510 confocal microscope (0 min). Next, a region of interest (ROI, 100 × 100 pixels) at the proximal axon was completely photobleached by a 488 nm laser. Another single frame image was taken 10 min after photobleaching. The intensity within the ROI was measured at 0 min (F0min), immediately after photobleaching (F’), and 10 min after photobleaching (F10min) by Metamorph 7.0. Background noise was subtracted from all images when measuring the fluorescent intensity. The fraction of GFP recovered in 10 min was calculated as (F10min − F’)/F0min.
Photoconversion of Dendra
Late L1 to early L2 worms expressing SAX-3::Dendra were immobilized by 1mM levamisole solution on agar pads and illuminated by UV (350 nm, DAPI excitation) for 20 sec under 63x lens on a Zeiss LSM510 confocal microscope to photoconvert Dendra. Z-stack images covering the neuronal soma and proximal axon of AVM neurons were immediately captured using a 543 nm laser. Worms were then recovered in M9 buffer and transferred to seeded NGM plates. Seven hours after photoconversion, AVM neurons were imaged again. The fluorescence intensity of Dendra at 7 h post photoconversion was measured by Metamorph 7.0 and normalized to that at 0 hr.
Mass Spectrometry and Biochemistry
HEK293T cell lines stably expressing Flag-tagged mouse ZSWIM8 full-length or ΔBox cDNA in a pQCXIP vector or the empty vector were generated by retroviral infection and puromycin selection and lysed for immunoprecipitation. Immunoprecipitants pulled down by mouse anti-Flag M2 Agarose (Sigma) were separated by SDS-PAGE (Invitrogen). Distinct bands pulled down by mouse ZSWIM8 but not by the controls were excised and analyzed by David King at the HHMI Mass Spectrometry lab (UC Berkeley). See the Supplemental Experimental Procedures for details.
To co-immunoprecipitate EBAX-1 and SAX-3, HEK293T cells transfected with indicated constructs for ~24 h were treated with 1 μM bortezomib or 25 μM MG132 for 2 h and collected in lysis buffer (50 mM Tris pH=7.0, 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-50) supplied with protease inhibitors. The lysates were incubated on ice for 30 min and then cleared by centrifugation at 4°C (14,000 rpm, 10 min). The supernatants were incubated with mouse anti-Flag M2 antibodies (Sigma) or mouse IgG for 4 h at 4°C, followed by incubation with Protein G agarose beads (Pierce) for 1 h at 4°C. Protein G beads were then washed with 5 × 1.5 ml lysis buffer and boiled in SDS loading buffer for 15 min at 85°C. Rabbit anti-V5 and HRP-conjugated mouse anti-Flag antibodies (Sigma) were used for immunoblotting.
To pulse chase the degradation of SAX-3(WT) and SAX-3(P37S), HEK293T cells were transfected with indicated constructs and treated with 1 μM cycloheximide to block protein synthesis for 0, 2, 4 or 6 h next day. Cells were collected in lysis buffer ~24 h after transfection and incubated on ice for 30 min before clearing by centrifugation at 4°C (14,000 rpm, 10 min). Equal amounts of supernatants from different time points were denatured by SDS loading buffer for immunoblotting.
Statistics Analysis
A two-tailed Student’s t test was used for comparisons of two groups. One-way ANOVA with a Bonferroni or Dunnett post test was used to compare multiple groups.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Larry Zipursky for generously sharing unpublished results of Drosophila CG34401 mutants, which led us to work on ebax-1 in C. elegans, and to Aljoscha Nern and Marta Morey-Ramonell for personal communications. We thank David King at the HHMI Mass Spectrometry lab at UC Berkeley for mass spectrometry analyses. Shohei Mitani, the C. elegans gene knockout consortium and the Caenorhabditis Genetics Center provided mutant strains. We value discussions with Randy Hampton, James Moresco, and our lab members. Z.W. was supported by a postdoctoral fellowship from the Jane Coffin Childs Memorial Fund for Medical Research, and X.G. was supported by a postdoctoral fellowship from the Susan G. Komen breast cancer foundation. X.G. and Y. H. contributed equally to this work. This work was partly supported by awards from the NIH (R01 NS057317 to A.D.C. and Y.J., and R01 NS 035546 to Y.J.). M.B. was supported by an NWO Vidi grant. Y. J. and J.E.D. are Investigators of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Birnby DA, Link EM, Vowels JJ, Tian H, Colacurcio PL, Thomas JH. A transmembrane guanylyl cyclase (DAF-11) and Hsp90 (DAF-21) regulate a common set of chemosensory behaviors in Caenorhabditis elegans. Genetics. 2000;155:85–104. doi: 10.1093/genetics/155.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell. 2010;40:238–252. doi: 10.1016/j.molcel.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Burga A, Casanueva MO, Lehner B. Predicting mutation outcome from early stochastic variation in genetic interaction partners. Nature. 2011;480:250–253. doi: 10.1038/nature10665. [DOI] [PubMed] [Google Scholar]
- Claessen JH, Kundrat L, Ploegh HL. Protein quality control in the ER: balancing the ubiquitin checkbook. Trends Cell Biol. 2012;22:22–32. doi: 10.1016/j.tcb.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai C, Garriga G, McIntire SL, Horvitz HR. A genetic pathway for the development of the Caenorhabditis elegans HSN motor neurons. Nature. 1988;336:638–646. doi: 10.1038/336638a0. [DOI] [PubMed] [Google Scholar]
- Dickson BJ, Gilestro GF. Regulation of commissural axon pathfinding by slit and its Robo receptors. Annu Rev Cell Dev Biol. 2006;22:651–675. doi: 10.1146/annurev.cellbio.21.090704.151234. [DOI] [PubMed] [Google Scholar]
- Edwards MJ, Saunders RD, Shiota K. Effects of heat on embryos and foetuses. Int J Hyperthermia. 2003;19:295–324. doi: 10.1080/0265673021000039628. [DOI] [PubMed] [Google Scholar]
- Evgrafov OV, Mersiyanova I, Irobi J, Van Den Bosch L, Dierick I, Leung CL, Schagina O, Verpoorten N, Van Impe K, Fedotov V, et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet. 2004;36:602–606. doi: 10.1038/ng1354. [DOI] [PubMed] [Google Scholar]
- Fleming T, Chien SC, Vanderzalm PJ, Dell M, Gavin MK, Forrester WC, Garriga G. The role of C. elegans Ena/VASP homolog UNC-34 in neuronal polarity and motility. Dev Biol. 2010;344:94–106. doi: 10.1016/j.ydbio.2010.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisawa K, Wrana JL, Culotti JG. The slit receptor EVA-1 coactivates a SAX-3/Robo mediated guidance signal in C. elegans. Science. 2007;317:1934–1938. doi: 10.1126/science.1144874. [DOI] [PubMed] [Google Scholar]
- Gardner RG, Nelson ZW, Gottschling DE. Degradation-mediated protein quality control in the nucleus. Cell. 2005;120:803–815. doi: 10.1016/j.cell.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Gelsthorpe ME, Baumann N, Millard E, Gale SE, Langmade SJ, Schaffer JE, Ory DS. Niemann-Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding. J Biol Chem. 2008;283:8229–8236. doi: 10.1074/jbc.M708735200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiou M, Tear G. Commissureless is required both in commissural neurones and midline cells for axon guidance across the midline. Development. 2002;129:2947–2956. doi: 10.1242/dev.129.12.2947. [DOI] [PubMed] [Google Scholar]
- Gidalevitz T, Prahlad V, Morimoto RI. The stress of protein misfolding: from single cells to multicellular organisms. Cold Spring Harb Perspect Biol. 2011;3:a009704. doi: 10.1101/cshperspect.a009704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurskaya NG, Verkhusha VV, Shcheglov AS, Staroverov DB, Chepurnykh TV, Fradkov AF, Lukyanov S, Lukyanov KA. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol. 2006;24:461–465. doi: 10.1038/nbt1191. [DOI] [PubMed] [Google Scholar]
- Hao JC, Yu TW, Fujisawa K, Culotti JG, Gengyo-Ando K, Mitani S, Moulder G, Barstead R, Tessier-Lavigne M, Bargmann CI. C. elegans slit acts in midline, dorsal-ventral, and anterior-posterior guidance via the SAX-3/Robo receptor. Neuron. 2001;32:25–38. doi: 10.1016/s0896-6273(01)00448-2. [DOI] [PubMed] [Google Scholar]
- Heck JW, Cheung SK, Hampton RY. Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc Natl Acad Sci U S A. 2010;107:1106–1111. doi: 10.1073/pnas.0910591107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Z, Vierstra RD. The cullin-RING ubiquitin-protein ligases. Annu Rev Plant Biol. 2011;62:299–334. doi: 10.1146/annurev-arplant-042809-112256. [DOI] [PubMed] [Google Scholar]
- Irobi J, Van Impe K, Seeman P, Jordanova A, Dierick I, Verpoorten N, Michalik A, De Vriendt E, Jacobs A, Van Gerwen V, et al. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat Genet. 2004;36:597–601. doi: 10.1038/ng1328. [DOI] [PubMed] [Google Scholar]
- Jarosz DF, Taipale M, Lindquist S. Protein homeostasis and the phenotypic manifestation of genetic diversity: principles and mechanisms. Annu Rev Genet. 2010;44:189–216. doi: 10.1146/annurev.genet.40.110405.090412. [DOI] [PubMed] [Google Scholar]
- Jen JC, Chan WM, Bosley TM, Wan J, Carr JR, Rub U, Shattuck D, Salamon G, Kudo LC, Ou J, et al. Mutations in a human ROBO gene disrupt hindbrain axon pathway crossing and morphogenesis. Science. 2004;304:1509–1513. doi: 10.1126/science.1096437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keleman K, Rajagopalan S, Cleppien D, Teis D, Paiha K, Huber LA, Technau GM, Dickson BJ. Comm sorts robo to control axon guidance at the Drosophila midline. Cell. 2002;110:415–427. doi: 10.1016/s0092-8674(02)00901-7. [DOI] [PubMed] [Google Scholar]
- Kjaer S, Ibanez CF. Intrinsic susceptibility to misfolding of a hot-spot for Hirschsprung disease mutations in the ectodomain of RET. Hum Mol Genet. 2003;12:2133–2144. doi: 10.1093/hmg/ddg227. [DOI] [PubMed] [Google Scholar]
- Lee DY, Brown EJ. Ubiquilins in the crosstalk among proteolytic pathways. Biol Chem. 2012;393:441–447. doi: 10.1515/hsz-2012-0120. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Lim J, Hao T, Shaw C, Patel AJ, Szabo G, Rual JF, Fisk CJ, Li N, Smolyar A, Hill DE, et al. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;125:801–814. doi: 10.1016/j.cell.2006.03.032. [DOI] [PubMed] [Google Scholar]
- Lukacs GL, Verkman AS. CFTR: folding, misfolding and correcting the DeltaF508 conformational defect. Trends Mol Med. 2012;18:81–91. doi: 10.1016/j.molmed.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahrour N, Redwine WB, Florens L, Swanson SK, Martin-Brown S, Bradford WD, Staehling-Hampton K, Washburn MP, Conaway RC, Conaway JW. Characterization of Cullin-box sequences that direct recruitment of Cul2-Rbx1 and Cul5-Rbx2 modules to Elongin BC-based ubiquitin ligases. J Biol Chem. 2008;283:8005–8013. doi: 10.1074/jbc.M706987200. [DOI] [PubMed] [Google Scholar]
- Makarova KS, Aravind L, Koonin EV. SWIM, a novel Zn-chelating domain present in bacteria, archaea and eukaryotes. Trends Biochem Sci. 2002;27:384–386. doi: 10.1016/s0968-0004(02)02140-0. [DOI] [PubMed] [Google Scholar]
- Morlot C, Thielens NM, Ravelli RB, Hemrika W, Romijn RA, Gros P, Cusack S, McCarthy AA. Structural insights into the Slit-Robo complex. Proc Natl Acad Sci U S A. 2007;104:14923–14928. doi: 10.1073/pnas.0705310104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najarro EH, Wong L, Zhen M, Carpio EP, Goncharov A, Garriga G, Lundquist EA, Jin Y, Ackley BD. Caenorhabditis elegans flamingo cadherin fmi-1 regulates GABAergic neuronal development. J Neurosci. 2012;32:4196–4211. doi: 10.1523/JNEUROSCI.3094-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen CB, Bross P, Winter VS, Corydon TJ, Bolund L, Bartlett K, Vockley J, Gregersen N. Misfolding, degradation, and aggregation of variant proteins. The molecular pathogenesis of short chain acyl-CoA dehydrogenase (SCAD) deficiency. J Biol Chem. 2003;278:47449–47458. doi: 10.1074/jbc.M309514200. [DOI] [PubMed] [Google Scholar]
- Rosenbaum JC, Fredrickson EK, Oeser ML, Garrett-Engele CM, Locke MN, Richardson LA, Nelson ZW, Hetrick ED, Milac TI, Gottschling DE, et al. Disorder targets misorder in nuclear quality control degradation: a disordered ubiquitin ligase directly recognizes its misfolded substrates. Mol Cell. 2011;41:93–106. doi: 10.1016/j.molcel.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth J, Zuber C, Park S, Jang I, Lee Y, Kysela KG, Le Fourn V, Santimaria R, Guhl B, Cho JW. Protein N-glycosylation, protein folding, and protein quality control. Mol Cells. 2010;30:497–506. doi: 10.1007/s10059-010-0159-z. [DOI] [PubMed] [Google Scholar]
- Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–774. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- Sharma HS, Hoopes PJ. Hyperthermia induced pathophysiology of the central nervous system. Int J Hyperthermia. 2003;19:325–354. doi: 10.1080/0265673021000054621. [DOI] [PubMed] [Google Scholar]
- Singh N, Zanusso G, Chen SG, Fujioka H, Richardson S, Gambetti P, Petersen RB. Prion protein aggregation reverted by low temperature in transfected cells carrying a prion protein gene mutation. J Biol Chem. 1997;272:28461–28470. doi: 10.1074/jbc.272.45.28461. [DOI] [PubMed] [Google Scholar]
- Skovronsky DM, Lee VM, Trojanowski JQ. Neurodegenerative diseases: new concepts of pathogenesis and their therapeutic implications. Annu Rev Pathol. 2006;1:151–170. doi: 10.1146/annurev.pathol.1.110304.100113. [DOI] [PubMed] [Google Scholar]
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11:515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, Lindquist S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell. 2012;150:987–1001. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol. 2010;11:777–788. doi: 10.1038/nrm2993. [DOI] [PubMed] [Google Scholar]
- Vollrath D, Liu Y. Temperature sensitive secretion of mutant myocilins. Exp Eye Res. 2006;82:1030–1036. doi: 10.1016/j.exer.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Voss AK, Thomas T, Gruss P. Mice lacking HSP90β fail to develop a placental labyrinth. Development. 2000;127:1–11. doi: 10.1242/dev.127.1.1. [DOI] [PubMed] [Google Scholar]
- Yan S, Sun X, Xiang B, Cang H, Kang X, Chen Y, Li H, Shi G, Yeh ET, Wang B, et al. Redox regulation of the stability of the SUMO protease SENP3 via interactions with CHIP and Hsp90. EMBO J. 2010;29:3773–3786. doi: 10.1038/emboj.2010.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yochem J, Gu T, Han M. A new marker for mosaic analysis in Caenorhabditis elegans indicates a fusion between hyp6 and hyp7, two major components of the hypodermis. Genetics. 1998;149:1323–1334. doi: 10.1093/genetics/149.3.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa-Kawada J, Kinoshita-Kawada M, Wu G, Rao Y, Wu JY. Midline crossing and Slit responsiveness of commissural axons require USP33. Nat Neurosci. 2009;12:1087–1089. doi: 10.1038/nn.2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallen JA, Yi BA, Bargmann CI. The conserved immunoglobulin superfamily member SAX-3/Robo directs multiple aspects of axon guidance in C. elegans. Cell. 1998;92:217–227. doi: 10.1016/s0092-8674(00)80916-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.