

Abstract
Paracrine function is a major mechanism of cell-cell communication within tissue microenvironment in normal development and disease. In vitro cell culture models simulating tissue or tumor microenvironment are necessary tools to delineate epithelial-stromal interactions including paracrine function, yet an ideal three-dimensional (3D) tumor model specifically studying paracrine function is currently lacking. In order to fill this void we developed a novel 3D co-culture model in double-layered alginate hydrogel microspheres, incorporating prostate cancer epithelial and stromal cells in separate compartments of the microspheres. The cells remained confined and viable within their respective spheres for over 30 days. As a proof of principle regarding paracrine function of the model, we measured shedded component of E-cadherin (sE-cad) in the conditioned media, a major membrane bound cell adhesive molecule that is highly dysregulated in cancers including prostate cancer. In addition to demonstrating that sE-cad can be reliably quantified in the conditioned media, the time course experiments also demonstrated that the amount of sE-cad is influenced by epithelial-stromal interaction. In conclusion, the study establishes a novel 3D in vitro co-culture model that can be used to study cell-cell paracrine interaction.
Introduction
Several of the in vitro cells co-culture models available to study cell-cell interactions use two-dimensional (2D) Petri dishes or plates [1,2,3]. Yet in most living organisms cells are embedded in a three-dimensional (3D) microenvironment, surrounded by other cells and influenced by soluble factors secreted in the extracellular environment. Alternatively sandwich models can be used for multilayer growth of cells, but limitations are obvious, as cells would alter their morphological features, metabolism and gene expression patterns in 2D culture, especially when they are from higher organisms [4,5]. In addition, conventional 2D cell cultures limit cellular communications and transportation of soluble factors, oxygen and nutrients, removal of wastes and cellular metabolism as present in native biological environments [6,7]. Therefore, it is critical to develop in vitro model systems that simulate tissue microenvironments to produce reliable and biologically meaningful experimental results.
3D modeling systems simulating tissue microenvironment were developed to address limitations associated with 2D models [8]. While 3D in vitro cell culture models overcome several limitations of 2D models, improvement in 3D modeling is necessary to discriminate specific types of cell-cell interaction such as cell-cell direct, autocrine or paracrine functions. Advances in biomaterials and bioengineering techniques allow use of novel materials such as collagen gels, laminin and Matrigel™ in cell culture, develop synthetic extracellular matrix and create a variety of 3D models [5,9,10,11,12,13,14,15]. Among the biomaterials available, alginate hydrogel possesses preferred properties for cell transplantation, drug delivery and tissue engineering. Alginate is a polysaccharide and a biocompatible polymer derived from brown seaweed. By addition of divalent cations such as calcium or barium, alginate polymers can be ionically cross-linked to form a hydrogel. The hydrophilic nature of the alginate scaffolds enables high cell loading that remain viable and functional in culture [16,17,18]. In addition, the production of alginate hydrogel is relatively simple and encapsulation can be achieved under non-stringent conditions. Various cell types including neuronal cells, osteoblasts, chondrocytes, myoblasts, have been encapsulated, cultured and expanded in alginate hydrogels [19,20,21,22,23].
In this study we established a 3D prostate cancer epithelial-stromal interaction in alginate hydrogel microspheres by co-culturing prostate cancer C4-2 cells (stably transfected with Protein Kinase D1 (PKD1) or control vector) and normal prostate stromal cells (WPMY-1 cells) in the same microcapsule, but in separate sub-layers. This system is ideal to study paracrine influence between the two cell types because direct interaction between epithelial and stromal cells is not allowed. As a proof of principle to study paracrine function, we measured shedding of E-cadherin (sE-cad) in soluble media. The sE-cad is an 80 kDa cleaved fragment of E-cadherin, a transmembrane cell adhesive protein that is dysregulated in several cancers including prostate [3,24,25,26]. Elevated sE-cad has been reported in fluids and serum of patients with a variety of cancers and other diseases [25,27,28,29,30] and serum levels have been shown to correlate positively with metastatic prostate cancer and disease recurrence. Thus, sE-cad is suggested to be a novel biomarker for cancer prognosis. We previously described the down regulation of PKD1 in advanced prostate cancer [31], and that PKD1 promotes the E-cadherin shedding through increased matrix metalloproteinases (MMPs) -2 and -9 secretion [24].
Materials and Methods
Cell Culture
C4-2 cells stably transfected with pcDNA3.1 vector (vector cells) or PKD1-GFP (PKD1 cells) were developed in our laboratory as previously described [31]. Normal prostate stromal cells (WPMY-1) were obtained from ATCC. Cells were grown in DMEM medium (high glucose) (HyClone, Cat# SH30243.01) with 10% FBS and 1% Antibioltic-antimycotics (HyClone Cat# SV30079.01) in 15-cm sterile culture plate, and incubated at 37°C with 5% CO2. When cells reached 80% confluence, media were removed from each plate and cells were washed with sterile PBS three times, treated with trypsin (HyClone, Cat#SH30236.01) for 20 minutes and transferred to sterile centrifuge tubes. They were washed with PBS again, and then resuspended in DMEM medium for encapsulation.
Fabrication of microcapsules
The stromal, vector and PKD1 cells were encapsulated in alginate hydrogel using a micro-fluidic device with some modifications of encapsulation as described by Tendulkar et al. and Khanna et al. [32,33]. Briefly, first cell type (stromal or vector or PKD1) was mixed with 1.5% (w/v) ultrapure low-viscosity high-mannuronate alginate (LVM) (NovaMatrix, Sandvika Norway) and extruded through the micro-fluidic encapsulation device. The droplets generated were collected in 100 mM calcium chloride solution. After the cross linking of alginate for five minutes in CaCl2, the microcapsules were washed with 0.9% NaCl containing 20 mM CaCl2. The microcapsules were then incubated with poly-L-Ornithine (PLO) (0.1% w/v) for 20 minutes to create a PLO layer, which serves as a perm selective basement membrane. The PLO-coated microcapsules were then mixed with the second cell type (stromal or vector or PKD1) suspended in 1.5% (w/v) LVM and encapsulated again using the micro-fluidic device in order to obtain multi-layered microcapsules. The microcapsules were washed with 0.9% NaCl containing 20 mM CaCl2 and cultured in DMEM containing fetal bovine serum (10% v/v) at 37°C with 5% CO2.
Viability Assay
For viability assessment of encapsulated cells, a few capsules from each transfected group were taken out and transferred to clean 24-well plates, media were aspirated out carefully and cells stained. Single cell type control staining: CFDA SE (Vybrant® CFDA SE Cell Tracer Kit, Invitrogen) reconstituted in serum-free DMEM (SFM, HyClone) (1:400) was added to each well (500 µl/well) and incubated for 15 minutes at 37°C, 5% CO2 in the incubator. Then SFM with CFDA SE was replaced by DMEM with 10% FBS and incubated again for another 30 minutes at 37°C, 5% CO2 in the incubator. The serum-containing medium was then replaced with 50 µg/ml of propidium iodide (PI) (Life Technologies/Invitrogen, Grand Island, NY) and incubated at room temperature for 2 min and the microcapsules were washed 3 times to remove excess PI. The microcapsules were then observed under inverted fluorescence microscope (Zeiss Axiovert 200M) and imaged. The number of live and dead cells was determined qualitatively from the composite image acquired using Image-Pro plus software (version 6.3.1.542). Double cell type staining: To demonstrate the differential compartmentalization of different cell types in the multi-layered micro-capsules, the inner core cells were pre-stained with Cell Tracker green (Invitrogen, cat# C2925) and the outer layer cells were pre-stained with Cell-tracker Orange (Invitrogen), prior to the synthesis of the multi-layered micro-capsules. Before observation, the microcapsules were stained with SYTOX Blue Nucleic Acid Stain (Invitrogen, cat# S11348) for dead cells. The multi-layered micro-capsules were then imaged using the fluorescent microscope (Zeiss Axiovert 200M).
Enzyme Linked Immunoabsorbent Assay (ELISA)
To evaluate the paracrine functions of encapsulated cells, the levels of sE-cad was measured in the culture media. Each group of microcapsule was cultured in quadruplets and the spent media was collected every alternate day. While collecting the spent media, the media in each well was mixed thoroughly; 1ml (half of the total volume) was taken out from each well and replaced with 1 ml of fresh complete DMEM with 10% FBS and 1% antibiotics. The sample media was stored in a clean Eppendorf tube at -20 °C. For cells growing in 2D tissue culture treated petri dishes, media were collected when the cells reached 90-100% confluence (three-day incubation). The cell number was counted for each cell line for later normalization. The levels of sE-cad were quantified using ELISA kit from R&D systems, Quantikine (human sE-cadherin) as per manufacturers’ instructions.
Results
Cell viability maintained for at least a month in microspheres
Based on the previous reports of multi-layer microcapsules produced for protein delivery [32,33] we designed double-layer microcapsules which were composed of an alginate inner core and outer layer of alginate (Figure 1) separated by an electrostatically -linked polycationic permselective poly-L-ornithine (PLO) coat with a pore size <150 kDa [32]. Whereas the model prevented direct cell-cell interaction, cellular secretions could permeate through the PLO coat allowing for paracrine activity. As an initial step towards validating the model for paracrine function, we assessed cell viability.
Figure 1. Model of double-layered alginate hydrogel microsphere.
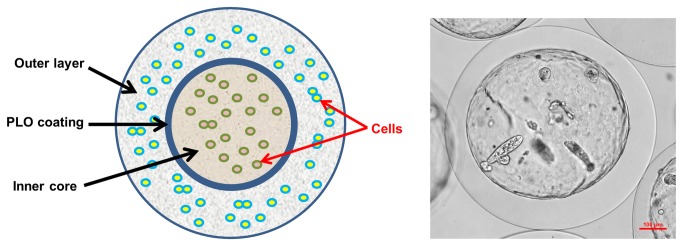
Left is a cartoon model of double-layered microsphere. Inner core and outer layer were separated by PLO coating as shown. Cells growing in separate layers were indicated with red arrows. Right is an actual microsphere observed under light microscope. Stromal cells were grown in the inner core for 7 days, and outer layer is blank.
The stromal cells (WPMY-1), C4-2 vector cells (C4-2 cells stably transfected with pcDNA3.1) and C4-2 PKD1 cells (C4-2 cells stably transfected and expressing PKD1) remained viable within inner core or outer layer in the double layered microcapsules for 4 weeks, and remained confined to their respective layers without migration through the PLO (Figure 2). Next, two different cell types were co-cultured in inner core or outer layer of the same microcapsule, and both cell types remained viable in co-culture for at least 4 weeks regardless of the layers or cell type combination (Figure 2). The results demonstrate that alginate microspheres can be used to grow epithelial or stromal cells compartmentalized in different layers in vitro for at least a month.
Figure 2. Different lines of prostate cells remained viable in microcapsules for over a month.
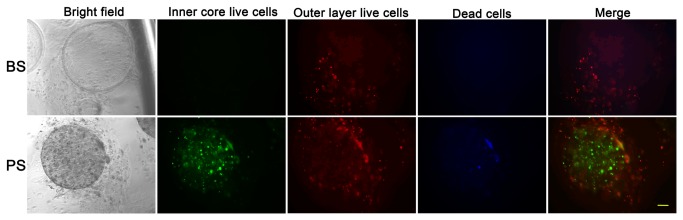
BS, microcapsules with blank inner core and stromal cells at outer layer. PS, microcapsules with C4-2 PKD1 cells at inner core and stromal cells at outer layer. Green, live cells at inner core. Red, live cells at outer layer. Blue, dead cells. Scale bar, 100µm.
Cells grown in microspheres demonstrated secretory function
In order to prove that viable cells in microspheres also remain functional, we measured the concentration of soluble E-cadherin fragment (sE-cad, or shedded E-cadherin) in the conditioned media as a marker of secretory function. E-cadherin is an important transmembrane cell-cell adhesion molecule that is dysregulated in several human cancers [3,34]. The extracellular domain of E-cadherin is cleaved by several extracellular proteases. The cleavage results in shedding of an approximately 80 kDa fragment, which has been shown to be a biomarker of aggressive phenotype in several human cancers including prostate [25,27,28,29,30]. Our previous studies demonstrated that dysregulation of PKD1 influences E-cadherin shedding in prostate cancer cells [24]. Therefore, we used stably transfected C4-2 PKD1 cells to quantify shedded E-cadherin level. In order to include a variety of positive and negative controls, we tested the sE-cad level in conditioned media of different human cell lines, including both normal and neoplastic cells. While some cell types did not show much evidence of E-cad shedding reflected by the low/trace level of sE-Cad, others secreted high levels of sE-cad in the conditioned media (Figure 3). The results showed that metastatic cell lines (MCF-7, PC3 and LnCap cells) have high sE-Cad level, and benign cell lines (HEK293T cells, 3T3 cells and MS1 cells) showed low/trace level of sE-Cad. There were significant differences between sE-Cad levels of metastatic cells and benign cells, which were confirmed by Student’s t-test (Figure 3). These results are consistent with published literature that sE-cad level correlates with cell invasiveness [25].
Figure 3. sE-Cad secretions in different human cell types.

sE-Cad level was measured by ELISA and was normalized to reflect the secretion of 107 cells for each cell line. sE-Cad level of metastatic cell lines (MCF-7, LnCap and PC3) were compared to HEK293T (as a representative of normal cell lines) and P values were calculated by Student’s t-test. Each column represents the average of three parallel experiments. Error bar represents the standard error.
Co-culture of epithelial and stromal cells influences shedded E-cadherin secretion as an evidence of paracrine function
To test paracrine interaction between two different cell lines, stromal cells, C4-2 vector cells and C4-2 PKD1 cells were grown in microcapsules in a variety of combinations and sE-cad in the conditioned media was quantified. When a single cell type was grown in either inner core or outer layer of the microcapsules, stromal cells did not secrete sE-cad, whereas both C4-2 vector cells and C4-2 PKD1 cells did when encapsulated together in separate compartments (Figure 4A). Quantification of sE-Cad was performed for cells cultured in 2D environment and similar results were observed (Figure 4B). The results validate reliability of the model to discriminate epithelial from stromal cells’ functions.
Figure 4. Whereas normal stromal cells do not secret sE-Cad in microcapsules, prostate cancer epithelial cells do.

A, cells were cultured in 3D environment for 6 days. BS, microcapsules with blank inner core and stromal cells in outer layer. BV, microcapsules with blank inner core and C4-2 vector cells in outer layer. BP, microcapsules with blank inner core and C4-2 PKD1 cells in outer layer. sE-Cad level was measured by ELISA. B, cells were cultured in 2D environment (petri dish). sE-Cad level was measured by ELISA and normalized to reflect the secretion of 107 cells for each cell line P values were calculated by Student’s t-test. Each column represents the average of three (2D culture) or four (3D culture) parallel experiments. Error bar represents the standard error.
C4-2 PKD1 cells increased secretion of sE-cad compared to C4-2 vector cells (control) (Figure 4), which is consistent with our prior publications [24]. We have also previously demonstrated that PKD1 up-regulated E-cadherin expression [24]. In the current experimental model C4-2 PKD1 cells but not C4-2 vector cells aggregate to form compact colonies (after cultured for three weeks in microcapsules) (Figure 5), which is a well-established phenotype of increased E-cadherin expression. When epithelial and stromal cells were co-cultured in independent layers within the same microcapsule, the amount of sE-cad secreted by C4-2 cells (both vector and PKD1 transfected cells) was decreased, suggesting stromal cells influence on epithelial sE-cad secretion (Figure 6). Because the stromal and epithelial cells are compartmentalized and unable to interact directly, we postulate that stromal cells influence epithelial cell secretory function via paracrine mechanism.
Figure 5. PKD1 expression induced formation of compact cell colonies.

Microcapsules incubated for 23 days are shown. PB, microcapsules with C4-2 PKD1 cells at inner core and blank outer layer. VB, microcapsules with C4-2 vector cells at inner core and blank outer layer. Green, CFDA staining for live cells. Red, Propidium iodide (PI) staining for dead cells. Arrows indicate cell colonies. Scale bar, 100µm.
Figure 6. Co-culture of prostate cancer epithelial cells and normal prostate stromal cells decreased sE-cad secretion by the epithelial cells.

sE-cad level was measured by ELISA following co-culture for 30 days. SB, microcapsules with stromal cells at inner core and blank outer layer. BV, microcapsules with blank inner core and C4-2 vector cells in outer layer. BP, microcapsules with blank inner core and C4-2 PKD1 cells in outer layer. SV, microcapsules with stromal cells at inner core and C4-2 vector cells in outer layer. SP, microcapsules with stromal cells at inner core and C4-2 PKD1 transfected cells in outer layer. P values were calculated by Student’s t-test. Each column represents the average of four parallel experiments. Error bar represents the standard error.
Discussion
The double layered microsphere model described in this study is a 3D environment which simulates the in vivo microenvironment, amenable to easy and scalable material production, purification and processing, no discernible cytotoxicity, and is chemically compatible with aqueous solutions and physiological conditions. Unlike other currently available 3D in vitro model, the microsphere model in this study allows to specifically analyze paracrine interaction between different types of cells. However, this model also exhibits certain limitations. The amount of quantifiable secretion of sE-cad of cells growing in inner core is lower than that of the same number and type of cells grown in outer layer of the microcapsule (data not shown). This decreased secretion may be due to the fact that sE-cad secreted by cells in the inner core needs to diffuse through two layers of hydrogel before entering the conditioned media. It is also possible that the permselectivity of PLO coat decreased the secretion of sE-Cad. To optimize this model for detection of other specific proteins, the chemical features of cross-linked PLO coat may need to be optimized for specific study needs. The influence of exosome should also be taken into consideration, as E-Cadherin was identified in microvesicles purified from normal murine dendritic cells and human cancer cells [35,36]. It remains possible that sE-Cad could be trapped in the exosomes, yet little is known about how microvesicles regulate cell-cell interaction in the paracrine system.
Another concern is the sustained mechanical property of alginate hydrogel, as ionically cross-linked alginates showed decreased gel strength after 90 days in vitro [37]. To solve this problem, stable covalent cross-links may be introduced into alginate hydrogels using bi-functional cross-linkers. Also, it is possible that an extended period of in vitro incubations may result in an imbalance in the prevailing ion concentrations (which is necessary to maintain microcapsule stability), but not under in vivo conditions, as no degradation of these alginate microspheres was observed for at least three months in our recent in vivo studies (unpublished data). In spite of these limitations, the model as described can be used effectively to study the paracrine epithelial-stromal interaction.
Epithelial-stromal interactions play a major role in normal development and neoplastic transformation. In normal human prostate the stromal cells are mainly composed of smooth muscle cells and fibroblasts, while the remainder are made of endothelial cells, pericytes, lymphocytes and macrophages [38]. It has been reported that carcinoma-associated stroma (CAS) could change the cell morphology/growth rate/aggressiveness of neoplastic human prostatic epithelial cells, but not normal prostatic epithelial cells [39]. Similar effects were reported using prostate carcinoma cell in co-culture with pleuripotent bone stromal cells [40]. Normal fibroblasts do not demonstrate such transformative effect on epithelial cells, suggesting a characteristic epithelial-stromal interaction during neoplastic process cells [39]. In this study, we demonstrate that one possible mechanism of stromal influence on epithelial cancer cells is through a paracrine mechanism. As a proof of principle we demonstrated reduction in sE-cad secretion by prostate cancer cells in presence of stromal cells and the model could presumably be used to study other cellular functions influenced by paracrine mechanism.
Stromal interaction influences proliferation, apoptosis and metastasis of epithelial cells. In pancreatic cancer, an antagonist of Hedgehog (Hh) inhibited tumor growth only when cancer-associated stroma exists, indicating signaling pathway dependent on stroma [41]. In prostate, it was reported that stromal factors, such as caveolin-1 and thymidine phosphorylase were related with tumor aggressiveness [42]. Other novel proteins that play a role in epithelial-stromal interaction have been identified by transcriptional profiling in prostate [43]. One of them, Decorin, was downregulated in prostate cancer [43]. Decreased Decorin resulted in a significant reduction of E-Cadherin both in vivo and in vitro, and physical interaction between Decorin and E-Cadherin was confirmed by co-immunoprecipitation [34]. Decorin expression is strictly limited in mesenchymal/stromal cells, but not in epithelial cells in prostate [43], and its expression is significantly decreased in carcinoma-associated stroma [43]. Whether Decorin also plays a role in shedding of E-cadherin remains unknown.
Another important group of extracellular matrix proteins, MMPs, could be possible candidates of stromal regulators that influenced the E-cad shedding. We previously showed that PKD1-induced secretion of MMP-2 and -9 increases E-cadherin shedding and suppresses cell proliferation [24].
In summary, the study demonstrates the feasibility and utility of using 3D alginate microsphere model to study paracrine function of cells. In particular, we used the model to explore epithelial-stromal interaction and demonstrated that normal prostate stromal cells influence secretion of sE-Cad by prostate cancer epithelial cells by paracrine interaction. The model could be used to validate additional cell lines and paracrine interaction of different types of cells.
Funding Statement
This work is supported by Wake Forest University institutional funds to KC Balaji and by NIH grant CA079448 to X. Fang. Funder's website is http://www.wakehealth.edu/. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Shen Q, Goderie SK, Jin L, Karanth N, Sun Y et al. (2004) Endothelial Cells Stimulate Self-Renewal and Expand Neurogenesis of Neural Stem Cells. Science 304: 1338-1340. doi:10.1126/science.1095505. PubMed: 15060285. [DOI] [PubMed] [Google Scholar]
- 2. Rajashekhar G, Traktuev DO, Roell WC, Johnstone BH, Merfeld-Clauss S et al. (2008) IFATS Collection: Adipose Stromal Cell Differentiation Is Reduced by Endothelial Cell Contact and Paracrine Communication: Role of Canonical Wnt Signaling. Stem Cells 26: 2674-2681. doi:10.1634/stemcells.2008-0277. PubMed: 18669909. [DOI] [PubMed] [Google Scholar]
- 3. Yates CC, Shepard CR, Stolz DB, Wells A (2007) Co-culturing human prostate carcinoma cells with hepatocytes leads to increased expression of E-cadherin. Br J Cancer 96: 1246-1252. doi:10.1038/sj.bjc.6603700. PubMed: 17406365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang F, Weaver VM, Petersen OW, Larabell CA, Dedhar S et al. (1998) Reciprocal interactions between β1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: A different perspective in epithelial biology. Proc Natl Acad Sci USA 95: 14821-14826. doi:10.1073/pnas.95.25.14821. PubMed: 9843973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bissell MJ, Rizki A, Mian IS (2003) Tissue architecture: the ultimate regulator of breast epithelial function. Curr Opin Cell Biol 15: 753-762. doi:10.1016/j.ceb.2003.10.016. PubMed: 14644202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sun T, Jackson S, Haycock JW, MacNeil S (2006) Culture of skin cells in 3D rather than 2D improves their ability to survive exposure to cytotoxic agents. J Biotechnol 122: 372-381. doi:10.1016/j.jbiotec.2005.12.021. PubMed: 16446003. [DOI] [PubMed] [Google Scholar]
- 7. Bhadriraju K, Chen CS (2002) Engineering cellular microenvironments to improve cell-based drug testing. Drug Discov Today 7: 612-620. doi:10.1016/S1359-6446(02)02273-0. PubMed: 12047872. [DOI] [PubMed] [Google Scholar]
- 8. Rhee HW, Zhau HE, Pathak S, Multani AS, Pennanen S et al. (2001) Permanent phenotypic and genotypic changes of prostate cancer cells cultured in a three-dimensional rotating-wall vessel. In Vitro Cell Dev Biol Anim 37: 127-140. doi:10.1290/1071-2690(2001)037. PubMed: 11370803. [DOI] [PubMed] [Google Scholar]
- 9. Weaver VM, Howlett AR, Langton-Webster B, Petersen OW, Bissell MJ (1995) The development of a functionally relevant cell culture model of progressive human breast cancer. Semin Cancer Biol 6: 175-184. doi:10.1006/scbi.1995.0021. PubMed: 7495986. [DOI] [PubMed] [Google Scholar]
- 10. Spancake KM, Anderson CB, Weaver VM, Matsunami N, Bissell MJ et al. (1999) E7-transduced Human Breast Epithelial Cells Show Partial Differentiation in Three-dimensional Culture. Cancer Res 59: 6042-6045. PubMed: 10626787. [PubMed] [Google Scholar]
- 11. Zhau HE, Goodwin TJ, Chang SM, Baker TL, Chung LW (1997) Establishment of a three-dimensional human prostate organoid coculture under microgravity-simulated conditions: Evaluation of androgen-induced growth and psa expression. In Vitro Cell Dev Biol Anim 33: 375-380. doi:10.1007/s11626-997-0008-3. PubMed: 9196896. [DOI] [PubMed] [Google Scholar]
- 12. Schmeichel KL, Bissell MJ (2003) Modeling tissue-specific signaling and organ function in three dimensions. J Cell Sci 116: 2377-2388. doi:10.1242/jcs.00503. PubMed: 12766184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lelièvre SA, Weaver VM, Nickerson JA, Larabell CA, Bhaumik A et al. (1998) Tissue phenotype depends on reciprocal interactions between the extracellular matrix and the structural organization of the nucleus. Proc Natl Acad Sci USA 95: 14711-14716. doi:10.1073/pnas.95.25.14711. PubMed: 9843954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cukierman E, Pankov R, Stevens DR, Yamada KM (2001) Taking Cell-Matrix Adhesions to the Third Dimension. Science 294: 1708-1712. doi:10.1126/science.1064829. PubMed: 11721053. [DOI] [PubMed] [Google Scholar]
- 15. Härmä V, Virtanen J, Mäkelä R, Happonen A, Mpindi JP et al. (2010) A Comprehensive Panel of Three-Dimensional Models for Studies of Prostate Cancer Growth, Invasion and Drug Responses. PLOS ONE 5: e10431. doi:10.1371/journal.pone.0010431. PubMed: 20454659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Glicklis R, Shapiro L, Agbaria R, Merchuk JC, Cohen S (2000) Hepatocyte behavior within three-dimensional porous alginate scaffolds. Biotechnol Bioeng 67: 344-353. doi:10.1002/(SICI)1097-0290(20000205)67:3. PubMed: 10620265. [DOI] [PubMed] [Google Scholar]
- 17. Dar A, Shachar M, Leor J, Cohen S (2002) Optimization of cardiac cell seeding and distribution in 3D porous alginate scaffolds. Biotechnol Bioeng 80: 305-312. doi:10.1002/bit.10372. PubMed: 12226863. [DOI] [PubMed] [Google Scholar]
- 18. Perets A, Baruch Y, Weisbuch F, Shoshany G, Neufeld G et al. (2003) Enhancing the vascularization of three-dimensional porous alginate scaffolds by incorporating controlled release basic fibroblast growth factor microspheres. J Biomed Mater Res A 65A: 489-497. doi:10.1002/jbm.a.10542. PubMed: 12761840. [DOI] [PubMed] [Google Scholar]
- 19. Holmes TC, de Lacalle S, Su X, Liu G, Rich A et al. (2000) Extensive neurite outgrowth and active synapse formation on self-assembling peptide scaffolds. Proc Natl Acad Sci USA 97: 6728-6733. doi:10.1073/pnas.97.12.6728. PubMed: 10841570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bokhari MA, Akay G, Zhang S, Birch MA (2005) The enhancement of osteoblast growth and differentiation in vitro on a peptide hydrogel—polyHIPE polymer hybrid material. Biomaterials 26: 5198-5208. doi:10.1016/j.biomaterials.2005.01.040. PubMed: 15792547. [DOI] [PubMed] [Google Scholar]
- 21. Rowley JA, Madlambayan G, Mooney DJ (1999) Alginate hydrogels as synthetic extracellular matrix materials. Biomaterials 20: 45-53. doi:10.1016/S0142-9612(98)00107-0. PubMed: 9916770. [DOI] [PubMed] [Google Scholar]
- 22. Augst AD, Kong HJ, Mooney DJ (2006) Alginate Hydrogels as Biomaterials. Macromol Biosci 6: 623-633. doi:10.1002/mabi.200600069. PubMed: 16881042. [DOI] [PubMed] [Google Scholar]
- 23. Alsberg E, Anderson KW, Albeiruti A, Rowley JA, Mooney DJ (2002) Engineering growing tissues. Proc Natl Acad Sci USA 99: 12025-12030. doi:10.1073/pnas.192291499. PubMed: 12218178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Biswas MHU, Du C, Zhang C, Straubhaar J, Languino LR et al. (2010) Protein Kinase D1 Inhibits Cell Proliferation through Matrix Metalloproteinase-2 and Matrix Metalloproteinase-9 Secretion in Prostate Cancer. Cancer Res 70: 2095-2104. doi:10.1158/0008-5472.CAN-09-4155. PubMed: 20160036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kuefer R, Hofer MD, Gschwend JE, Pienta KJ, Sanda MG et al. (2003) The Role of an 80 kDa Fragment of E-cadherin in the Metastatic Progression of Prostate Cancer. Clin Cancer Res 9: 6447-6452. PubMed: 14695147. [PubMed] [Google Scholar]
- 26. Syed V, Mak P, Du C, Balaji KC (2008) β-catenin mediates alteration in cell proliferation, motility and invasion of prostate cancer cells by differential expression of E-cadherin and protein kinase D1. J Cell Biochem 104: 82-95. doi:10.1002/jcb.21603. PubMed: 17979146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang H, Guan G, Zhang R, Liu G, Cheng J et al. (2009) Identification of urinary soluble E-cadherin as a novel biomarker for diabetic nephropathy. Diabetes/Metabolism Research and Reviews 25: 232-241. doi:10.1002/dmrr.940. PubMed: 19177462. [DOI] [PubMed] [Google Scholar]
- 28. Okugawa Y, Toiyama Y, Inoue Y, Iwata T, Fujikawa H et al. (2012) Clinical Significance of Serum Soluble E-cadherin in Colorectal Carcinoma. J Surg Res 175: e67-e73. doi:10.1016/j.jss.2011.11.009. PubMed: 22277332. [DOI] [PubMed] [Google Scholar]
- 29. Weiß JV, Klein-Scory S, Kübler S, Reinacher-Schick A, Stricker I et al. (2011) Soluble E-cadherin as a serum biomarker candidate: Elevated levels in patients with late-stage colorectal carcinoma and FAP. Int J Cancer 128: 1384-1392. doi:10.1002/ijc.25438. PubMed: 20473926. [DOI] [PubMed] [Google Scholar]
- 30. Matsumoto K, Shariat SF, Casella R, Wheeler TM, Slawin KM et al. (2003) Preoperative Plasma Soluble E-Cadherin Predicts Metastases to Lymph Nodes and Prognosis in Patients Undergoing Radical Cystectomy. J Urol 170: 2248-2252. doi:10.1097/01.ju.0000094189.93805.17. PubMed: 14634390. [DOI] [PubMed] [Google Scholar]
- 31. Jaggi M, Rao PS, Smith DJ, Hemstreet GP, Balaji KC (2003) Protein kinase C μ is down-regulated in androgen-independent prostate cancer. Biochem Biophys Res Commun 307: 254-260. doi:10.1016/S0006-291X(03)01161-6. PubMed: 12859948. [DOI] [PubMed] [Google Scholar]
- 32. Khanna O, Moya ML, Opara EC, Brey EM (2010) Synthesis of multilayered alginate microcapsules for the sustained release of fibroblast growth factor-1. J Biomed Mater Res A 95A: 632-640. doi:10.1002/jbm.a.32883. PubMed: 20725969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tendulkar S, Mirmalek-Sani SH, Childers C, Saul J, Opara EC et al. (2012) A three-dimensional microfluidic approach to scaling up microencapsulation of cells. Biomed Microdevices 14: 461-469. doi:10.1007/s10544-011-9623-6. PubMed: 22245953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bi X, Pohl NM, Qian Z, Yang GR, Gou Y et al. (2012) Decorin-mediated inhibition of colorectal cancer growth and migration is associated with E-cadherin in vitro and in mice. Carcinogenesis 33: 326-330. doi:10.1093/carcin/bgr293. PubMed: 22159220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Choi DS, Lee JM, Park GW, Lim HW, Bang JY et al. (2007) Proteomic Analysis of Microvesicles Derived from Human Colorectal Cancer Cells. J Proteome Res 6: 4646-4655. doi:10.1021/pr070192y. PubMed: 17956143. [DOI] [PubMed] [Google Scholar]
- 36. Segura E, Nicco C, Lombard B, Véron P, Raposo G et al. (2005) ICAM-1 on exosomes from mature dendritic cells is critical for efficient naive T-cell priming. Blood 106: 216-223. doi:10.1182/blood-2005-01-0220. PubMed: 15790784. [DOI] [PubMed] [Google Scholar]
- 37. Shoichet MS, Li RH, White ML, Winn SR (1996) Stability of hydrogels used in cell encapsulation: An in vitro comparison of alginate and agarose. Biotechnol Bioeng 50: 374-381. doi:10.1002/(SICI)1097-0290(19960520)50:4. PubMed: 18626986. [DOI] [PubMed] [Google Scholar]
- 38. Kassen A, Sutkowski DM, Ahn H, Sensibar JA, Kozlowski JM et al. (1996) Stromal cells of the human prostate: Initial isolation and characterization. Prostate 28: 89-97. doi:10.1002/(SICI)1097-0045(199602)28:2. PubMed: 8604397. [DOI] [PubMed] [Google Scholar]
- 39. Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD et al. (1999) Carcinoma-associated Fibroblasts Direct Tumor Progression of Initiated Human Prostatic Epithelium. Cancer Res 59: 5002-5011. PubMed: 10519415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chung LWK (2003) Prostate carcinoma bone-stroma interaction and its biologic and therapeutic implications. Cancer 97: 772-778. doi:10.1002/cncr.11140. PubMed: 12548574. [DOI] [PubMed] [Google Scholar]
- 41. Hwang RF, Moore TT, Hattersley MM, Scarpitti M, Yang B et al. (2012) Inhibition of the Hedgehog Pathway Targets the Tumor-Associated Stroma in Pancreatic Cancer. Mol Cancer Res, 10: 1147–57. PubMed: 22859707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Giatromanolaki A, Koukourakis MI, Koutsopoulos A, Mendrinos S, Sivridis E (2012) The metabolic interactions between tumor cells and tumor-associated stroma (TAS) in prostatic cancer. Cancer Biol Ther 13: 0--1. PubMed: 22895074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Henke A, Grace OC, Ashley GR, Stewart GD, Riddick ACP et al. (2012) Stromal Expression of Decorin, Semaphorin6D, SPARC, Sprouty1 and Tsukushi in Developing Prostate and Decreased Levels of Decorin in Prostate Cancer. PLOS ONE 7: e42516. doi:10.1371/journal.pone.0042516. PubMed: 22880013. [DOI] [PMC free article] [PubMed] [Google Scholar]