

Abstract
Nitric oxide synthases (NOSs) synthesize nitric oxide (NO), a signaling molecule, from l-arginine, utilizing electrons from NADPH. NOSs are flavo-hemo proteins, with two flavin molecules (FAD and FMN) and one heme per monomer, which require the binding of calcium/calmodulin (Ca2+/CaM) to produce NO. It is therefore important to understand the molecular factors influencing CaM binding from a structure/function perspective. A crystal structure of the CaM-bound iNOS FMN-binding domain predicted a salt bridge between R536 of human iNOS and E47 of CaM. To characterize the interaction between the homologous Arg of rat nNOS (R753) and murine iNOS (R530) with CaM, the Arg was mutated to Ala and, in iNOS, to Glu. The mutation weakens the interaction between nNOS and CaM, decreasing affinity by ∼3-fold. The rate of electron transfer from FMN is greatly attenuated; however, little effect on electron transfer from FAD is observed. The mutated proteins showed reduced FMN binding, from 20% to 60%, suggesting an influence of this residue on FMN incorporation. The weakened FMN binding may be due to conformational changes caused by the arginine mutation. Our data show that this Arg residue plays an important role in CaM binding and influences FMN binding.
Keywords: Nitric oxide synthase, Calmodulin, Reductase, Flavoprotein, Electron transfer
Introduction
Nitric oxide synthases (NOSs)3 catalyze the conversion of l-arginine to l-citrulline and the signaling molecule nitric oxide (NO) using NADPH as the electron donor [1]. There are three known isoforms of NOS, neuronal NOS (nNOS), endothelial NOS (eNOS) and inducible NOS (iNOS) transcribed from three different genes. NOSs have two domains, the N-terminal oxygenase or heme domain, where catalysis occurs, and a C-terminal reductase or flavin domain with two flavin co-factors, flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN), which transfer electrons from NAPDH to the heme domain. NO plays critical roles in both physiological and pathological situations, necessitating that its production be highly regulated. Inter- and intra-molecular electron transfer in nitric oxide synthases (NOSs) is regulated by both intrinsic and extrinsic factors. Regulatory subdomains present in the enzyme, such as the autoregulatory insert (AR), the C-terminal tail (CT) and the beta loop, control the flow of electrons within the NOS protein [2]. Interactions of NOS with other proteins also influence electron flow, the most notable of which is its interaction with calmodulin (CaM), an obligatory step for the production of NO [3,4]. CaM interaction with nNOS and eNOS is dependent on intracellular Ca2+ levels, whereas it is bound to iNOS at very low Ca2+ levels, independent of Ca2+ influx.
The interaction of NOS with CaM triggers a series of structural rearrangements in NOS, positioning various co-factors in an optimum state for efficient electron transfer from the reductase domain to the heme domain for NO production [1,2,5]. The CaM binding site of NOSs interacts with the EF hands of CaM (the helix-loop-helix structures) in an antiparallel orientation [6,7]. The binding of Ca2+ to CaM, which allows CaM to interact with NOSs, is cooperative [8]. As the affinities of different lobes of CaM to calcium are different, calcium concentration dictates which lobe(s) interact with NOS. For example, the C-terminal lobe interacts with nNOS first, but binding of the N-terminal lobe is necessary for the enzyme activation [9,10]. Direct interaction between CaM and the small insertion (SI) in the reductase hinge region and autoregulatory regions of NOSs has been proposed, based on various NOS constructs and catalytic measurements. However, this interaction appears to be quite limited and direct interaction between the carbon backbones, at least in the closed conformation, seems to be restricted to a single arginine residue, which is conserved throughout the NOSs [11]. It has been proposed that the AR element in the FMN domain of the constitutive NOSs acts as a conformation-stabilizing element, stabilizing the closed conformation in the absence of NADPH oxidation by binding to the CT or a nearby region [3]. When NADPH is oxidized, the CT shifts [12], breaking the contact of the AR on the FAD domain and freeing it to stabilize the open conformation potentially by interacting directly with CaM [3]. However, as no structure has been solved of the complete CaM-bound reductase domain of either of the constitutive NOSs, direct proof of such interactions is still elusive.
The structure of the human iNOS-FMN domain bound to CaM reported by Xia et al. [11] sheds light for the first time on the interacting residues between NOS and CaM [11]. Most notable is the salt bridge between iNOS Arg536, which is conserved across isoforms and species, and Glu47 of CaM. Arg536 also makes hydrogen bonds with the main chain carbonyl oxygens of Ser562, Cys563, Ala564, and Phe565 of the iNOS FMN domain, as well as with Asn42 of CaM. It was proposed that Arg536 in the iNOS FMN domain, along with its salt bridge with Glu47 of CaM, acts as a hinge around which the two domains (FMN- and CaM-binding) of iNOS swivel, at least partially enabling the structural movement of the FMN domain necessary for catalysis to occur. A mutational study involving the homologous Arg752 residue of nNOS and Glu47 of CaM endorsed the pivoting motion of CaM and NOS around the conserved arginine of NOSs to orient FAD, FMN and heme for efficient electron transfer [13]. Furthermore, the apparent Kd of the NOS and CaM interaction was unaffected in these studies by the mutation of Arg752 of nNOS or Glu47 of CaM, as measured by rates of NO produced at different CaM concentrations [13].
Herein, we report that mutation of the conserved arginine residue in reductase domain (red) constructs of rat nNOS (R753A) and murine iNOS (R530A/E; mouse R530 is analogous to human R536) leads to destabilization of FMN binding. The reductase domain was used to elucidate the role of this conserved arginine residue as it allowed us to draw conclusions specific to the domain and its interacting partner, CaM, without interference from the heme domain. The structural alteration triggered by the mutation does not allow for reconstitution of the enzyme with FMN either permanently or transiently, as judged by catalytic measurements. This FMN depletion is much more severe in the case of nNOSred than in iNOSred, which is co-expressed with CaM, perhaps indicating a role for CaM in stabilization or incorporation of FMN in nNOS. We also report the direct binding constant for the nNOSred/CaM interaction derived from analytical ultracentrifugation. Thus, the conserved arginine plays a role in FMN binding and its mutation weakens, but does not obliterate, the nNOSred interaction with CaM, suggesting that other interactions remain intact.
Experimental procedures
Clone generation and protein purification
Plasmids
The plasmids nNOSpCW and iNOSpCW were constructed by Roman et al. (1995) [14] and Roman et al. (2000) [15] using pCWori+[16]. The plasmid encoding calmodulin, CaM pACMIP, was kindly provided by Anthony Persechini of the University of Missouri – Kansas City.
Recombinant DNA manipulations
iNOSred R530A, R530E, and nNOSred R753A mutations were created using a nested PCR technique. Primers were designed to amplify the regions of recombinant iNOS and nNOS from the start of the CaM binding site, incorporating a 6-His sequence just after the initiating ATG, to the mutation (PCR #1), and from the mutation to the STOP codon (PCR #2). Products from this amplification were then used as template for a third PCR reaction (PCR #3), spanning the entire reductase domain sequence.
The primers used were:
iNOSred R530A PCR#1: Upstream – 5′ GGA GGT CAT ATG GCT CAC CAC CAC CAC CAC CAC AAG CTG AGG CCC AGG AGG; Downstream – 5′ GAG GAC TGT GGC TCT GAC CGC TGA AGC CAT GAC CTT TCG.
iNOSred R530A PCR#2: Upstream – 5′ CGA AAG GTC ATG GCT TCA GCG GTC AGA GC CACA GTC CTC; Downstream – 5′ TCA TCG ATA AGC TTA GAG ACG CGT.
iNOSred R530A PCR#3: Upstream – 5′ GGA GGT CAT ATG GCT CAC CAC CAC CAC CAC CAC AAG CTG AGG CCC AGG AGG; Downstream – 5′ TCA TCG ATA AGC TTA GAG ACG CGT.
iNOSred R530E PCR#1: Upstream – 5′ GGA GGT CAT ATG GCT CAC CAC CAC CAC CAC CAC AAG CTG AGG CCC AGG AGG; Downstream – 5′ GAG GAC TGT GGC TCT GAC TTC TGA AGC CAT GAC CTT TCG.
iNOSred R530E PCR#2: Upstream – 5′ CGA AAG GTC ATG GCT TCA GAA GTC AGA GC CACA GTC CTC; Downstream – 5′ TCA TCG ATA AGC TTA GAG ACG CGT.
iNOSred R530E PCR#3: Upstream – 5′ GGA GGT CAT ATG GCT CAC CAC CAC CAC CAC CAC AAG CTG AGG CCC AGG AGG; Downstream – 5′ TCA TCG ATA AGC TTA GAG ACG CGT.
nNOSred R753A PCR#1: Upstream – 5′ TAC GTA CAT ATG CAC CAC CAC CAC CAC CAC AAA CGG CGA GCT ATC GGC; Downstream – 5′ GAG AAT GGT CGC CTT GAC AGC CTT GGC CAT GGC CTG CCC.
nNOSred R753A PCR#2: Upstream – 5′ GGG CAG GCC ATG GCC AAG GCT GTC AAG GCG ACC ATT CTC; Downstream – 5′ GTC GAC TCT AGA TTA GGA GCT GAA.
nNOSred R753A PCR#3: Upstream – 5′ TAC GTA CAT ATG CAC CAC CAC CAC CAC CAC AAA CGG CGA GCT ATC GGC; Downstream – 5′ GTC GAC TCT AGA TTA GGA GCT GAA.
The resultant product is a NOS reductase domain coding sequence with an N-terminal 6-His tag and the indicated mutation. The PCR products from PCR #3 were restriction digested with Ndel/Xbal (nNOS) or Ndel/HindIII (iNOS) and ligated with Ndel/Xbal- or Ndel/HindIII-digested pCW vector, respectively. This ligation was used to transform XL10-gold cells (Stratagene) and colonies were screened by restriction digest. The correct construct was then used to transform E. coli BL21 cells.
Purification
Transformed BL21 cells with the plasmid were grown to an O.D.600nm of 0.8–1 prior to induction of protein expression by an induction cocktail containing 0.25 mM IPTG and riboflavin. Cells were grown at room temperature (22 °C–24 °C) for 24 h in the dark. Harvested cells were suspended in buffer with lysozyme (100 μg/ml) and protease inhibitor cocktail prior to sonication. Sonicated cells were centrifuged at 32,000 rpm for 1 h and the supernatant was collected and loaded onto a 2′5′-ADP-Sepharose column. Protein was eluted with 5 mM 2′ AMP after extensive washing on the column. Eluted protein was loaded onto a Ni–NTA column for further purification and eluted with 100 mM imidazole. Protein eluted from the Ni-column was dialyzed extensively to eliminate residual imidazole prior to its use in experiments. Notably, the nNOSred R753A protein was less stable than the other reductase domain constructs as evidenced by its proclivity to precipitation.
Activity measurements
Steady state kinetic measurements of reduction of external electron acceptors such as cytochrome c and K3Fe(CN)6 as well as NADPH oxidation were performed following published protocols [17].
Flavin content measurement
Flavin adenine nucleotide (FAD) and flavin mononucleotide (FMN) contents of proteins were measured using HPLC as described previously [18]. In short, proteins of known concentrations were boiled for 5 min. to release the flavin, and the protein was precipitated by immediate chilling. Precipitated proteins were removed by centrifugation for 10 min at high speed (17,000g). Aliquots (10 μl) of the supernatant containing the released flavins were analyzed using a Hewlett–Packard (Palo Alto, CA) Series 1100 high-performance liquid chromatography (HPLC) equipped with a G1316A diode-array detector. Flavins (FAD and FMN) were separated using a Hypersil Gold column (Dimension: 150 × 2.1 mm, 5 μm) from Thermo Fisher Scientific (Waltham, MA), fitted with a guard column packed with Nova-Pak C18 Guard-Pak inserts (Waters, Milford, MA). Integration and analysis were performed using Agilent Technologies (Santa Clara, CA) ChemStation software (Rev. B.03.02).
Modification of CaM by Alexa Fluor 488®
CaM was fluorescently-labeled with Alexa Fluor488 (Cat # A20000, Molecular Probes, Eugene, OR) using the vendor-supplied protocol. In short, 1 mg of CaM was mixed well with 10 μl of Alexa Fluor (10 mg/ml, in dimethylformamide) and incubated on ice for 4 h in the dark. Protein was dialyzed twice (1.5 h each) against 0.1 M sodium bicarbonate, 3 mM DTT, pH 8.3 to remove unbound fluorophore prior to binding experiments using analytical ultracentrifugation.
Binding measurements using analytical ultracentrifugation (AUC)
All sedimentation experiments were performed with a Beckman Optima XL-I at the Center for Analytical Ultracentrifugation of Macromolecular Assemblies at the University of Texas Health Science Center at San Antonio. Sedimentation velocity data were analyzed with the UltraScan-II software [19] version 9.9 [20]. All calculations were performed on the Lonestar and Ranger clusters at the Texas Advanced Computing Center at the University of Texas at Austin, and on the Jacinto cluster in the Bioinformatics Core Facility at the University of Texas Health Science Center at San Antonio. All measurements were made with the Aviv fluorescence detector with 488 nm excitation in a buffer containing 50 mM Tris–HCl, pH 7.5, 500 mM NaCl, 2 mM CaCl2, and 270 mM glycerol (2%). The experimental data were collected at 20 °C, and at 50,000 rpm, using standard epon 2-channel centerpieces. Hydrodynamic corrections for buffer density, viscosity and partial specific volume were made according to methods outlined in Laue et al. [21] and as implemented in UltraScan [19]. All data were first analyzed by 2-dimensional spectrum analysis (2DSA) [22]with simultaneous removal of time- and radial-invariant noise and fitting of the meniscus position, followed by van Holde–Weischet analysis [23] and a combined 2DSA-Monte Carlo analysis (MC) [24]. The van Holde–Weischet approach provides sedimentation coefficient distributions from which an assessment of composition can be obtained. The 2DSA and GA analyses further provide molecular weight and anisotropy for any species found in the mixture. From the known amount of CaM and nNOSred, and the results obtained from the SV analysis, precise molar amounts of free CaM, free nNOS and the amount of complex can be derived.
Results
Protein expression and purification
Both the wild-type (wt) and mutant proteins were expressed in E. coli and purified to virtually 100% homogeneity as analyzed by SDS PAGE (Fig. 1(a)). This level of purity was achieved by employing two affinity columns: one utilizing the NADPH binding site on the C-terminus and the other using an engineered His-tag at the N-terminal, thus insuring that both ends of the protein were intact. The iNOSred proteins were co-expressed with CaM, and presence of CaM in the purified protein can be seen as a low molecular weight band (Fig. 1(a)). Spectral analysis was also performed on these purified proteins using a UV-Vis spectrophotometer. Spectra of all purified proteins were taken between 300 nm and 600 nm to examine the maxima for flavins at 455 nm and other signatures of NOS reductase-bound flavins. Both wt and mutant proteins showed similar characteristic peaks. Spectra of nNOSred wt and R753A are presented to show the similarities between the wt and mutant protein spectra (Fig. 1(b)). Purity of these proteins was an important factor in downstream solution binding experiments.
Fig. 1.
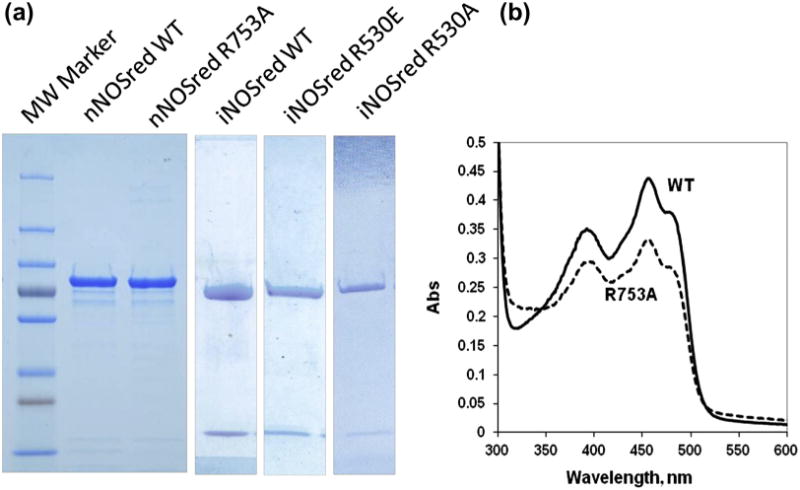
Characterization of purified proteins. (a) SDS PAGE analyses were performed for purified nNOSred and iNOSred wt proteins as well as arginine mutants. One primary band was present in both nNOSred wt and nNOSred R753A. Two bands are present for iNOSred wt, iNOSred R530E and iNOSred R530A. The higher molecular weight band is the reductase domain and the lower molecular weight band is CaM (iNOSred proteins were co-expressed with CaM). (b) Visible spectra of nNOSred wt and nNOSred R753A were taken between 300 nm and 600 nm, showing no difference in the maxima attributed to flavins between the wt and the mutant.
Flavin complementation of purified proteins
The integrity of the proteins and the effect of the mutations were assessed by measuring the flavin content using HPLC (Fig. 2). Mutation of the arginine residue in both nNOSred and iNOSred resulted in decreased binding of FMN. The loss of FMN is more pronounced in nNOSred R753A, which is only 30% replete as wt, than in either the Ala or Glu mutant of iNOSred. The FMN in iNOSred R530A was 78% replete, whereas iNOSred R530E was only 55% replete as compared to the wt iNOSred. That iNOSred R530E shows less FMN content than that of iNOSred R530A mutant is most likely due to the charge reversal of the Arg to Glu mutation, which would presumably cause a larger distortion in structure than the mutation to Ala. The lower FMN content in the Arg mutants of both nNOSred and iNOSred suggests a possible link between FMN binding affinity and the arginine residue, which was shown to form a salt bridge with CaM and is within hydrogen bonding distance from other residues in the reductase domain itself [11].
Fig. 2.
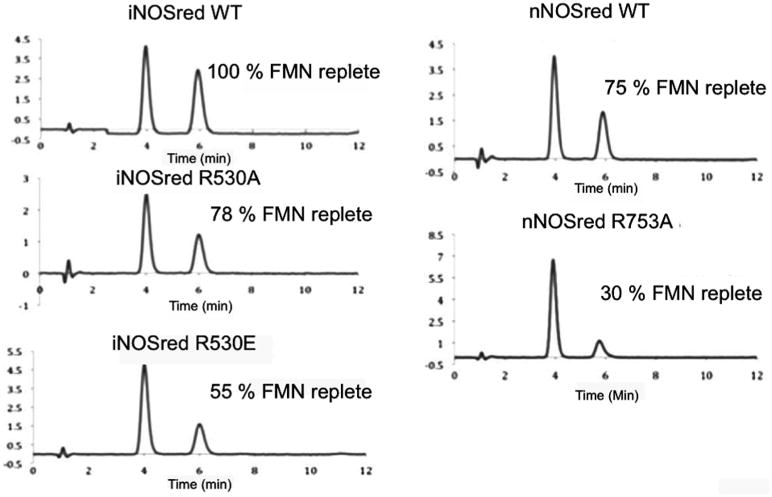
FAD and FMN content measurement: Protein-bound FAD and FMN content were measured using HPLC. Flavins were extracted from 50 μM of wt or arginine mutant preparations of both nNOSred and iNOSred by boiling and pelleting the precipitated protein by centrifugation. The supernatant, which contains the extracted flavins, was analyzed using HPLC (Experimental Procedures).
Steady-state kinetics measurements using external electron acceptors
Cytochrome c reduction
The rate of cytochrome c reduction was measured for wt iNOSred, nNOSred, and their arginine mutant proteins, both in the absence and presence of CaM (Fig. 3(a)). Cytochrome c is reduced by electrons transferred from the FMN cofactor, but not from the FAD cofactor, of these proteins. As expected, the addition of CaM to nNOSred wt increased the rate of cytochrome c reduction by at least 4-fold. As the FMN binding to the nNOSred R753A mutant is compromised, a much lower cytochrome c reduction activity was measured. No increase in the activity was monitored upon addition of CaM to the reaction mixture despite the observation that 30% of the enzyme was replete with flavins, suggesting that the binding of CaM or, at least, the structural changes in nNOSred that result from CaM binding, might also be affected by the R753A mutation.
Fig. 3.
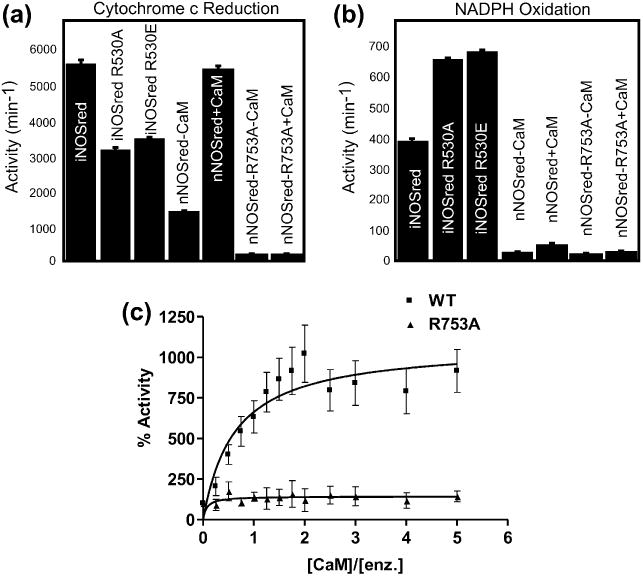
Rates of cytochrome c reduction and NADPH oxidation: (a) The rates of cytochrome c reduction by wt and arginine mutants for nNOSred and iNOSred were measured as an increase in absorbance at 550 nm. The reaction mixture contained 5 nM enzyme and 80 uM cytochrome c, and the reaction was initiated by the addition of 100 uM NADPH. Calmodulin (25 nM) was added to nNOSred preparations. (b) The rates of NADPH oxidation were monitored as a decrease in absorbance at 340 nm. (c) Rates of cytochrome c reduction by nNOSred wt and nNOSred R753A were measured at different CaM to enzyme ratios.
The rate of cytochrome c reduction by nNOSred wt and R753A proteins was measured with varying CaM concentrations to determine whether stimulation by CaM in the mutant could be rescued by increased CaM levels (Fig. 3(c)). Although nNOSred showed an optimum rate in cytochrome c reduction at a CaM concentration ∼2-fold higher than the enzyme, the nNOSred R753A mutant did not show any substantial increase in rate even at ∼50-fold excess of CaM (not shown in the graph). Similarly, both iNOSred R530A and iNOSred R530E demonstrated lower cytochrome c reductase activity compared to the iNOSred wt protein. nNOSred R753A protein was incubated with 10-fold excess FMN to determine whether cytochrome c reduction activity could be recovered (Fig. S1). That the flavin-incubated protein was unable to restore cytochrome c reductase activity suggests that the mutant protein cannot be reconstituted with FMN. FMN reconstitution was also attempted in the presence of CaM to determine whether CaM could facilitate FMN incorporation, but cytochrome c activity did not increase considerably in comparison with wt enzymes. As the nNOSred R753A mutant is significantly deficient in FMN content, it is hard to speculate about changes in the binding affinity of CaM from these steady state kinetic measurements. Therefore, direct measurement of CaM binding to the nNOSred was necessary and is described below. Potassium ferricyanide reduction typically measures electron flow from the FAD and thus is usually used to verify the integrity of the FAD/NADPH binding domain. No difference (or a small increase) was observed in the rate of ferricyanide reduction between wild type and the arginine mutants of both nNOSred and iNOSred (data not shown). Similar rates or increases in ferricyanide reduction rates by the mutants suggest that the electron flow in the FAD domain is most likely unaffected by Arg mutation. This is consistent with the flow of electrons to ferricyanide emanating from the FAD domain, supported by previous evidence [25] with the FAD-/NADPH-binding domain, and with the premise that the flavin-binding domains act independently except when they are brought into spatial contiguity. Furthermore, the flavin analysis also showed the FAD content to be the same between wild type and the Arg mutants of both nNOSred and iNOSred.
NADPH oxidation
The NOS reductase domain has been shown to reduce O2 using electrons from NADPH, so NADPH oxidation activity can be used as a measure of the tendency of NOS isoforms to form superoxide . The rate of NADPH oxidation was measured for nNOSred wt and nNOSred R753A, both in the absence and presence of CaM, as well as for iNOSred wt and its arginine mutants (R530A and R530E). The rate of NADPH oxidation by iNOSred wt is higher compared to nNOSred wt, as the rate of electron transfer in the iNOSred wt is known to be highest among NOS isoforms (Fig. 3(b)). Mutation of the arginine residue in iNOSred to either Ala or Glu increased the rate of NADPH oxidation even higher, thus increasing the rate of oxygen reduction. However, the R753A mutation in nNOSred reduced the rate of NADPH oxidation to the level of activity in the absence of CaM.
Analytical ultracentrifugation
To identify the relative binding strength of CaM to the nNOSred wt and the R753A mutant, sedimentation velocity (SV) experiments were performed in which nNOSred was titrated against a fixed amount of CaM that was fluorescently tagged with Alexa-488 as described [26]. Thus, when observed in the fluorescence detector of the analytical ultracentrifuge, only the CaM is visible. In the absence of any nNOSred, CaM sediments at 2S. If CaM binds to nNOSred, that portion of the labeled CaM complexed with nNOS will sediment at an increased sedimentation speed due to the increased molecular weight of the complex. Since only CaM is visible in the fluorescence optics, only CaM and complex are visible, and there is no background signal from free nNOSred. Since the nNOSred complex with CaM has a higher molecular weight than free CaM, it is easily distinguishable from free CaM, and Kd values can be readily calculated from the relative amounts.
A van Holde–Weischet integral distribution plot of SV experiments indicated an increase in the amount of complex formed with increasing titration of nNOSred (Fig. 4). The molar ratios examined overlapped the concentration of the Kd, since at the lowest ratio only free CaM was visible, while at the highest ratio, the CaM was essentially fully complexed (data not shown). An intermediate ratio (1:0.5 CaM:nNOSred) was chosen to derive the binding constant for this association according to the equation, Kd = ([Free CaM]* [Free nNOSred domain])/([CaM-nNOSred complex]). Amounts were determined by integration of the 2S species from 2DSA-Monte Carlo results, and indicated 64.6% free calmodulin for nNOS mutant R753A vs. 30.9% for wild-type nNOS. From these measurements, Kd values of 167 ± 82 nM for nNOSred/CaM and 468 ± 113 nM for mutant R753A/CaM were derived.
Fig. 4.

Analytical Ultracentrifugation data for CaM and nNOSred binding: van Holde-Weischet integral distribution graphs for CaM (2S) binding to nNOS-red (5S and above) for a 1:0.5 M ratio of CaM to nNOS. A clear difference in binding strength is evident from the relative amount of free CaM for wild type nNOS (grey circles, about 40% free CaM) versus mutant R753A nNOSred (black circles, about 65% free CaM). The loading concentration of CaM was 250 nM. The sedimentation coefficient reported is corrected to standard conditions at 20 °C and in water.
Discussion
The NOS reductase domain is absolutely essential due to its role in catalysis in regulating electron transfer from NADPH to the oxygenase domain. The NADPH-binding site present in the reductase domain selectively binds NADPH over NADH and regulates post-hydride transfer dissociation of NADP+[27,28]. The reduced FAD transfers one electron at a time to FMN and only fully reduced FMN swings away to interact with the oxygenase domain to facilitate electron transfer to the heme, the site of catalysis [29]. The binding of CaM also influences structural elements within the reductase domain, whose concerted movements also regulate electron flow [2,3]. The various structural elements present within constitutive NOS isoforms are the autoregulatory insert (AR), C-terminal tail (CT), small insertion (SI) in the FAD domain and a hinge region [2], iNOS has neither an AR nor an SI. These various elements in the reductase domain make it very flexible while, due to its critical nature, the domain is tightly coordinated. The electron densities of these regions are poorly defined in the published crystal structure of the nNOS reductase domain, confirming the flexible nature of these elements [30].
The structure of the iNOS FMN domain with bound CaM is the only published structure of the reductase domain showing interaction of CaM with any of the NOS isoforms [11]. This structure sheds light on possible interactions between NOS and CaM, showing that residues from both proteins form hydrogen bonds and a salt bridge. The Arg536 of iNOS forms a salt bridge with Glu47 of CaM, and it is suggested that this interaction forms a hinge between NOS and CaM allowing these proteins to pivot around the arginine residue [11]. The arginine residue is conserved among all NOS isoforms, suggesting that this interaction may also occur with the constitutive isoforms as well. Tejero et al. (2010) [13] examined the role of the homologous arginine of nNOS (Arg752) and the glutamate of CaM (Glu47) by mutating these two residues. Mutation of the conserved arginine residue to either glutamate or glutamine in the nNOS full-length protein obliterated CaM-dependent stimulation of cytochrome c reduction activity. The rate of cytochrome c reduction by the nNOS Arg753 mutants in the presence of wt CaM or by wt nNOS in the presence of CaM Glu47 mutants is higher compared to the cytochrome c reduction by nNOS wt only, i.e., in the absence of any CaM. An increase in the cytochrome c reduction rate in the presence of mutant CaM or mutant nNOS compared to nNOS wt in the absence of CaM raises the possibility of stimulation due to CaM interaction, albeit very weak binding. In other words, the residues of nNOS and CaM that play a role in the salt bridge interaction are not solely responsible for the nNOS and CaM interaction. In fact, binding measurements carried out between nNOS-red wt or nNOS-red Arg753Ala with wt CaM, using AUC, demonstrated that the mutant protein interacts with CaM with a Kd of 468 ± 113 nM compared to the wild type Kd of 167 ± 82 nM, indicating an almost 3-fold decrease in binding affinity of the mutant reductase for CaM.
The flexibility of the reductase domain is well documented, although its movement is very tightly regulated. It can be speculated that a mutation in this domain may affect the structural integrity of the protein. (Indeed, the nNOSred Arg753Ala mutant tended to be less stable than the wt nNOSred protein.) The role of the residue becomes more critical if it interacts with other residues within the protein through H-bonding. The iNOS FMN-CaM crystal structure ([11]; Fig. 1) showed that the conserved Arg residue not only forms a salt bridge with CaM, but also potentially forms several H-bonds with the main chain oxygen residues of Ser562, Cys563, Ala564 and Phe565 of the FMN domain. Mutation of this conserved arginine residue will affect this H-bonding network, and may trigger a backbone rearrangement causing the FMN to bind weakly. A mutation involving charge reversal compared to a neutral mutation would presumably have a larger impact on the protein structural integrity.
In the presence of CaM, the wild-type proteins have the iNOS R530 (nNOS R753) hinge, and the CaM-binding peptide bound to CaM is a rigid rod of ∼30 Å length. This rod is fixed at iNOS R530 (nNOSR753) and the movement of this rod is restricted because of the interactions between R530 and the backbones of S556, Y557, and F559. Since CaM is always bound to iNOS, even in the mutant R530A/E, the rod is still rigid and, presumably, does not disrupt the FMN binding to the protein. In the absence of CaM, as is the case in nNOS, the nNOS CaM-binding region has a relatively random structure and is flexible (i.e., no rigid rod). Since the wt peptide presumably maintains contacts with K778, H779 and F781 (analogous to S556, Y557, and F559 of iNOS) in the FMN domain, the peptide movement is limited, resulting in 75% bound FMN (less than that of iNOS). However, in the mutant nNOSred, the CaM-binding peptide, which is not bound to K778, H779 and F781, can move more freely (i.e., can reach to a wider range) and it may bind, most likely via hydrophobic interactions, as the peptide is very hydrophobic, near the phosphate moiety of FMN, resulting in a weaker FMN binding.
The data presented herein corroborate this conjecture, as the iNOSred Arg530Glu mutant showed decreased FMN binding affinity compared to iNOSred Arg530Ala mutation, although both the mutants showed lower FMN binding than iNOSred wt. The FMN content of nNOSred Arg753Ala was lower than that of both the iNOSred Arg530 mutants. The FMN content of the previously published work by Tejero et al. [13] did not seem to be affected by mutation of this arginine as inferred from UV-Vis spectral analysis of purified proteins. The difference may be attributed to the fact that Tejero et al. [13] used full-length enzyme, whereas the reductase domain was utilized in these studies. In this study, the reductase domain was used to elucidate the role of the conserved arginine residue, as it allowed us to draw conclusions specific to the domain and its interacting partner, CaM, without any potential interference from the heme domain in the measurements. Although UV-Vis spectral analyses of our mutant proteins did not reveal significant differences from wild-type, direct flavin measurements revealed that all mutant proteins were partially depleted of FMN.
Several mutations in cytochrome P450 oxidoreductase (POR), which has ∼58% sequence homology [31] and a high degree of structural homology with the NOS reductase domains [30], have been shown to destabilize the flavin incorporation into the protein to varying degrees [18,32–34]. POR Y181D, an FMN-deficient full-length POR mutant was able to recover its activity partially by exogenous addition of FMN; however, it could not retain the FMN [18]. The ability of flavin-deficient POR mutants to be reconstituted suggests that these mutations did not drastically affect the flavin binding site. It is also likely that the flavin binding sites of POR are more accessible, allowing added flavins to bind back, as POR lacks the various regulatory elements present in NOSs. However, the arginine mutant of nNOSred was not able to be reconstituted by the addition of FMN. In this case, arginine could be playing a structural role and, in addition, the structural alteration caused by the mutation may not provide access for FMN to bind back to the protein. Higher FMN content in the case of iNOSred arginine mutations compared to the nNOSred arginine mutant may suggest that FMN can bind, albeit more weakly, to the protein during translation and that further loss is prevented due to the compact nature of iNOSred and tight CaM binding. However, preincubation of nNOSred R753A with CaM did not allow for FMN reconstitution, measured by recovery of cytochrome c reduction activity, perhaps because CaM is not bound to the nascent protein under basal conditions.
These studies examine the critical, homologous Arg residue that forms a flexible hinge between the CaM-binding and FMN-binding domains of both murine iNOS and rat nNOS reductase domain constructs. Mutation of this residue to either Ala or Glu permitted the assessment of both CaM binding affinity and FMN avidity. The critical nature of this residue, first reported by Xia et al. [11], relates to its role in conformational integrity and in the stimulation of activity in the nNOS constructs. The compactness and tight affinity for CaM binding by iNOS suggests an influence on the internal conformation of this isoform that contrasts with that of nNOS, in which there is a profound inhibitory effect on cytochrome c reduction in the mutants, R753A and R753E, which the addition of CaM cannot rescue. The intrinsic factors that regulate the NOS isoforms contribute importantly to NOS function and, indeed, the extrinsic factors, i.e., CaM, have their effects through these structural elements. Understanding these factors is crucial to deciphering the ultimate function and regulation of NOS isoforms in vivo.
Supplementary Material
Acknowledgments
The authors wish to thank Dr. Jung-Ja Kim, Department of Biochemistry, Medical College of Wisconsin, Milwaukee, WI-53226 for her critical reading of the manuscript and suggestions. Supported by NIH GM052419 to BSSM and LJR. BSSM is the Robert A. Welch Distinguished Professor in Chemistry (AQ-0012). The development of the UltraScan software is supported by the National Institutes of Health through grant RR022200 (to BD). Supercomputer time allocations were provided through National Science Foundation grant TG-MCB070038 (to BD). We acknowledge the support of the San Antonio Cancer Institute grant P30 CA054174 for the Center for Analytical Ultracentrifugation of Macromolecular Assemblies at the University of Texas Health Science Center at San Antonio.
Footnotes
Abbreviations used: NOSs, Nitric oxide synthases; NO, nitric oxide; nNOS, neuronal NOS; eNOS, endothelial NOS; iNOS, inducible NOS; FAD, flavin adenine dinucleotide; FMN, flavin mononucleotide; CaM, calmodulin; HPLC, high-performance liquid chromatography; AUC, analytical ultracentrifugation; 2DSA, 2-dimensional spectrum analysis; SV, sedimentation velocity; AR, autoregulatory insert; CT, C-terminal tail; SI, small insertion.
Appendix A. Supplementary data: Supplementary data associated with this article can be found, in the online version, at http://dx.doi.Org/10.1016/j.abb.2013.03.004.
References
- 1.Feng C. Coord Chem Rev. 2012;256:393–411. doi: 10.1016/j.ccr.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roman LJ, Martasek P, Masters BS. Chem Rev. 2002;102:1179–1190. doi: 10.1021/cr000661e. [DOI] [PubMed] [Google Scholar]
- 3.Roman LJ, Masters BSS. J Biol Chem. 2006;281:23111–23118. doi: 10.1074/jbc.M603671200. [DOI] [PubMed] [Google Scholar]
- 4.Bredt DS, Snyder SH. Proc Natl Acad Sci USA. 1990;87:682–685. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daff S. Nitric Oxide. 2010;23:1–11. doi: 10.1016/j.niox.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Liu Q, Gross SS. Methods Enzymol. 1996;268:311–324. doi: 10.1016/s0076-6879(96)68033-1. [DOI] [PubMed] [Google Scholar]
- 7.Spratt DE, Taiakina V, Palmer M, Guillemette JG. Biochemistry. 2007;46:8288–8300. doi: 10.1021/bi062130b. [DOI] [PubMed] [Google Scholar]
- 8.Linse S, Chazin WJ. Protein Sci. 1995;4:1038–1044. doi: 10.1002/pro.5560040602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Persechini A, Gansz KJ, Paresi RJ. Biochemistry. 1996;35:224–228. doi: 10.1021/bi952383x. [DOI] [PubMed] [Google Scholar]
- 10.Spratt DE, Newman E, Mosher J, Ghosh DK, Salerno JC, Guillemette JG. FEBS J. 2006;273:1759–1771. doi: 10.1111/j.1742-4658.2006.05193.x. [DOI] [PubMed] [Google Scholar]
- 11.Xia C, Misra I, Iyanagi T, Kim JJ. J Biol Chem. 2009;284:30708–30717. doi: 10.1074/jbc.M109.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tiso M, Tejero J, Panda K, Aulak KS, Stuehr DJ. Biochemistry. 2007;46:14418–14428. doi: 10.1021/bi701646k. [DOI] [PubMed] [Google Scholar]
- 13.Tejero J, Haque MM, Durra D, Stuehr DJ. J Biol Chem. 2010;285:25941–25949. doi: 10.1074/jbc.M110.126797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roman LJ, Sheta EA, Martásek P, Gross SS, Liu Q, Masters BSS. Proc Natl Acad Sci USA. 1995;92:8428–8432. doi: 10.1073/pnas.92.18.8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roman LJ, Miller RT, de La Garza MA, Kim JJ, Masters BS. J Biol Chem. 2000;275:21914–21919. doi: 10.1074/jbc.M002449200. [DOI] [PubMed] [Google Scholar]
- 16.Gegner JA, Dahlquist FW. Proc Natl Acad Sci USA. 1991;88:750–754. doi: 10.1073/pnas.88.3.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martásek P, Miller RT, Roman LJ, Shea T, Masters BS. Methods Enzymol. 1999;301:70–78. doi: 10.1016/s0076-6879(99)01070-8. [DOI] [PubMed] [Google Scholar]
- 18.Marohnic CC, Panda SP, McCammon K, Rueff J, Masters BS, Kranendonk M. Drug Metab Dispos. 2010;38:332–340. doi: 10.1124/dmd.109.030445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Demeler B. In: Modern Analytical Ultracentrifugation. Scott DJ, Harding SE, Rowe AJ, editors. Techniques and Methods Royal Society of Chemistry; UK: 2005. pp. 210–219. [Google Scholar]
- 20.Demeler B. A comprehensive data analysis software package for analytical ultracentrifugation experiments Ultra Scan II, Dept of Biochemistry. University of Texas Health Science Center at San Antonio; San Antonio: 2011. [Google Scholar]
- 21.Laue TM, Shah BD, Ridgeway TM, Pelletier SL. In: Analytical Ultracentrifugation in Biochemistry and Polymer Science. Harding SE, Rowe AJ, Horton JC, editors. Royal Society of Chemistry; Cambridge: 1992. pp. 90–125. [Google Scholar]
- 22.Brookes E, Cao W, Demeler B. Eur Biophys J. 2010;39:405–414. doi: 10.1007/s00249-009-0413-5. [DOI] [PubMed] [Google Scholar]
- 23.Demeler B, van Holde KE. Anal Biochem. 2004;335:279–288. doi: 10.1016/j.ab.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 24.Demeler B, Brookes E. Colloid Polym Sci. 2008;286:129–137. [Google Scholar]
- 25.Zhang J, Martasek P, Paschke R, Shea T, Siler Masters BS, Kim JJ. J Biol Chem. 2001;276:37506–37513. doi: 10.1074/jbc.M105503200. [DOI] [PubMed] [Google Scholar]
- 26.Zhuo Y, Ilangovan U, Schirf V, Demeler B, Sousa R, Hinck AP, Lafer EM. J Mol Biol. 2010;404:274–290. doi: 10.1016/j.jmb.2010.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Craig DH, Chapman SK, Daff S. J Biol Chem. 2002;277:33987–33994. doi: 10.1074/jbc.M203118200. [DOI] [PubMed] [Google Scholar]
- 28.Daff S. Biochem Soc Trans. 2003;31:502–505. doi: 10.1042/bst0310502. [DOI] [PubMed] [Google Scholar]
- 29.Masters BS. Annu Rev Nutr. 1994;14:131–145. doi: 10.1146/annurev.nu.14.070194.001023. [DOI] [PubMed] [Google Scholar]
- 30.Garcin ED, Bruns CM, Lloyd SJ, Hosfield DJ, Tiso M, Gachhui R, Stuehr DJ, Tainer JA, Getzoff ED. J Biol Chem. 2004;279:37918–37927. doi: 10.1074/jbc.M406204200. [DOI] [PubMed] [Google Scholar]
- 31.Bredt DS, Hwang PM, Glatt CE, Lowenstein C, Reed RR, Snyder SH. Nature. 1991;351:714–718. doi: 10.1038/351714a0. [DOI] [PubMed] [Google Scholar]
- 32.Marohnic CC, Panda SP, Martasek P, Masters BS. J Biol Chem. 2006;281:35975–35982. doi: 10.1074/jbc.M607095200. [DOI] [PubMed] [Google Scholar]
- 33.Shen AL, Porter TD, Wilson TE, Kasper CB. J Biol Chem. 1989;264:7584–7589. [PubMed] [Google Scholar]
- 34.Xia C, Panda SP, Marohnic CC, Martasek P, Masters BS, Kim JJ. Proc Natl Acad Sci USA. 2011;108:13486–13491. doi: 10.1073/pnas.1106632108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.